Hypophosphatasia
Hypophosphatasia is a rare genetic condition that causes abnormal development of the bones and teeth 1. In patients with hypophosphatasia, low alkaline phosphatase (ALP) activity results in the accumulation of phosphorylated substrates, specifically inorganic pyrophosphate, pyridoxal 5′-phosphate (the active form of vitamin B6) and phosphoethanolamine 2. Elevated inorganic pyrophosphate levels inhibit mineralization of the bone matrix, leading to hypomineralization of the skeleton 3. Mineralization is critical for the formation of bones that are strong and rigid and teeth that can withstand chewing and grinding. Hypophosphatasia is characterized by defective mineralization in which minerals such as calcium and phosphorus are deposited in developing bones and teeth in the presence of low activity of serum and bone alkaline phosphatase (ALP). The inability of pyridoxal 5′-phosphate, the circulating form of vitamin B6, to cross the blood-brain barrier likely contributes to the seizures observed in some infants with hypophosphatasia 4.
The signs and symptoms of hypophosphatasia vary widely and can appear anywhere from before birth to adulthood. Clinical features range from stillbirth without mineralized bone at the severe end to pathologic fractures of the lower extremities in later adulthood at the mild end. Although hypophosphatasia disease spectrum is a continuum, six clinical forms are usually recognized based on age at diagnosis and severity of features 5:
- Perinatal (severe) hypophosphatasia characterized by respiratory insufficiency and hypercalcemia
- Perinatal (benign) hypophosphatasia with prenatal skeletal manifestations that slowly resolve into one of the milder forms
- Infantile hypophosphatasia with onset between birth and age six months of rickets without elevated serum alkaline phosphatase activity
- Childhood (juvenile) hypophosphatasia that ranges from low bone mineral density for age with unexplained fractures to rickets, and premature loss of primary teeth with intact roots
- Adult hypophosphatasia characterized by stress fractures and pseudofractures of the lower extremities in middle age, sometimes associated with early loss of adult dentition
- Odontohypophosphatasia characterized by premature exfoliation of primary teeth and/or severe dental caries without skeletal manifestations
The most severe forms of hypophosphatasia tend to occur before birth and in early infancy. Hypophosphatasia weakens and softens the bones, causing skeletal abnormalities similar to another childhood bone disorder called rickets. Affected infants are born with short limbs, an abnormally shaped chest, and soft skull bones. Additional complications in infancy include poor feeding and a failure to gain weight, respiratory problems, and high levels of calcium in the blood (hypercalcemia), which can lead to recurrent vomiting and kidney problems. These complications are life-threatening in some cases.
The forms of hypophosphatasia that appear in childhood or adulthood are typically less severe than those that appear in infancy. Early loss of primary (baby) teeth is one of the first signs of the condition in children. Affected children may have short stature with bowed legs or knock knees, enlarged wrist and ankle joints, and an abnormal skull shape. Adult forms of hypophosphatasia are characterized by a softening of the bones known as osteomalacia. In adults, recurrent fractures in the foot and thigh bones can lead to chronic pain. Affected adults may lose their secondary (adult) teeth prematurely and are at increased risk for joint pain and inflammation.
The overall incidence and prevalence of all forms of hypophosphatasia is unknown.
The mildest form of hypophosphatasia is called odontohypophosphatasia, only affects the teeth. People with odontohypophosphatasia typically experience abnormal tooth development and premature tooth loss, but do not have the skeletal abnormalities seen in other forms of hypophosphatasia.
Severe forms of hypophosphatasia affect an estimated 1 in 100,000 newborns. Milder cases can go undiagnosed or misdiagnosed, making it difficult to determine the true frequency of hypophosphatasia in the general population.
Hypophosphatasia affects males and females in equal numbers.
Hypophosphatasia has been reported worldwide in people of various ethnic backgrounds. Hypophosphatasia appears to be most common in white populations 1. Hypophosphatasia occurs with greatest frequency in a Mennonite population in Manitoba, Canada, where about 1 in 2,500 infants is born with severe features of hypophosphatasia 1. Hypophosphatasia is also relatively prevalent in Japan, and is relatively rare in black individuals.
Hypophosphatasia causes
Mutations in the ALPL gene cause hypophosphatasia. The ALPL gene provides instructions for making an enzyme called tissue-nonspecific alkaline phosphatase, which plays an essential role in mineralization of the skeleton and teeth. Mutations in the ALPL gene lead to the production of an abnormal version of tissue-nonspecific alkaline phosphatase that cannot participate effectively in the mineralization process. A shortage of tissue-nonspecific alkaline phosphatase allows several other substances, which are normally processed by the enzyme, to build up abnormally in the body. Researchers believe that a buildup of one of these compounds, inorganic pyrophosphate (PPi), underlies the defective mineralization of bones and teeth in people with hypophosphatasia.
