What is blood coagulation
Blood is a necessary component of the human body, and the loss of this fluid may be life-threatening. The human body protects against loss of blood through the clotting mechanism. Vascular mechanisms, platelets, coagulation factors, prostaglandins, enzymes, and proteins are the contributors to the clotting mechanism which act together to form clots and stop a loss of blood 1. Through vasoconstriction, adhesion, activation, and aggregation, the contributors form a transient plug to act as the cork to the leaking blood flow. Soon after, fibrin, the functioning form of fibrinogen, stabilizes this weak platelet plug.
The cellular components of the clotting mechanism include platelets, endothelial cells, and a series of proteins, enzymes, and ions.
The clotting mechanism is broken into 2 stages:
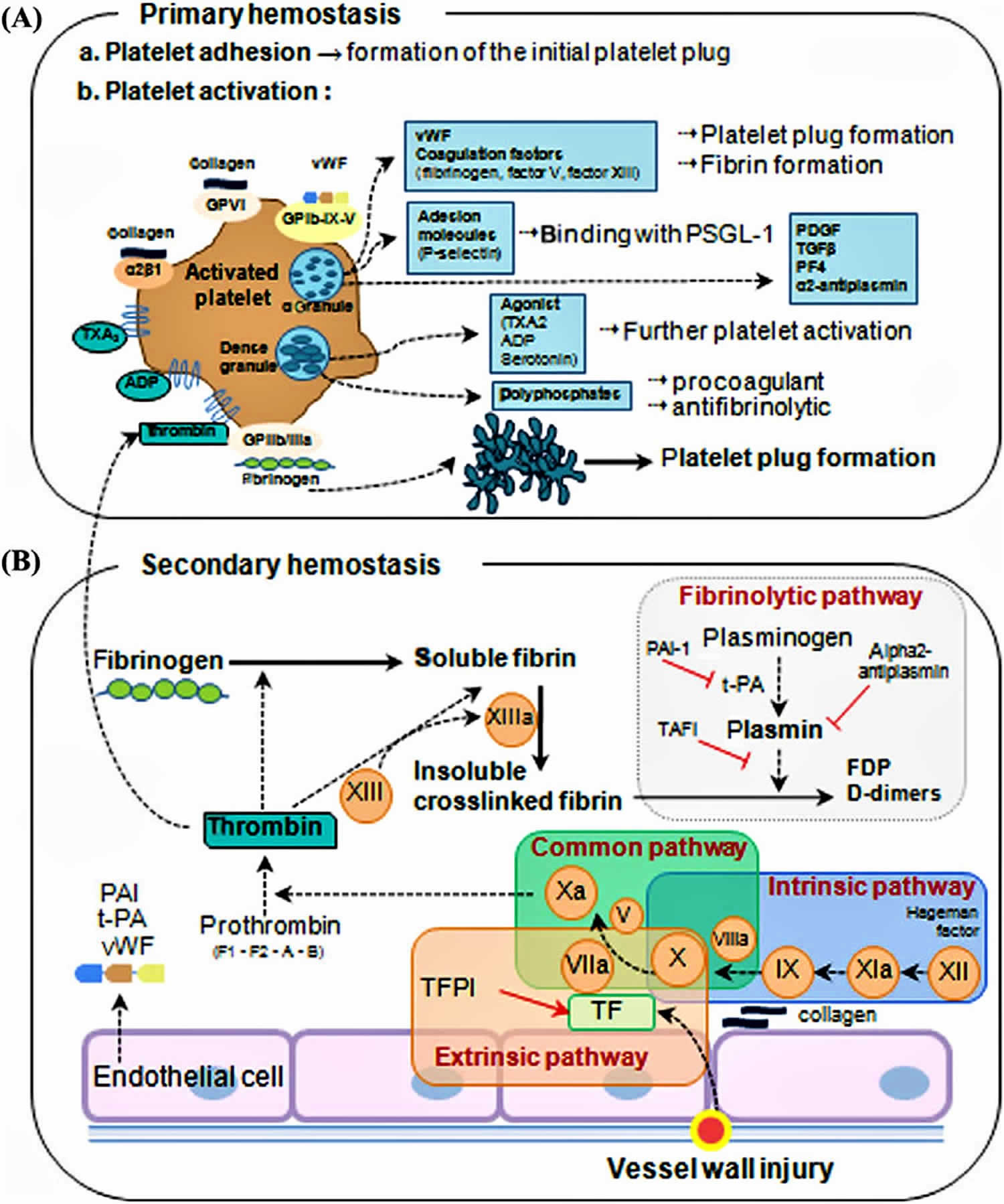
- Primary hemostasis: Formation of a weak platelet plug
- Secondary hemostasis: Stabilizing the weak platelet plug into a clot by the fibrin network
Figure 1. Overview of blood coagulation
Primary hemostasis
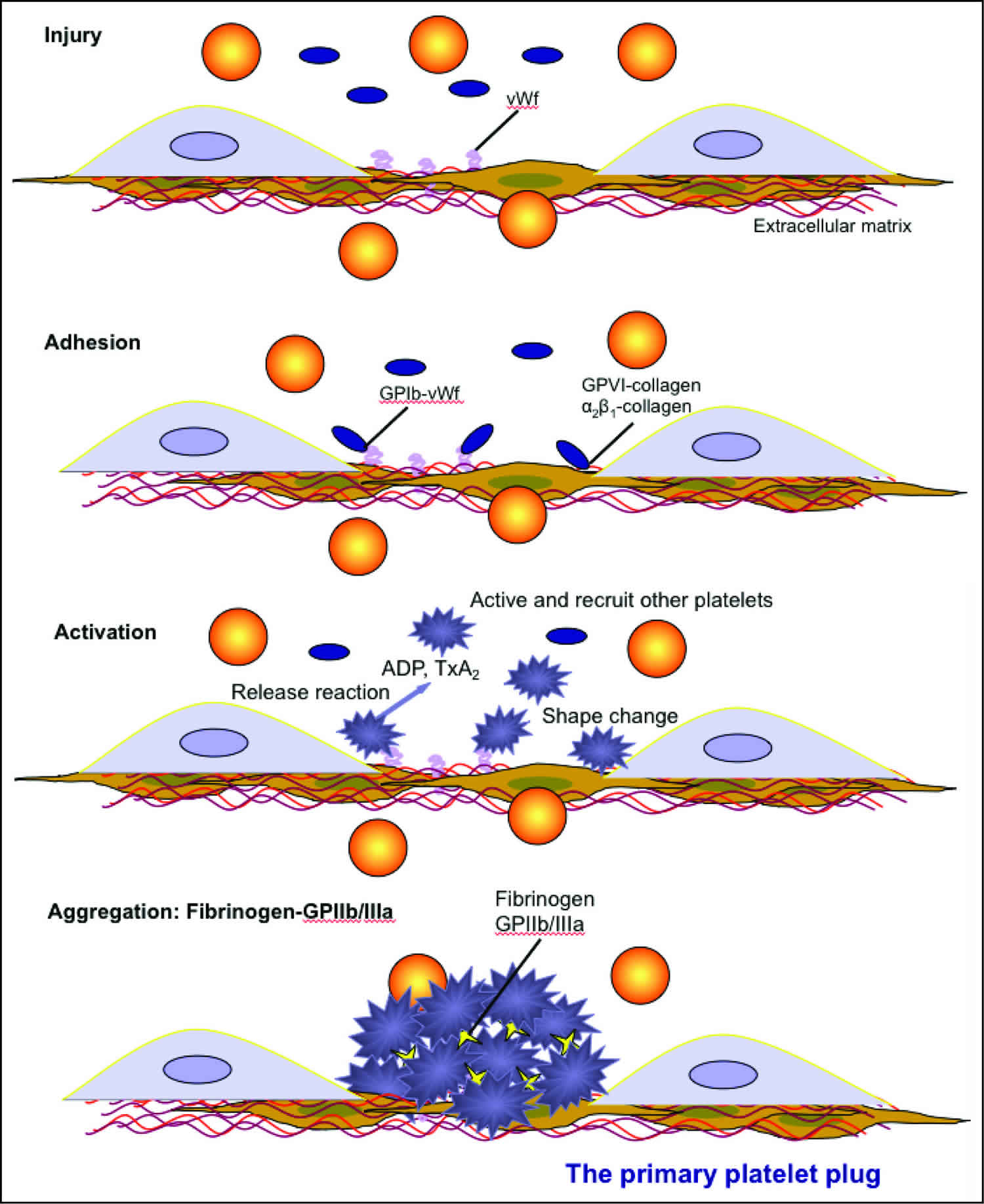
Primary hemostasis is the formation of a weak platelet plug which is achieved in four phases 1:
- Vasoconstriction,
- Platelet adhesion,
- Platelet activation, and
- Platelet aggregation.
