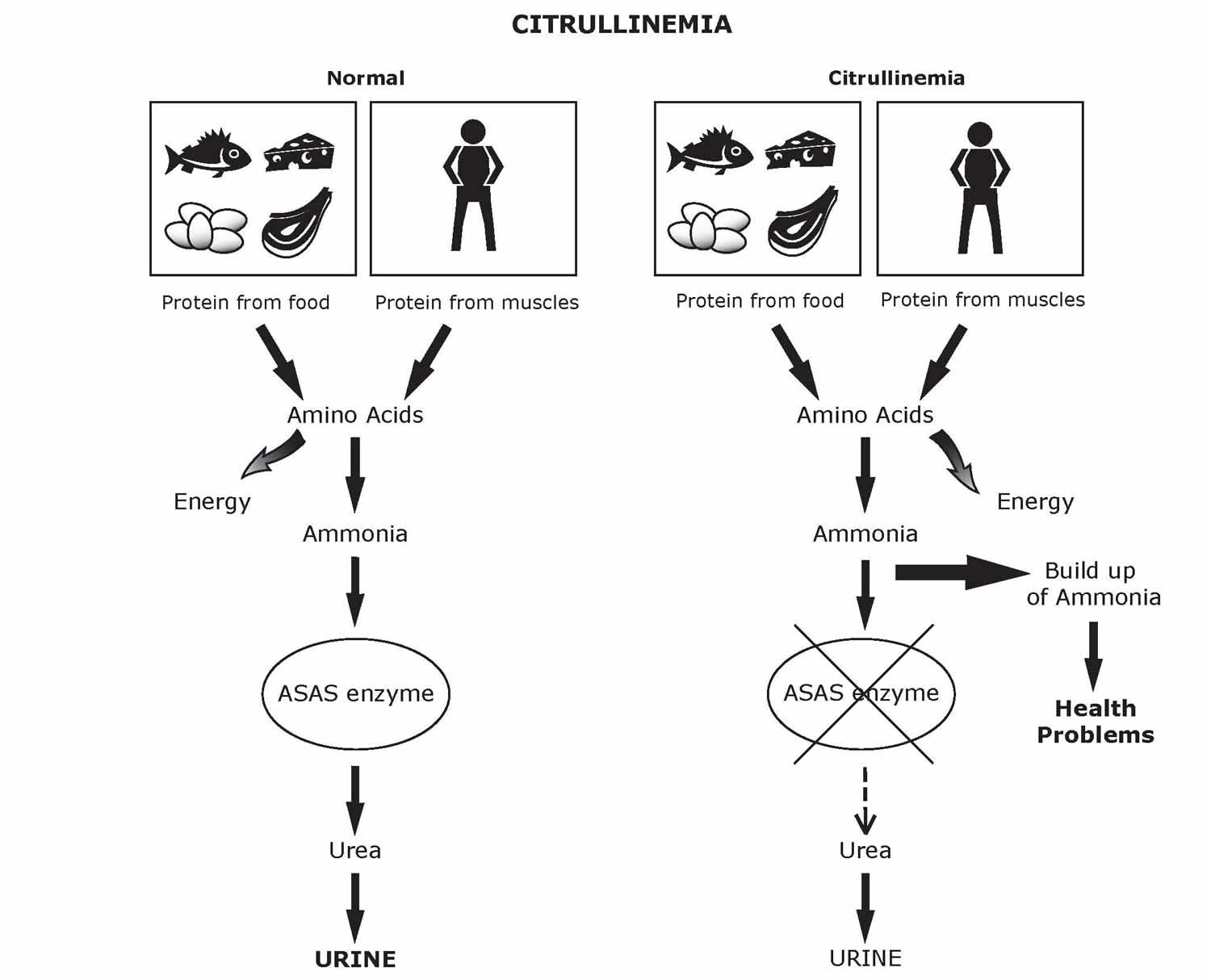
Citrullinemia
Citrullinemia is a rare inherited disorder that causes ammonia and other toxic substances to accumulate in the blood 1. Ammonia is a harmful substance. Ammonia is made when protein and its building blocks, amino acids, are broken down for use by the body. Normally, your body changes ammonia into a substance called “urea”. Urea is then safely removed in the urine. If ammonia is not changed to urea, high levels build up in the blood. This can be very harmful. If ammonia levels stay high for too long, severe brain damage can occur.
Because citrullinemia is caused by problems with the urea cycle, it belongs to a class of genetic diseases called urea cycle disorders.
Two types of citrullinemia have been described; they have different signs and symptoms and are caused by mutations in different genes.