ALPL gene mutations that almost completely eliminate the activity of tissue-nonspecific alkaline phosphatase usually result in the more severe forms of hypophosphatasia. Other mutations, which reduce but do not eliminate the activity of the enzyme, often cause the milder forms of the condition.
Hypophosphatasia inheritance pattern
The severe forms of hypophosphatasia that appear early in life are inherited in an autosomal recessive pattern. Autosomal recessive inheritance means that two copies of the gene in each cell are altered. Most often, the parents of an individual with an autosomal recessive disorder each carry one copy of the altered gene but do not show signs and symptoms of the disorder. This means that to be affected, a person must have a mutation in both copies of the responsible gene (ALPL) in each cell. Affected people inherit one mutated copy of the gene from each parent, who is referred to as a carrier. Carriers of an autosomal recessive condition typically do not have any signs or symptoms (they are unaffected). When 2 carriers of an autosomal recessive condition have children, each child has a (see Figure 1 below):
- 25% (1 in 4) chance to be affected
- 50% (1 in 2) chance to be an unaffected carrier like each parent
- 25% chance to be unaffected and not be a carrier.
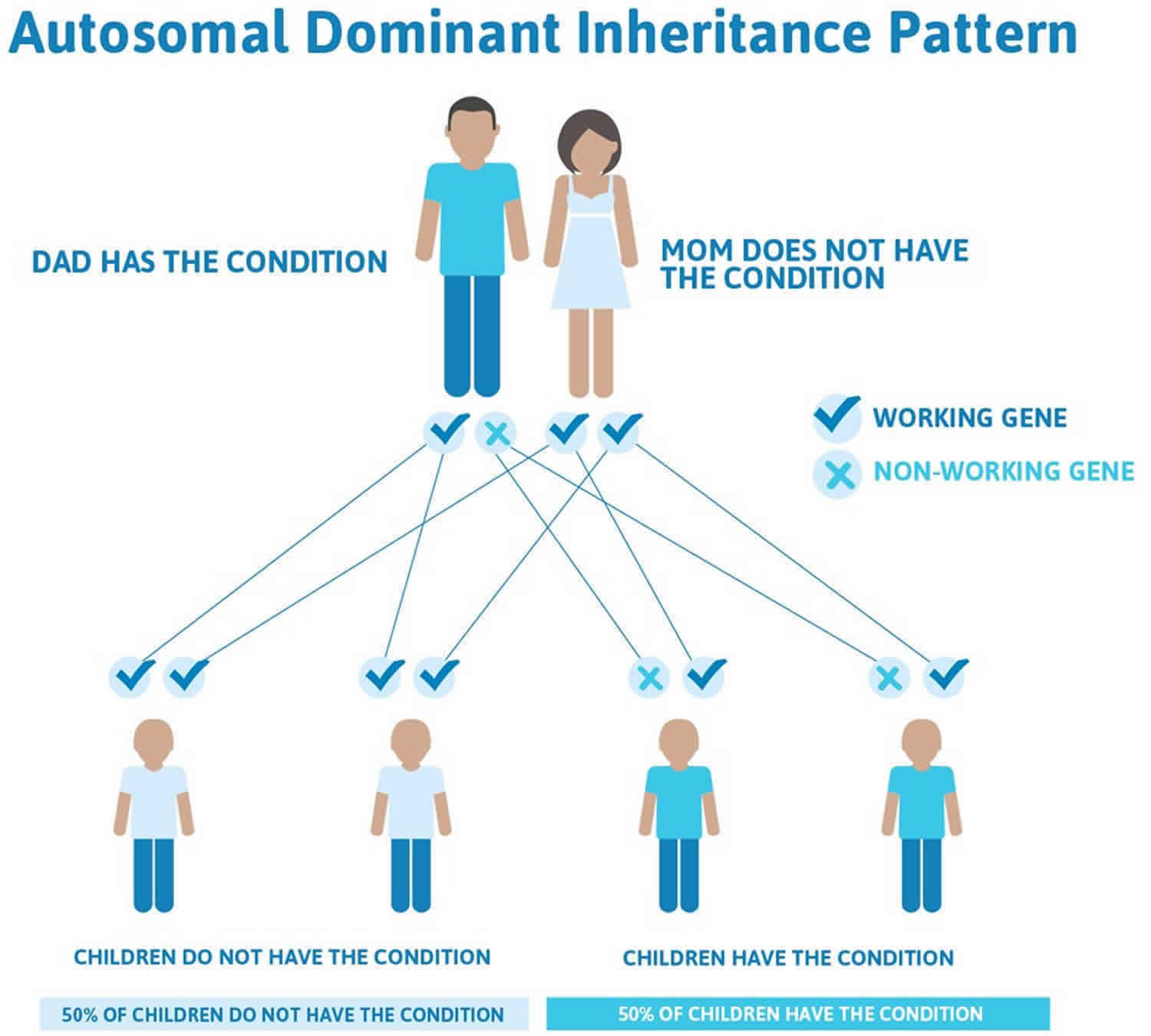