Vasoconstriction is the initial response whenever there is vessel injury. Vasospasm of the blood vessels occurs first in response to injury of the vasculature. This vasospasm, in turn, stimulates vasoconstriction. Vasoconstriction is primarily mediated by endothelin-1, a potent vasoconstrictor, which is synthesized by the damaged endothelium. Damaged endothelium exposes sub-endothelial collagen, von Willebrand factor (vWF), releases ATP, and inflammatory mediators. von Willebrand factor is synthesized by megakaryocytes which later gets stored in a-granules of platelets. Weibel-Palade bodies of the endothelium also synthesize von Willebrand factor (vWF). It is the combination of exposure of von Willebrand factor, subendothelial collagen, ATP, and inflammatory mediators which provide the gateway into the second phase of primary hemostasis, platelet adhesion 1. The von Willebrand factor (vWF) is responsible for helping platelets stick to the injured blood vessel wall. von Willebrand factor (vWF) is also the carrier protein for factor VIII. A deficiency in von Willebrand factor can cause von Willebrand disease, an inherited bleeding disorder. While von Willebrand factor may be ordered along with coagulation factors if an inherited factor deficiency is suspected, it is usually considered separately because it is mainly associated with platelets and not part of the classic coagulation cascade.
Platelet adhesion is the process by which platelets attach to the exposed subendothelial von Willebrand factor (vWF). Post vascular damage, platelets begin to roll along vessel walls and adhere to areas of exposed subendothelial collagen and von Willebrand factor (vWF). Platelet membranes are rich in G protein (Gp) receptors located within the phospholipid bilayer. Specifically, it is Gp Ib-IX receptor on platelets that bind to vWF within the endothelium that creates the initial connection between the two. Once bound, a variety of events can occur in the third phase of primary hemostasis to activate the platelet.
Platelet activation consists of platelets undergoing two specific events once they have adhered to the exposed von Willebrand factor (i.e. the damaged vessel site). First, platelets will undergo an irreversible change in shape from smooth discs to multi-pseudopodal plugs, which greatly increases their surface area. Second, platelets secrete their cytoplasmic granules.
Platelet activation is mediated via thrombin by two mechanisms. Thrombin directly activates platelets via proteolytic cleavage by binding the protease-activated receptor. Thrombin also stimulates platelet granule release which includes serotonin, platelet activating factor, and Adenosine Diphosphate (ADP). ADP is an important physiological agonist which is stored specifically in the dense granules of platelets. When Adenosine Diphosphate (ADP) is released, it binds to P2Y1 and P2Y12 receptors on platelet membranes. P2Y1 induces the pseudopod shape change and aids in platelet aggregation. P2Y12 plays a major role in inducing the clotting cascade. When Adenosine Diphosphate (ADP) binds to its receptors, it induces Gp IIb/IIIa complex expression at the platelet membrane surface. The Gp IIb/IIIa complex is a calcium-dependent collagen receptor which is necessary for platelet-to-endothelial adherence and platelet-to-platelet aggregation. Simultaneously, platelets synthesize Thromboxane A2 (TXA2). Thromboxane A2 (TXA2) further intensifies vasoconstriction and platelet aggregation (next step in the primary hemostasis process). The process of platelet activation readies the local environment for platelet aggregation.