Citrullinemia type 1 also known as classic citrullinemia, usually becomes evident in the first few days of life. Citrullinemia type 1 is the most common form of citrullinemia, affecting about 1 in 57,000 people worldwide. Affected infants typically appear normal at birth, but as ammonia builds up, they experience a progressive lack of energy (lethargy), poor feeding, vomiting, seizures, and loss of consciousness. Some affected individuals develop serious liver problems. The health problems associated with citrullinemia type 1 are life-threatening in many cases. Less commonly, a milder form of citrullinemia type 1 can develop later in childhood or adulthood. This later-onset form is associated with intense headaches, blind spots (scotomas), problems with balance and muscle coordination (ataxia), and lethargy. Some people with gene mutations that cause citrullinemia type 1 never experience signs and symptoms of the disorder.
Citrullinemia type 2 also known as adult-onset citrullinemia type 2, chiefly affects the nervous system, causing confusion, restlessness, memory loss, abnormal behaviors (such as aggression, irritability, and hyperactivity), seizures, and coma. These signs and symptoms can be life-threatening. The signs and symptoms appear during adulthood and are triggered by certain medications, infections, surgery, and alcohol intake. Citrullinemia type 2 is found primarily in the Japanese population, where it occurs in an estimated 1 in 100,000 to 230,000 individuals. Citrullinemia type 2 also has been reported in other populations, including other people from East Asia, the Middle East, the United States, and the United Kingdom. Affected individuals often have specific food preferences, preferring protein-rich and fatty foods and avoiding carbohydrate-rich foods. The signs and symptoms of this disorder typically appear during adulthood (adult-onset) and can be triggered by certain medications, infections, surgery, and alcohol intake. These signs and symptoms can be life-threatening in people with adult-onset citrullinemia type 2.
Adult-onset citrullinemia type 2 may also develop in people who as infants had a liver disorder called neonatal intrahepatic cholestasis caused by citrin deficiency. This liver condition is also known as neonatal-onset citrullinemia type 2. Neonatal intrahepatic cholestasis caused by citrin deficiency blocks the flow of bile (a digestive fluid produced by the liver) and prevents the body from processing certain nutrients properly. In many cases, the signs and symptoms of neonatal intrahepatic cholestasis caused by citrin deficiency go away within a year. In rare cases, affected individuals develop other signs and symptoms in early childhood after seeming to recover from neonatal intrahepatic cholestasis caused by citrin deficiency, including delayed growth, extreme tiredness (fatigue), specific food preferences (mentioned above), and abnormal amounts of fats (lipids) in the blood (dyslipidemia). This condition is known as failure to thrive and dyslipidemia caused by citrin deficiency. Years or even decades later, some people with neonatal intrahepatic cholestasis caused by citrin deficiency or failure to thrive and dyslipidemia caused by citrin deficiency develop the features of adult-onset citrullinemia type 2.
Citrullinemia type 1
Citrullinemia type 1 also known as classic citrullinemia or argininosuccinate synthetase deficiency, is a rare autosomal recessive genetic disorder that includes a neonatal acute (classic) citrullinemia, a milder late-onset citrullinemia, a form that begins during or after pregnancy, and an asymptomatic citrullinemia. Citrullinemia type 1 occurs in approximately 1 per 57,000 births.
Citrullinemia type 1 is caused by deficiency or absence of the enzyme argininosuccinate synthetase. Argininosuccinate synthetase is one of six enzymes that play a role in the removal of nitrogen from the body, a process known as the urea cycle. The lack of this enzyme results in excessive accumulation of nitrogen, in the form of ammonia (hyperammonemia), in the blood and all body fluids.
Infants with the classic form may experience vomiting, refusal to eat, progressive lethargy, and show signs of increased intracranial pressure. Prompt treatment can prolong survival, but neurologic deficits are usually present. The course of the late-onset form is sometimes milder but episodes of hyperammonemia are similar to the classic form.
Citrullinemia type 1 causes
Citrullinemia type 1 is caused by mutations in the ASS1 gene that is responsible for production of the argininosuccinate synthetase enzyme. The symptoms of citrullinemia type 1 develop due to deficiency of this enzyme, which is needed to detoxify ammonia in the body. Failure to properly remove ammonia via synthesis of urea leads to the abnormal accumulation of ammonia in the blood (hyperammonemia).
Citrullinemia type 1 is inherited as an autosomal recessive genetic condition. Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
All individuals carry 4-5 abnormal genes. Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
Citrullinemia type 1 symptoms
The severity of citrullinemia type 1 varies from patient to patient. The classic form, characterized by profound lack of argininosuccinate synthetase enzyme activity, displays symptoms shortly after birth (neonatal period). A milder form of the disorder, which is characterized by partial lack of the argininosuccinate synthetase enzyme, affects some infants later during infancy or childhood.
The symptoms of citrullinemia type 1 are caused by the accumulation of ammonia in the blood and cerebrospinal fluid (CSF). The classic form occurs within 24-72 hours after birth, usually following a protein feeding and is initially characterized by refusal to eat, lethargy, lack of appetite, vomiting, and irritability. Affected infants may also experience seizures, diminished muscle tone (hypotonia), respiratory distress, accumulation of fluid in the brain (cerebral edema), and liver failure.