Milder forms of hypophosphatasia, especially adult hypophosphatasia and odontohypophosphatasia, can have either an autosomal recessive or an autosomal dominant pattern of inheritance. In autosomal dominant inheritance, having a mutation in only one copy of the ALPL gene in each cell is enough to cause features of the condition. When a person with a mutation that causes an autosomal dominant hypophosphatasia has children, each child has a 50% (1 in 2) chance to inherit that mutation (see Figure 2 below). Most people with autosomal dominant hypophosphatasia have inherited the mutation from a parent who may or may not have symptoms. Not all people with a mutation that causes autosomal dominant hypophosphatasia develop symptoms of the condition. While it is possible to have autosomal dominant hypophosphatasia due to a new mutation that was not inherited (a de novo mutation), this has never been reported in hypophosphatasia.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Figure 1. Hypophosphatasia autosomal recessive inheritance pattern
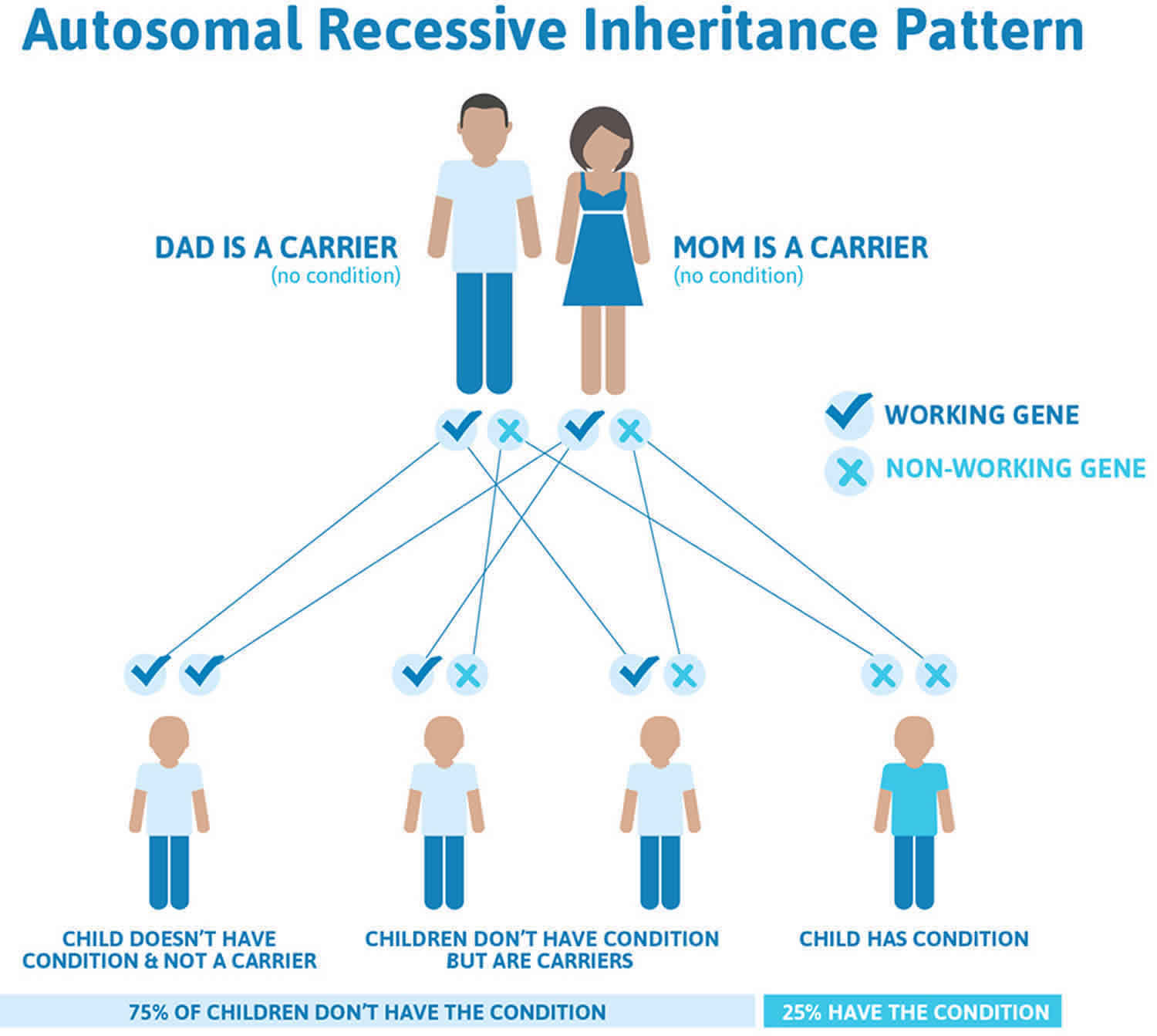
Figure 2. Hypophosphatasia autosomal dominant inheritance pattern
Hypophosphatasia symptoms
Hypophosphatasia should be suspected in individuals with:
- Defective mineralization of bone and/or teeth;
- Premature loss of teeth with intact roots;
- Reduced serum alkaline phosphatase (ALP) activity.