Platelet aggregation begins once platelets have been activated. Once activated, the Gp IIb/IIIa receptors adhere to von Willebrand factor (vWF) and fibrinogen. Fibrinogen is found in the circulation and forms a connection between the Gp IIb/IIIa receptors of platelets to interconnect them with each other. This ultimately forms the weak platelet plug.
Ultimately, primary hemostasis allows the culmination of a weak platelet plug to temporarily protect from hemorrhage until further stabilization of fibrinogen to fibrin via thrombin occurs in secondary hemostasis.
Figure 2. Primary hemostasis
Secondary hemostasis
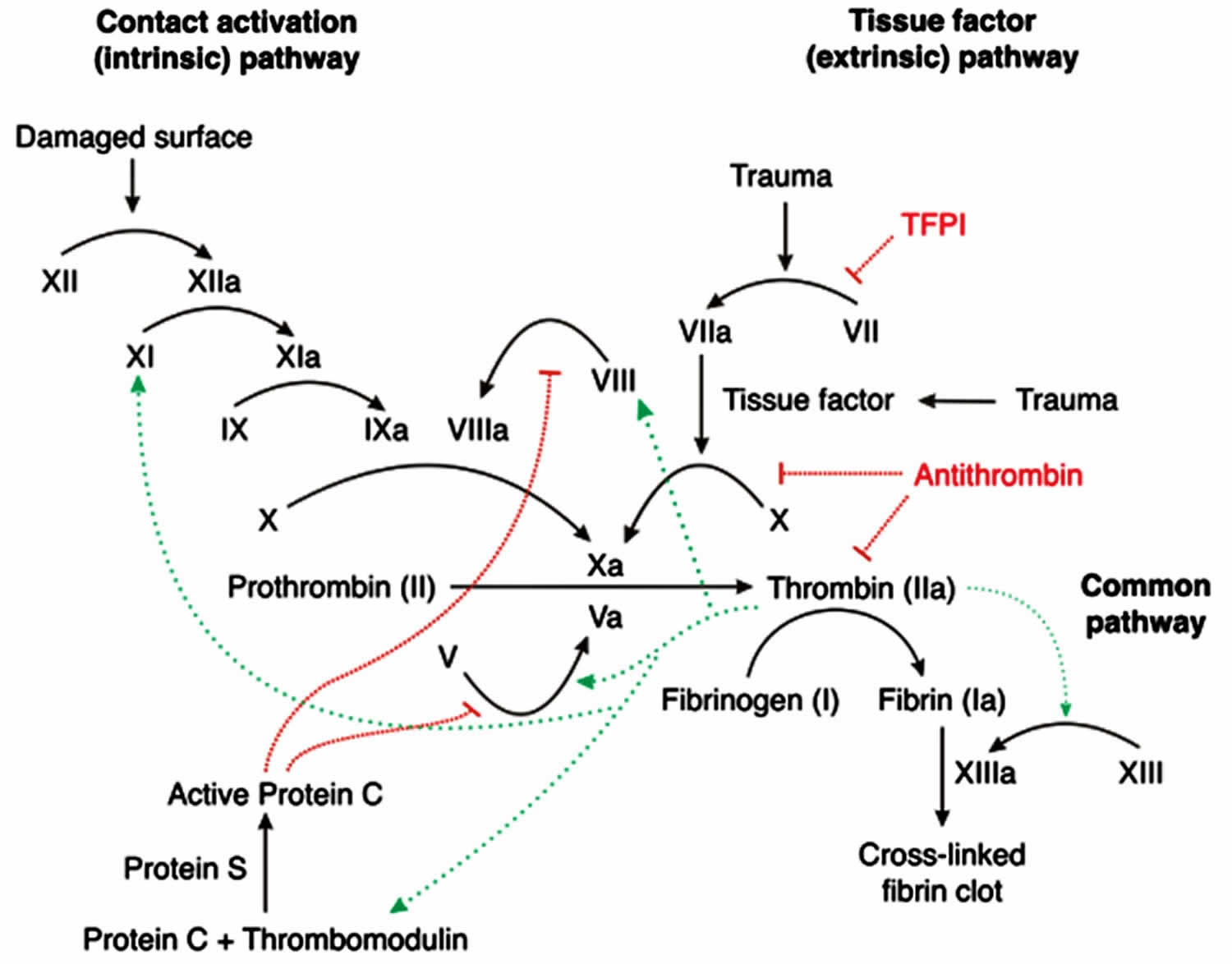
Secondary hemostasis involves the clotting factors acting in a cascade to ultimately stabilize the weak platelet plug. This is accomplished by completing three tasks: (1) triggering activation of clotting factors, (2) conversion of prothrombin to thrombin, and (3) conversion of fibrinogen to fibrin. These tasks are accomplished initially by 1 of 2 pathways; the extrinsic and intrinsic pathway, which converge at the activation of factor X and then complete their tasks via the common pathway. Please note that calcium ions are required for the entire process of secondary hemostasis.