If untreated, citrullinemia type 1 may progress to coma due to high levels of ammonia in the CSF (hyperammonemic coma). Neurological abnormalities including developmental delays, intellectual disability, and cerebral palsy may occur and are more severe in infants who are in hyperammonemic coma for more than three days. Increased intracranial pressure can result in increased muscle tone, spasticity, abnormal reflex movements of the foot (ankle clonus), and seizures. If left untreated, the disorder will result in life-threatening complications.
In some patients, including those with partial enzyme deficiency, onset of the disorder may not occur until later during infancy or childhood. Symptoms may include failure to grow and gain weight at the expected rate (failure to thrive), avoidance of high-protein foods from the diet, inability to coordinate voluntary movements (ataxia), progressive lethargy, and vomiting. Infants with the mild form may alternate between periods of wellness and hyperammonemia. Infants and children with this form of citrullinemia type 1 may also develop hyperammonemic coma and life-threatening complications.
Another form of citrullinemia type 1 occurs during and after pregnancy. Affected women may experience repeated episodes of vomiting, lethargy, seizures, confusion, hallucinations, and potentially coma. Behavioral changes may also occur including manic episodes and psychosis. Affected women may also have accumulation of fluid in the brain (cerebral edema).
Some individuals with citrullinemia type 1 do not experience symptoms or hyperammonemia. The basis for these milder variants is not established.
Citrullinemia type 1 diagnosis
A diagnosis of citrullinemia can be confirmed by a detailed patient/family history, identification of characteristic findings, and a variety of specialized tests. Excessive amounts of ammonia and citrulline in the blood strongly suggests the diagnosis of citrullinemia type 1. Molecular genetic testing for ASS1 gene mutations is available to confirm the diagnosis.
Citrullinemia type 1 may also be diagnosed through newborn screening programs. Citrulline can be measured on the newborn blood spot by tandem mass spectroscopy. Every state in the U.S. screens every newborn for citrullinemia type 1. Early detection is important because prompt identification and treatment may prevent the hyperamonnemia that causes brain damage.
Carrier testing and prenatal diagnosis are available if the specific gene-causing mutation has been identified in the family.
Siblings of an affected child should be tested immediately after birth and those with elevated ammonia or citrulline should receive a low protein diet.
Citrullinemia type 1 treatment
Treatment of an individual with citrullinemia type 1 requires the coordinated efforts of a team of specialists. Biochemical geneticists, pediatricians, neurologists, and dieticians, are needed to work together to ensure a comprehensive approach to treatment. Management involves prompt diagnosis, control of hyperammonemia and control of intracranial pressure.
Prompt treatment is necessary when individuals have extremely high ammonia levels (severe hyperammonemic episode). Prompt treatment can avoid hyperammonemic coma and associated neurological symptoms. However, in some cases, especially those with complete enzyme deficiency, prompt treatment will not prevent recurrent episodes of hyperammonemia and the potential development of serious complications.
The treatment of citrullinemia type 1 is aimed at preventing excessive ammonia from being formed or for removing excessive ammonia during a hyperammonemic episode. Medications that assist in the removal of nitrogen from the body by providing an alternative means of waste nitrogen removal are employed. Available medications are Buphenyl, Ammonul, and Raviciti, as well as arginine.
Dietary restrictions are aimed at limiting the amount of protein intake to avoid the development of excess ammonia. However, enough protein must be taken in by an affected infant to ensure proper growth. Infants are placed a low protein, high calorie diet supplemented by essential amino acids. A combination of a high biological value natural protein such as breast milk or cow’s milk formulate, an essential amino acid formula, and a calorie supplement without protein is often used.
Multiple vitamins and calcium supplements may also be used.
Aggressive treatment including hospitalization and protein restriction is needed in hyperammonemic episodes that have progressed to vomiting and increased lethargy. Affected individuals may also receive treatment with intravenous administration of arginine and a combination of sodium benzoate and sodium phenylacetate. Non-protein calories may be also provided as glucose.
In cases where there is no improvement or in cases where hyperammonemic coma develops, the removal of ammonia by filtering an affected individual’s blood through a machine (hemodialysis) may be necessary. Hemodialysis is also used to treat infants, children, and adults who are first diagnosed with citrullinemia type 1 during hyperammonemic coma.
Affected children should be monitored to prevent increased intracranial pressure and to anticipate the onset of a hyperammonemic episode. Warning signs include mood changes, headaches, lethargy, nausea, vomiting, refusal to eat, and ankle clonus. Affected individuals should receive periodic blood tests to determine the level of ammonia in the blood and to determine the concentration of plasma amino acids to assist in the management of the protein restricted diet. Detection of elevated levels of ammonia may allow treatment before clinical symptoms appear.