At least six clinical forms are currently recognized based on age at diagnosis and severity of features (see Table 1).
Table 1. Clinical features of hypophosphatasia by type
| Type | Inheritance | Cardinal Features | Dental Features | Clinical Diagnosis |
|---|---|---|---|---|
| Perinatal (severe) | Autosomal recessive | Hypomineralization, osteochondral spurs | ± 1 | Radiographs, prenatal ultrasound examination |
| Perinatal (benign) | Autosomal recessive or Autosomal dominant | Long-bone bowing, benign postnatal course | ± | Prenatal ultrasound examination, clinical course |
| Infantile 2 | Mostly autosomal recessive | Craniosynostosis, Hypomineralization, rachitic ribs, hypercalciuria | Premature loss, deciduous teeth | Clinical course, radiographs, laboratory findings |
| Childhood (juvenile) | Autosomal recessive or Autosomal dominant | Short stature, skeletal deformity, bone pain/fractures | Premature loss, deciduous teeth (incisors) | Clinical course, radiographs, laboratory findings |
| Adult 3 | Autosomal recessive or Autosomal dominant | Stress fractures: metatarsal, tibia; chondrocalcinosis | ± | Clinical course, radiographs, laboratory findings |
| Odontohypo- phosphatasia | Autosomal recessive or Autosomal dominant | Alveolar bone loss | Exfoliation (incisors), dental caries | Clinical course, dental panorex, laboratory findings |
Footnote:
- In the past individuals with severe phenotypes have typically died before teeth erupted and could be lost. In the new “treated perinatal (severe) and infantile” category, the dental features are not precisely known but emerging data suggests the possibility of such features.
- Rare reported cases of infantile hypophosphatasia that have normal serum alkaline phosphatase activity (in vitro) have been designated “pseudohypophosphatasia.” The biochemical and molecular basis of pseudohypophosphasia remains unclear.
- Persons with adult hypophosphatasia may give a history of features typically reported in childhood (juvenile), infantile, and even prenatal hypophosphatasia.
The signs and symptoms of hypophosphatasia vary widely and can appear anywhere from before birth to adulthood. The most severe forms of hypophosphatasia tend to occur before birth and in early infancy. Hypophosphatasia weakens and softens the bones, causing skeletal abnormalities similar to another childhood bone disorder called rickets. Affected infants are born with short limbs, an abnormally shaped chest, and soft skull bones. Additional complications in infancy include poor feeding and a failure to gain weight, respiratory problems, and high levels of calcium in the blood (hypercalcemia), which can lead to recurrent vomiting and kidney problems. These complications are life-threatening in some cases.
The forms of hypophosphatasia that appear in childhood or adulthood are typically less severe than those that appear in infancy. Early loss of primary (baby) teeth is one of the first signs of the condition in children. Affected children may have short stature with bowed legs or knock knees, enlarged wrist and ankle joints, and an abnormal skull shape. Adult forms of hypophosphatasia are characterized by a softening of the bones known as osteomalacia. In adults, recurrent fractures in the foot and thigh bones can lead to chronic pain. Affected adults may lose their secondary (adult) teeth prematurely and are at increased risk for joint pain and inflammation.
The mildest form of this condition, called odontohypophosphatasia, only affects the teeth. People with this disorder typically experience abnormal tooth development and premature tooth loss, but do not have the skeletal abnormalities seen in other forms of hypophosphatasia.
Clinical features of hypophosphatasia include the following 5:
- Prenatal long-bone bowing with osteochondral spurs and pretibial dimpling
- Infantile rickets without elevated serum alkaline phosphatase activity. Features can include growth failure, craniotabes, craniosynostosis, blue sclerae, costochondral enlargement (“rachitic rosary”), scoliosis, thickening of wrists and ankles, bowing of long bones, lax ligaments, and hypotonia.
- Hypercalcemia and hypercalciuria particularly during the first year of life
- Pathologic fractures and bone pain. Growing children may have a predilection to metaphyseal fractures; however, epiphyseal and diaphyseal fractures are also seen. In adults, metatarsal stress fractures and femoral pseudofractures prevail.
- Premature loss of deciduous teeth beginning with the incisors. Unusually and characteristically, the dental root remains attached to the lost tooth. Dental caries and early loss or extraction of adult teeth is also seen.
- Family history of any of the forms of hypophosphatasia consistent with autosomal recessive inheritance or autosomal dominant inheritance with variable expressivity
The radiographic signs of hypophosphatasia vary with age and type, and may be quite distinctive. Perinatal lethal hypophosphatasia is radiographically distinct. In milder cases, the combination of clinical, laboratory, and radiographic findings are required for diagnosis because the radiographic signs are not pathognomonic.
- Osteopenia, osteoporosis, or low bone mineral content for age detected by dual-energy x-ray absorptiometry (DEXA). Bone mineral content increases with age, and there may be improvement during adolescence with recurrence in middle age.
- Infantile rickets. Findings include undermineralized bones, widened-appearing sutures, brachycephaly, flail chest, rachitic costochondral rib changes (see Figure 2A), flared metaphyses (resulting in enlarged wrists, knees, and ankles), poorly ossified epiphyses, and bowed legs.
- Alveolar bone loss resulting in premature loss of deciduous teeth. This most typically involves the anterior mandible, with the central incisors lost first. However, any tooth may be affected (see Figure 2B).
- Focal bony defects of the metaphyses resembling radiolucent “tongues” (see Figure 2C). This feature is fairly specific for childhood (juvenile) hypophosphatasia.
- Metatarsal stress fractures in childhood (juvenile) and adult hypophosphatasia
- Osteomalacia with lateral pseudofractures (Looser zones) in adult hypophosphatasia (see Figure 2D)
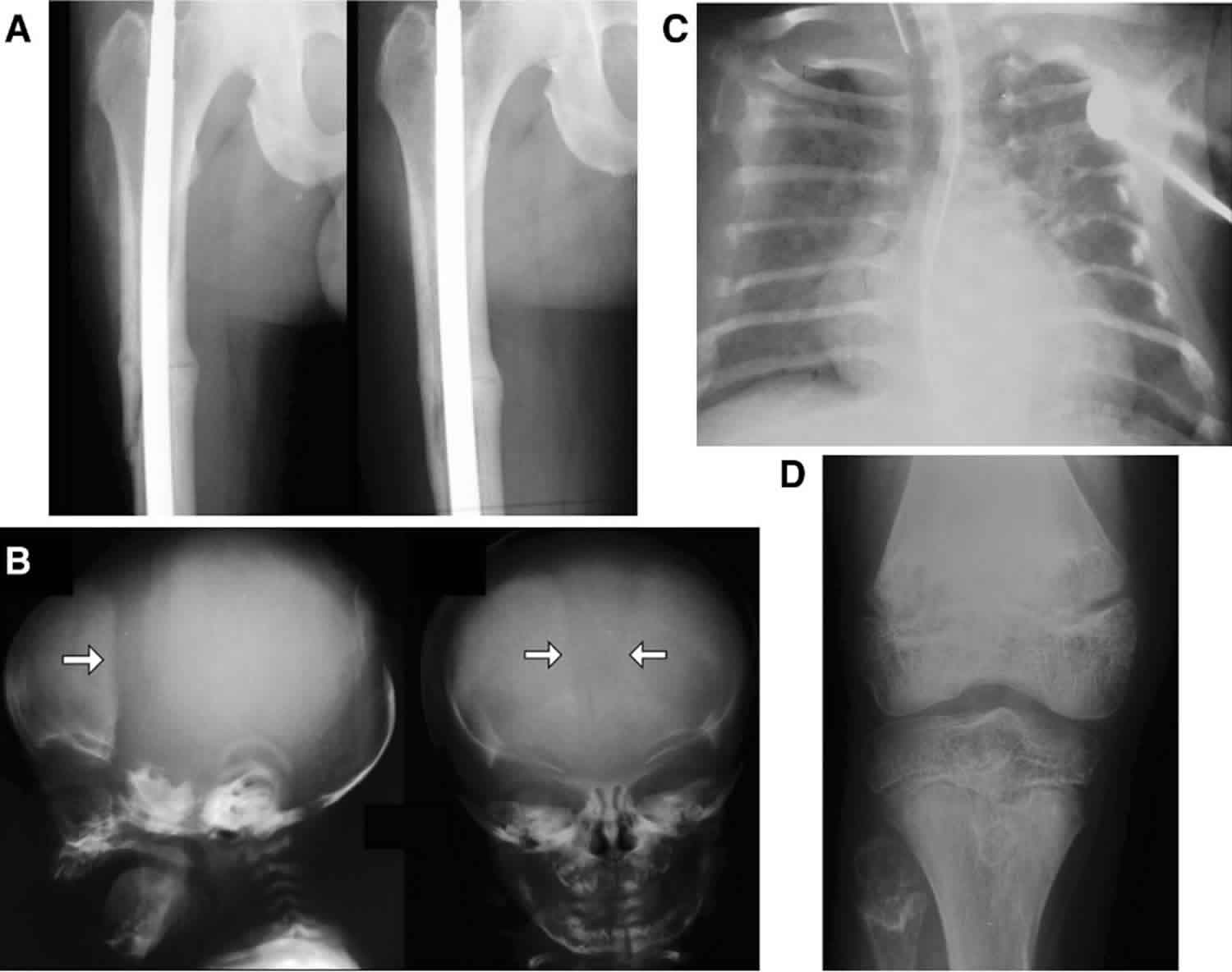
Figure 2. Radiographic signs of hypophosphatasia
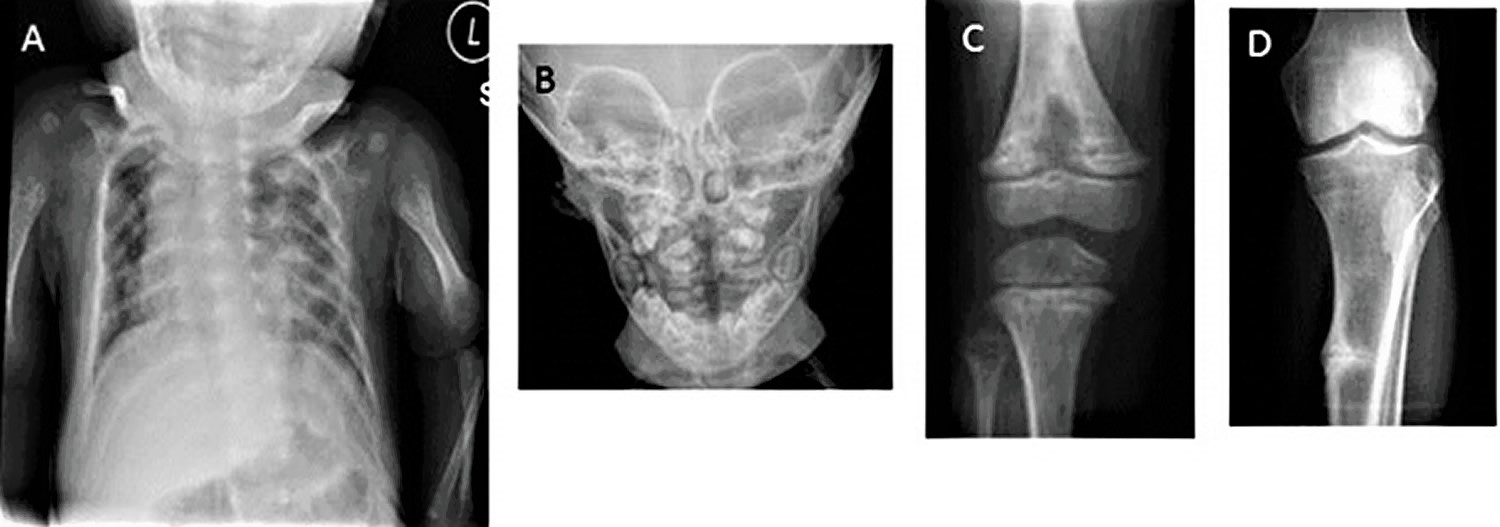
Footnote: Radiographic signs of hypophosphatasia
- A. Rachitic rib changes, flail chest, and metaphyseal dysplasia (proximal humerus) in infantile hypophosphatasia
- B. Alveolar bone loss surrounding molars in childhood (juvenile) hypophosphatasia
- C. Hypolucent “tongue” mid-metaphysis in childhood (juvenile) hypophosphatasia
- D. Looser zone (pseudofracture) in adult hypophosphatasia
Hypophosphatasia diagnosis