Liver transplantation has been reported to improve quality of life and prolong survival in some patients.
Citrullinemia type 2
Citrullinemia type 2 also known as adult-onset citrullinemia type 2, chiefly affects the nervous system, causing confusion, restlessness, memory loss, abnormal behaviors (such as aggression, irritability, and hyperactivity), seizures, and coma. These signs and symptoms can be life-threatening. The signs and symptoms appear during adulthood and are triggered by certain medications, infections, surgery, and alcohol intake. Adult-onset citrullinemia type 2 is found primarily in the Japanese population, where it occurs in an estimated 1 in 100,000 to 230,000 individuals. Adult-onset citrullinemia type 2 also has been reported in other populations, including other people from East Asia, the Middle East, the United States, and the United Kingdom. Affected individuals often have specific food preferences, preferring protein-rich and fatty foods and avoiding carbohydrate-rich foods. The signs and symptoms of this disorder typically appear during adulthood (adult-onset) and can be triggered by certain medications, infections, surgery, and alcohol intake. These signs and symptoms can be life-threatening in people with adult-onset citrullinemia type 2.
Adult-onset citrullinemia type 2 may also develop in people who as infants had a liver disorder called neonatal intrahepatic cholestasis caused by citrin deficiency. This liver condition is also known as neonatal-onset citrullinemia type 2. Neonatal intrahepatic cholestasis caused by citrin deficiency blocks the flow of bile (a digestive fluid produced by the liver) and prevents the body from processing certain nutrients properly. In many cases, the signs and symptoms of neonatal intrahepatic cholestasis caused by citrin deficiency go away within a year. In rare cases, years or even decades later, affected individuals develop other signs and symptoms in early childhood after seeming to recover from neonatal intrahepatic cholestasis caused by citrin deficiency, including delayed growth, extreme tiredness (fatigue), specific food preferences (mentioned above), and abnormal amounts of fats (lipids) in the blood (dyslipidemia). This condition is known as failure to thrive and dyslipidemia caused by citrin deficiency. Years or even decades later, some people with neonatal intrahepatic cholestasis caused by citrin deficiency or failure to thrive and dyslipidemia caused by citrin deficiency develop the features of adult-onset citrullinemia type 2.
Adult-onset citrullinemia type II is an inherited disorder that causes ammonia and other toxic substances to accumulate in the blood. The condition chiefly affects the nervous system, causing confusion, restlessness, memory loss, abnormal behaviors (such as aggression, irritability, and hyperactivity), seizures, and coma. These signs and symptoms can be life-threatening. The signs and symptoms appear during adulthood and are triggered by certain medications, infections, surgery, and alcohol intake. The features of adult-onset type II citrullinemia may also develop in people who as infants had a liver disorder called neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD). In many cases, the signs and symptoms of NICCD resolve within a year.
Citrullinemia type 2 causes
Citrullinemia type 2 is caused by mutations in the SLC25A13 gene resulting in a deficiency of the citrin protein. Citrin helps break down carbohydrates and transport certain amino acids, which are the building blocks of proteins. In infants, this can cause a liver disorder called neonatal intrahepatic cholestasis caused by citrin deficiency characterized by a block in the flow of bile that prevents the body from processing certain nutrients properly. Neonatal intrahepatic cholestasis caused by citrin deficiency often resolves within a year, but those affected can later develop features of adult-onset citrullinemia type 2.
Citrullinemia type 2 is inherited in an autosomal recessive pattern.
Citrullinemia type 2 symptoms
Signs and symptoms of citrullinemia type 2 can begin in infancy, adolescence, and adulthood. In babies, the signs of citrullinemia type 2 usually begin between one and five months of age.
Babies with citrullinemia type 2 show signs of:
- Yellowish skin and eyes (known as jaundice)
- Low birth weight
- Delayed growth
- Low blood sugar (called hypoglycemia)
Some of these signs may be seen especially when babies with citrullinemia type 2eat foods that their bodies cannot break down. They can be triggered by long periods of time without eating, illnesses, and infections.
If your baby shows any of these signs, be sure to contact your baby’s doctor immediately.
Citrullinemia type 2 chiefly affects the nervous system, causing neuropsychiatric symptoms including confusion, nocturnal delirium, aggression, irritability, hyperactivity, delusions, disorientation, restlessness, drowsiness, memory loss, flapping tremor, seizures, and coma. These signs and symptoms can be life-threatening. The signs and symptoms of this disorder appear suddenly during adulthood, usually between ages 20 and 50 years. The symptoms appear to be triggered by certain medications, infections, surgery, and alcohol intake. Many individuals with adult-onset citrullinemia type 2 are fond of protein-rich and/or fatty foods and have an aversion to carbohydrate-rich foods. Pathologic findings include fatty infiltration and mild fibrosis of the liver despite little or no liver dysfunction.
The features of adult-onset citrullinemia type 2 may also develop in people who as infants had a liver disorder called neonatal intrahepatic cholestasis caused by citrin deficiency. In many cases, the signs and symptoms of neonatal intrahepatic cholestasis caused by citrin deficiency resolve within a year. Years or even decades later, however, some of these people develop the characteristic features of adult-onset citrullinemia type 2.
Citrullinemia type 2 diagnosis
Citrullinemia can be diagnosed through newborn screening 2. Every state in the US offers newborn screening for citrullinemia 2. When a baby tests positive for citrullinemia on a newborn screen, the diagnosis needs to be confirmed through specific medical test. Genetic testing can help identify the gene mutation in the family.
Making a diagnosis for a genetic or rare disease can often be challenging. Healthcare professionals typically look at a person’s medical history, symptoms, physical exam, and laboratory test results in order to make a diagnosis.
Citrullinemia type 2 treatment
Babies with citrullinemia type 2 are often treated with a change to a low-carbohydrate diet. A nutritionist or dietician familiar with citrullinemia type 2 can help you plan an appropriate diet for your child. Your baby’s doctor may prescribe arginine supplements. Arginine is a substance naturally found in proteins, which can help lower blood ammonia concentration.
Reduction in calorie and/or carbohydrate intake can lessen high triglycerides. Individuals with citrullinemia type 2 are encouraged to consume a diet rich in lipids and protein and low in carbohydrates. This may help to prevent hyperammonemia.
Liver transplantation for adult-onset citrullinemia type 2 prevents hyperammonemic crises, corrects metabolic disturbances, and eliminates preferences for protein-rich foods.
Citrullinemia type 2 prognosis
The prognosis for individuals with adult-onset citrullinemia type 2 varies. In babies, the signs and symptoms usually resolve on their own. In adults, treatment may reduce the signs and symptoms of the condition. As adult-onset citrullinemia type 2 can cause severe liver problems, untreated adults may need a liver transplant 3.
Citrullinemia causes
Mutations in the ASS1 and SLC25A13 genes cause citrullinemia. The proteins produced from these genes play roles in the urea cycle. The urea cycle is a sequence of chemical reactions that takes place in liver cells. These reactions process excess nitrogen that is generated when protein is used by the body. The excess nitrogen is used to make a compound called urea, which is excreted in urine.
Mutations in the ASS1 gene cause citrullinemia type 1. This gene provides instructions for making an enzyme, argininosuccinate synthase 1, that is responsible for one step of the urea cycle. Mutations in the ASS1 gene reduce the activity of the enzyme, which disrupts the urea cycle and prevents the body from processing nitrogen effectively. Excess nitrogen (in the form of ammonia) and other byproducts of the urea cycle accumulate in the bloodstream. Ammonia is particularly toxic to the nervous system, which helps explain the neurologic symptoms (such as lethargy, seizures, and ataxia) that are often seen in citrullinemia type 1.
Mutations in the SLC25A13 gene are responsible for adult-onset citrullinemia type 2, neonatal intrahepatic cholestasis caused by citrin deficiency, and failure to thrive and dyslipidemia caused by citrin deficiency. This gene provides instructions for making a protein called citrin. Within cells, citrin helps transport molecules used in the production and breakdown of simple sugars, the production of proteins, and the urea cycle. Molecules transported by citrin are also involved in making nucleotides, which are the building blocks of DNA and its chemical cousin, RNA. Mutations in the SLC25A13 gene typically prevent cells from making any functional citrin, which inhibits the urea cycle and disrupts the production of proteins and nucleotides. The resulting buildup of ammonia and other toxic substances leads to the signs and symptoms of adult-onset citrullinemia type 1. A lack of citrin also leads to the features of neonatal intrahepatic cholestasis caused by citrin deficiency and failure to thrive and dyslipidemia caused by citrin deficiency, although ammonia does not build up in the bloodstream of individuals with these conditions.