A diagnosis of hypophosphatasia is based upon identification of characteristic signs and symptoms, a detailed patient history, a thorough clinical evaluation, and a variety of laboratory tests including routine x-ray and biochemical studies. Proper diagnosis of hypophosphatasia is easy for physicians who are familiar or experienced with this disorder. However, most physicians have little or no knowledge of hypophosphatasia. Consequently, affected individuals and families may face a frustrating delay in diagnosis. Now, mutation analysis of the ALPL gene is available from commercial laboratories.
Clinical testing and workup
The diagnosis is rarely first suspected from a routine panel of biochemical tests that includes measuring the activity of alkaline phosphatase in blood. Instead signs and symptoms have led to this routine test where the low levels of alkaline phosphatase (ALP) must be recognized. Individuals with hypophosphatasia have reduced serum alkaline phosphatase activity for their age, except for the extremely rare individual with pseudohypophosphatasia who has normal activity levels. Identification of deficient alkaline phosphatase activity is consistent with hypophosphatasia, but not conclusive since other conditions can result in this finding. Additionally, some individuals who are genetic carriers of hypophosphatasia, but who do not develop any symptoms of the disorder, may also have low blood ALP (alkaline phosphatase) levels.
Importantly, the range of serum ALP activity varies by age. Healthy children normally have higher ALP levels than healthy adults. If the laboratory doing the testing only gives the normal range of ALP activity in adults in its report, a diagnosis of hypophosphatasia in a child can be missed because the child’s ALP activity will mistakenly be believed to be normal.