Citrullinemia inheritance pattern
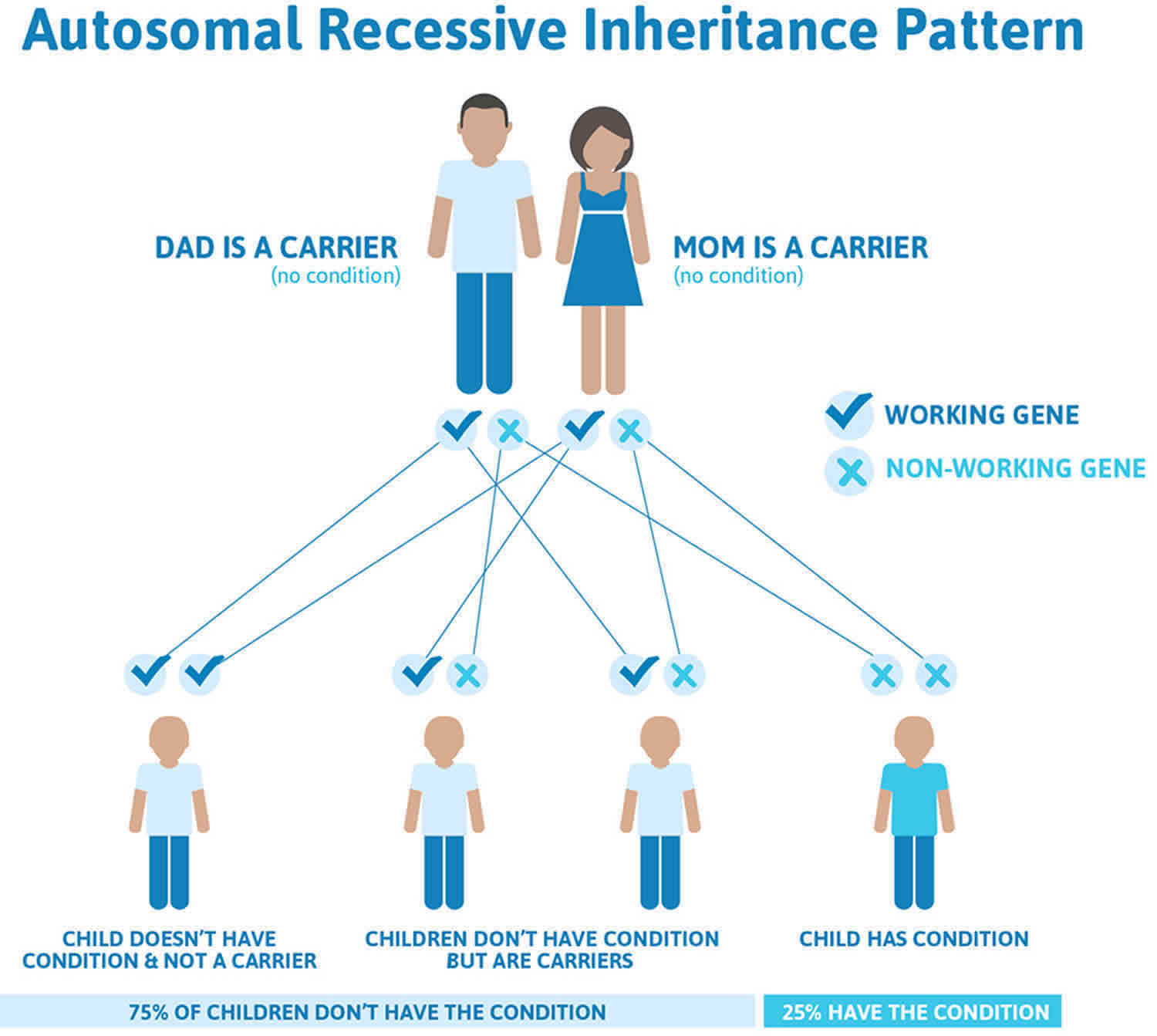
Both types of citrullinemia are inherited in an autosomal recessive pattern, which means both copies of the respective gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 1 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 1. Citrullinemia autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Citrullinemia symptoms
Citrullinemia symptoms and the age they start, vary from person to person. At least one half of newborns with citrullinemia present in the first several days of life. There are two main forms of citrullinemia. The most common is called “classic” citrullinemia type 1. It usually starts in infancy. There are also milder forms that start later in infancy or childhood. There is also a rare adult-onset citrullinemia type 2 more common in people from Japan. Some women only have symptoms during or after pregnancy.
Classic citrullinemia type 1
Infants seem healthy at birth but quickly develop symptoms. Within a few days of life, babies will have high levels of ammonia in their blood. Some of the first symptoms of high blood ammonia are:
- poor appetite
- extreme sleepiness or lack of energy
- irritability
- vomiting
If not treated, high ammonia levels cause:
- muscle weakness
- decreased or increased muscle tone
- breathing problems
- problems staying warm
- seizures
- swelling of the brain
- coma, and sometimes death
Other effects of citrullinemia can include:
- poor growth
- enlarged liver
- learning delays or intellectual disabilities
Without treatment, most babies die within the first few weeks of life.
Milder citrullinemia type 1
In the milder citrullinemia type 1, symptoms start later in infancy or childhood. Symptoms in untreated children can include:
- poor growth
- dry, brittle hair
- hyperactivity
- behavior problems
- learning problems or intellectual disabilities
- avoidance of meat and other high-protein foods
- spasticity
- stroke
- episodes of high levels of ammonia in the blood
- liver failure
Episodes of high blood ammonia often happen:
- after going without food for long periods of time
- during illness or infection
- after high-protein meals
Some of the first symptoms of high blood ammonia in children are:
- poor appetite
- severe headache
- vomiting
- extreme sleepiness or lack of energy
- slurred speech
- poor coordination or balance problems
If not treated, children with high blood ammonia levels may develop:
- breathing problems
- swelling of the brain
- seizures
- coma, sometimes leading to death
A rare form of citrullinemia occurs during and after pregnancy. Women may experience:
- vomiting
- lethargy
- seizures
- confusion and hallucinations
- changes in behavior including manic episodes and psychosis
- swelling of the brain
Some people have very mild or no symptoms and are only found to be affected after a brother or sister is diagnosed.
No routine laboratory studies provide a diagnostic clue, and only a high index of suspicion can prompt the physician to obtain a blood ammonia measurement. The need for a high index of suspicion cannot be sufficiently emphasized.
In the face of intercurrent illness, other affected children experience delayed development from infancy with exaggerated lethargy and vomiting. Again, only a high index of suspicion based on a thorough history can lead to proper diagnosis.
Citrullinemia type 2 symptoms
Citrullinemia type 2 chiefly affects the nervous system, causing neuropsychiatric symptoms including confusion, nocturnal delirium, aggression, irritability, hyperactivity, delusions, disorientation, restlessness, drowsiness, memory loss, flapping tremor, seizures, and coma. These signs and symptoms can be life-threatening. The signs and symptoms of this disorder appear suddenly during adulthood, usually between ages 20 and 50 years. The symptoms appear to be triggered by certain medications, infections, surgery, and alcohol intake. Many individuals with adult-onset citrullinemia type 2 are fond of protein-rich and/or fatty foods and have an aversion to carbohydrate-rich foods. Pathologic findings include fatty infiltration and mild fibrosis of the liver despite little or no liver dysfunction.
The features of adult-onset citrullinemia type 2 may also develop in people who as infants had a liver disorder called neonatal intrahepatic cholestasis caused by citrin deficiency. In many cases, the signs and symptoms of neonatal intrahepatic cholestasis caused by citrin deficiency resolve within a year. Years or even decades later, however, some of these people develop the characteristic features of adult-onset citrullinemia type 2.
Citrullinemia complications
Complications of citrullinemia are chiefly neurological, including mental retardation, acute hyperammonemic coma, and death.
Citrullinemia diagnosis
In patients with citrullinemia who are symptomatic, the measurement of blood ammonia levels is the primary laboratory test in diagnosis. No other routinely obtained study provides diagnostically useful information.
Quantitative measurement of blood amino acid levels is the next immediate step. Citrulline levels are unmistakably elevated in patients with citrullinemia. In such patients, urine amino acid, urine organic acid, and urine orotic acid levels should be analyzed. Orotic acid levels in urine are abnormally elevated in citrullinemia.
Measurement of argininosuccinic acid synthase in cultured skin fibroblasts can provide an unequivocal biochemical diagnosis.
Molecular analysis of the gene should be pursued to establish the nature of the mutation, which can be used for prenatal diagnostic studies.
In neonates who are jaundiced and have normal or mildly elevated ammonia levels, hypercholesterolemia suggests citrin deficiency 4.
Citrullinemia treatment
The following are treatments often recommended for babies and children with citrullinemia.
The treatment of citrullinemia type 1 is aimed at preventing excessive ammonia from being formed or for removing excessive ammonia during a hyperammonemic episode. Medications that assist in the removal of nitrogen from the body by providing an alternative means of waste nitrogen removal are employed. Available medications are Buphenyl, Ammonul, and Raviciti, as well as arginine.
Babies with citrullinemia type 2 are often treated with a change to a low-carbohydrate diet. A nutritionist or dietician familiar with citrullinemia type 2 can help you plan an appropriate diet for your child. Your baby’s doctor may prescribe arginine supplements. Arginine is a substance naturally found in proteins, which can help lower blood ammonia concentration.
Reduction in calorie and/or carbohydrate intake can lessen high triglycerides. Individuals with citrullinemia type 2 are encouraged to consume a diet rich in lipids and protein and low in carbohydrates. This may help to prevent hyperammonemia.
1. Low-protein diet and/or special medical foods and formula
As in all hyperammonemic states, immediately restrict dietary protein in patients with citrullinemia. Most children need to eat a diet made up of very low-protein foods, special medical foods, and sometimes, a special formula. However, enough protein must be taken in by an affected infant to ensure proper growth. Your dietician will create a food plan that contains the right amount of protein, nutrients, and energy to keep your child healthy. A combination of a high biological value natural protein such as breast milk or cow’s milk formulate, an essential amino acid formula, and a calorie supplement without protein is often used. A special food plan should be continued throughout your child’s life.
Low-protein diet
The most effective treatment for citrullinemia is a low-protein diet. Foods that need to be avoided or strictly limited include:
- milk, cheese, and other dairy products
- meat and poultry
- fish
- eggs
- dried beans and legumes
- nuts and peanut butter
Eating foods high in protein can cause ammonia to build up, causing severe illness. Many vegetables and fruits have only small amounts of protein and can be eaten in carefully measured amounts.