In the U.S. and elsewhere, a suspected diagnosis of hypophosphatasia can be further supported by measuring the serum level of vitamin B6. This test is performed by several commercial laboratories. Individuals with hypophosphatasia have elevated levels of pyridoxal 5’-phosphate (the active form of vitamin B6) in the blood because pyridoxal 5’-phosphate is normally broken down by tissue nonspecific alkaline phosphatase. Pyridoxal 5’-phosphate is elevated even in individuals with mild hypophosphatasia. However, some genetic carriers of hypophosphatasia who do not develop any symptoms can have an elevated pyridoxal 5’-phosphate level as well. In the past, blood or urine was tested for increased amounts of phosphoethanolamine, another chemical normally broken down by tissue nonspecific alkaline phosphatase. However, this finding is not specific to hypophosphatasia and can occur because of other metabolic bone diseases. Additionally, some individuals with hypophosphatasia have normal phosphoethanolamine levels. Screening for elevated pyridoxal 5’-phosphate is preferred over screening for phosphoethanolamine because it is more sensitive, more precise, and less expensive.
In the most severe cases of hypophosphatasia, specifically the perinatal and infantile forms, x-ray studies can reveal diagnostic changes within the bones. However, these changes may not be recognized as being associated with hypophosphatasia, except by radiologists familiar with the disorder.
Molecular genetic testing can support a diagnosis of hypophosphatasia. Molecular genetic testing can detect mutations in the ALPL gene known to cause the disorder, but it is only available as a diagnostic service at specialized laboratories. The test is often expensive and often not necessary to confirm a diagnosis of hypophosphatasia.
Evaluations following initial diagnosis
To establish the extent of disease and needs in an individual diagnosed with hypophosphatasia, the following evaluations are recommended:
- Blood urea nitrogen (BUN) and serum creatinine concentration to assess renal function
- Serum concentration of calcium, phosphorus, magnesium
- Serum concentration of 25(OH) and 1,25(OH)2 vitamin D, nPTH (parathyroid hormone, N-terminal part) to assess rickets
- Assessment of pulmonary function in infants with the perinatal type to assist in prognosis and distinguishing between the perinatal (severe) type and the perinatal (benign) type
- Radiographs of the skull to assess for craniosynostosis in young children with the infantile form of hypophosphatasia
- Baseline dental evaluation
- Baseline orthopedic evaluation
- Consultation with a clinical geneticist and/or genetic counselor
Hypophosphatasia treatment
Until recently, management of hypophosphatasia has mostly been aimed at addressing symptoms of the condition. For example 6:
- Hydration, restriction of dietary calcium, vitamin D, and sometimes thiazide diuretics for hypercalcemia
- Ventilatory support for severely affected infants, some of which need a tracheostomy, which can lead to problems with speech and language development and tolerance of oral feeds
- Physiotherapy, occupational therapy and chronic pain management for pain and motor difficulty
- Surgery for fractures that fail to heal
More recently, research has shown positive effects of human recombinant enzyme replacement therapy (ERT), called asfotase alfa, on people who began having symptoms before 6 months of age. There reportedly have been significant improvements in the X-ray appearances of bone tissue, along with improvements in growth, respiratory function, motor development and calcium homeostasis after 6–12 months of treatment. The children in the original study have now received more than three years of treatment, without apparent major side effects, and with continuing improvement in affected systems 6. Asfotase alfa appears to be a valuable emerging therapy for the treatment of bone manifestations in people with pediatric-onset hypophosphatasia 7. In October of 2015 the FDA approved asfotase alfa, sold as Strensiq 8.
Bone marrow and stem cell transplantation in infancy and childhood have improved the severity of the disease, but have not provided long term improvement 6. Bone marrow transplantation (hematopoietic cell transplantation) was used to treat an eight-month-old girl with severe hypophosphatasia with prolonged, significant clinical and radiologic improvement 9. Seven years after transplantation, the patient was reported to be active and growing, and to have the clinical phenotype of the more mild childhood (juvenile) form of hypophosphatasia 10. In another trial, both bone marrow and allogenic mesenchymal stem cells were implanted in an eight-month old patient, resulting in improvement of respiratory conditions 11. However,the patient developed therapy-related leukemia 12. Transplantation of ex vivo expanded mesenchymal stem cells for patients who had previously undergone bone marrow transplantation improved bone mineralization, muscle mass, respiratory function, intellectual development, and survival 13.
Prevention of primary manifestations
Low-impact physical activity and exercise may improve general bone health. Supervision by a physician specialist familiar with hypophosphatasia is suggested.
Prevention of secondary complications
Calcium supplementation and vitamin D therapy may prevent secondary hyperparathyroidism in adults. This should only be pursued with close monitoring by a physician specialist familiar with hypophosphatasia.
Surveillance
Children with hypophosphatasia should be seen by a pediatric dentist twice yearly, beginning at age one year.
Children with the infantile type of hypophosphatasia are at elevated risk for increased intracranial pressure secondary to craniosynostosis, and should be monitored for this complication.