Do not remove all protein from the diet. Your child still needs a certain amount of protein for normal growth and development. Any changes in the diet should be made under the guidance of a dietician.
Medical foods and formula
There are medical foods such as special low-protein flours, pastas, and rice that are made especially for people with amino acid disorders.
Your baby may need to drink a special medical formula that contains the correct amount of amino acids and nutrients. Your metabolic doctor and dietician will decide whether your child needs this treatment. Some states offer help with payment, or require private insurance to pay for the formula and other special medical foods.
Your child’s exact food plan will depend on many things such as his or her age, weight, and general health. Your dietician will fine-tune your child’s diet over time. Any diet changes should be made under the guidance of a dietician.
2. Medication
There are certain medications that can help the body get rid of ammonia. These are taken by mouth or by tube feeding to prevent high ammonia levels. Your doctor will decide whether your child needs these medications, which ones, and how much to use.
During episodes of high ammonia, children need to be treated in the hospital. Medications to remove ammonia are often given by IV. Dialysis is sometimes needed to remove ammonia from the blood.
An amino acid called arginine is often given by mouth to help prevent ammonia build-up. Your doctor will tell you whether your child needs arginine and how much to use. Do not use any supplements or medications without checking with your doctor.
Intravenous sodium benzoate and sodium phenylacetate are another important therapeutic avenues for reduction of blood ammonia levels. Intravenous benzoate and phenylacetate are investigational new drugs.
In severe cases, hemodialysis may be indicated to rapidly reduce the blood ammonia level.
Long-term management requires close dietary monitoring and oral administration of sodium phenylbutyrate and arginine.
3. Blood tests
Your child will have regular blood tests to measure ammonia and amino acid levels. Your child’s diet and medication may need to be adjusted based on blood test results. In every case, a biochemical geneticist should administer definitive short- and long-term treatment with sufficient laboratory backup to obtain rapid ammonia and amino acid levels.
4. Call your doctor at the start of any illness:
For some babies and children with citrullinemia, even minor illness can cause high ammonia levels. In order to prevent problems, call your doctor right away when your child has any of the following:
- loss of appetite
- low energy or extreme sleepiness
- vomiting
- fever
- infection or illness
- behavior or personality changes
- difficulty walking or balance problems
Symptoms of high ammonia often need to be treated in the hospital. Ask your metabolic doctor if you should carry a special travel letter with medical instructions for your child’s care.
5. Liver transplantation
Liver transplant surgery is an optional treatment for people with citrullinemia. The argininosuccinate synthetase enzyme that causes citrullinemia type 1 is located in the liver. Because of this, some children with citrullinemia type 1 have had liver transplantation surgery (removal of their liver and replacement with a donor liver) to treat their citrullinemia symptoms. Liver transplantation for adult-onset citrullinemia type 2 prevents hyperammonemic crises, corrects metabolic disturbances, and eliminates preferences for protein-rich foods.
Liver transplant is a major surgical procedure and is associated with risks, and individuals who have had a liver transplant must take medication for the rest of their lives to prevent their body from rejecting the donor liver. However, successful liver transplantation has been reported to improve quality of life and prolong survival in some cases.
Many factors must be considered before surgery and this option should be discussed very thoroughly with your or your child’s physicians.
Citrullinemia prognosis
With prompt and lifelong treatment, children with citrullinemia can often live healthy lives with typical growth and learning. Early treatment can help prevent high ammonia levels. With appropriate treatment, survival into adulthood is possible and has been documented.
Even with treatment, some children still have episodes of high ammonia. This can result in brain damage. This can cause lifelong learning problems, intellectual disabilities or spasticity. Consistent with the course of most urea cycle disorders, the degree of intellectual impairment is roughly parallel to the severity of initial presentation and frequency of subsequent hyperammonemic episodes. Subsequent hyperammonemic episodes predictably recur with any intercurrent infection 5.
References- Citrullinemia. https://ghr.nlm.nih.gov/condition/citrullinemia
- Amino Acid Disorder. http://www.newbornscreening.info/Parents/aminoaciddisorders/ASAS.html
- Saheki T, Song YZ. Citrin Deficiency. 2005 Sep 16 [Updated 2017 Aug 10]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1181
- Nagasaka H, Okano Y, Tsukahara H, et al. Sustaining hypercitrullinemia, hypercholesterolemia and augmented oxidative stress in Japanese children with aspartate/glutamate carrier isoform 2-citrin-deficiency even during the silent period. Mol Genet Metab. 2009 Jan 25.
- Waisbren SE, Cuthbertson D, Burgard P, Holbert A, McCarter R, Cederbaum S, et al. Biochemical markers and neuropsychological functioning in distal urea cycle disorders. J Inherit Metab Dis. 2018 Jul. 41 (4):657-667.





{kind=link}