Agents/Circumstances to Avoid
- Biphosphonates are relatively contraindicated in hypophosphatasia. Although adverse outcomes have not been identified in children with the severe infantile type 14, theoretic concern has long been raised based on the structure of bisphosphonates. The phosphate motifs in bisphosphonates have a similar conformation to inorganic pyrophosphate (PPi), the natural substrate of tissue-nonspecific alkaline phosphatase; thus, treatment with bisphosphonates is thought to be analogous to “adding fuel to the fire.” In adults with hypophosphatasia and osteomalacia treated with bisphosphonates, lateral subtrochanteric femoral pseudofractures have been described 15. As the prevalence of adult hypophosphatasia is not known and many undiagnosed adult patients undoubtedly are treated with bisphosphonates, the frequency of this unusual complication is not known.
- Excess vitamin D can exacerbate hypercalcemia/hypercalciuria in children with infantile hypophosphatasia who have hypercalcemia.
- Teriparatide (recombinant human parathyroid hormone fragment, amino acids 1-34) at high doses induces osteosarcoma in rats, and may increase the risk of radiation-induced osteosarcoma (a pediatric growth plate tumor) in humans. It is contraindicated in children with hypophosphatasia.
Hypophosphatasia prognosis
The most severe form is perinatal hypophosphatasia, which is considered lethal in most cases. The infantile form is fatal in approximately 30% of patients. Longevity studies have not been conducted for the infantile and childhood forms. Individuals with the adult and odontohypophosphatasic forms have normal lifespans.
References- Hypophosphatasia. https://ghr.nlm.nih.gov/condition/hypophosphatasia
- Offiah AC, Vockley J, Munns CF, Murotsuki J. Differential diagnosis of perinatal hypophosphatasia: radiologic perspectives. Pediatr Radiol. 2019;49(1):3–22. doi:10.1007/s00247-018-4239-0 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6313373
- Whyte MP (2018) Hypophosphatasia and how alkaline phosphatase promotes mineralization. In: Thakker RV, Whyte MP, Eisman J, Igarashi T (eds) Genetics of bone biology and skeletal disease, 2nd edn. Elsevier (Academic Press, London), San Diego, CA, pp 481–504.
- Linglart A, Biosse-Duplan M. Hypophosphatasia. Curr Osteoporos Rep. 2016;14:95–105. doi: 10.1007/s11914-016-0309-0
- Mornet E, Nunes ME. Hypophosphatasia. 2007 Nov 20 [Updated 2016 Feb 4]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1150
- Bishop N. Clinical management of hypophosphatasia. Clin Cases Miner Bone Metab. May-August 2015; 12(2):170-173.
- Scott LJ. Asfotase Alfa: A Review in Paediatric-Onset Hypophosphatasia. Drugs. February, 2016; 76(2):255-262.
- Morrow T. Expensive New Biologic Helps Children Fight Hypophosphatasia. Manag Care. December, 2015; 24(12):25-26. https://www.managedcaremag.com/archives/2015/12/expensive-new-biologic-helps-children-fight-hypophosphatasia
- Whyte MP, Kurtzberg J, McAlister WH, Mumm S, Podgornik MN, Coburn SP, Ryan LM, Miller CR, Gottesman GS, Smith AK, Douville J, Waters-Pick B, Armstrong RD, Martin PL. Marrow cell transplantation for infantile hypophosphatasia. J Bone Miner Res. 2003;18:624–36.
- Cahill RA, Wenkert D, Perlman SA, Steele A, Coburn SP, McAlister WH, Mumm S, Whyte MP. Infantile hypophosphatasia: transplantation therapy trial using bone fragments and cultured osteoblasts. J Clin Endocrinol Metab. 2007;92:2923–30.
- Tadokoro M, Kanai R, Taketani T, Uchio Y, Yamaguchi S, Ohgushi H. New bone formation by allogeneic mesenchymal stem cell transplantation in a patient with perinatal hypophosphatasia. J Pediatr. 2009;154:924–30.
- Taketani T, Kanai R, Abe M, Mishima S, Tadokoro M, Katsube Y, Yuba S, Ogushi H, Fukuda S, Yamaguchi S. Therapy-related Ph+ leukemia after both bone marrow and mesenchymal stem cell transplantation for hypophosphatasia. Pediatr Int. 2013;55:e52–5.
- Taketani T, Oyama C, Mihara A, Tanabe Y, Abe M, Hirade T, Yamamoto S, Bo R, Kanai R, Tadenuma T, Michibata Y, Yamamoto S, Hattori M, Katsube Y, Ohnishi H, Sasao M, Oda Y, Hattori K, Yuba S, Ohgushi H, Yamaguchi S. Ex vivo expanded allogeneic mesenchymal stem cells with bone marrow transplantation improved osteogenesis in infants with severe hypophosphatasia. Cell Transplant. 2015;24:1931–43.
- Deeb AA, Bruce SN, Morris AA, Cheetham TD. Infantile hypophosphatasia: disappointing results of treatment. Acta Paediatr. 2000;89:730–3.
- Whyte MP. Atypical femoral fractures, bisphosphonates, and adult hypophosphatasia. J Bone Miner Res. 2009;24:1132–4.





{kind=link}