The extrinsic pathway includes tissue factor (TF) and factor VII (FVII). It is initiated when TF binds to FVII, activating FVII to factor VIIa (FVIIa), forming a TF-FVIIa complex. This complex, in turn, activates factor X (FX). Note, the TF-FVIIa complex can also activate factor IX of the intrinsic pathway, which is called the alternate pathway. Once Factor X is activated to FXa by TF-FVIIa complex, the cascade continues down the common pathway (see below).
The intrinsic pathway includes Hageman factor (FXII), factor I (FXI), factor IX (FIX), and factor VIII (FVIII). The process is initiated when FXII comes into contact with exposed subendothelial collagen and becomes activated to FXIIa. Subsequently, FXIIa activates FXI to FXIa, and FXIa activates FIX to FIXa. FIXa works in combination with activated factor VIII (FVIIIa) to activate factor X. Once Factor X is activated by FIXa-FVIIIa complex, the cascade continues down the common pathway (see below).
The common pathway is initiated via activation of Factor Xa. Factor Xa combines with Factor Va and calcium on phospholipid surfaces to create a prothrombinase complex ultimately activating prothrombin (aka Factor II) into thrombin. This activation of thrombin occurs via serine protease cleaving of prothrombin. Now, thrombin activates factor XIIIa (FXIIIa). FXIIIa crosslinks with fibrin forming the stabilized clot.
Figure 3. Coagulation cascade
Coagulation factors
Coagulation factors are proteins circulating in the blood that are essential for proper blood clot formation. Coagulation factor tests measure the function of or sometimes the amount of these proteins in the blood.
Blood clotting is a complex process that involves numerous coagulation factors, which are produced by the liver and blood vessels. Each coagulation factor is evaluated with one or more tests. When factor levels are low, it can cause blood clotting to fail, leading to unexplained bleeding episodes. Measuring coagulation factors can help a healthcare practitioner determine the cause of the bleeding and the best treatment.
Coagulation factors are usually tested by measuring the factor’s activity level in the blood. Activity assays can detect reduced levels of protein or proteins that don’t function properly. Rarely, the amount (antigen level) of a coagulation factor may also be measured. Coagulation factor antigen tests can tell how much of the protein is present, but not whether its function is normal.
When someone bleeds (e.g., with an injury), the coagulation system is activated, plugging the leaking blood vessel with a clot. The coagulation system consists of a series of coagulation factors that activate in a step-by-step process called the coagulation cascade. The end result is the formation of insoluble fibrin threads that link together at the site of injury, along with aggregated cell fragments called platelets, to form a stable blood clot. The clot prevents additional blood loss and remains in place until the injured area has healed.
Blood clotting is dynamic; once a clot is formed, other factors are activated that slow clotting or dissolve the clot in a process called fibrinolysis. The clot is eventually removed after the injury site heals. In normal healthy individuals, this balance between clot formation and removal ensures that bleeding does not become excessive and that clots are removed once they are no longer needed.
For people with bleeding disorders, clotting does not work properly because they lack platelets or coagulation factors, or their platelets or factors don’t work properly. There are a variety of bleeding disorders that may be passed through families (inherited) or acquired after birth. If a person has signs and symptoms of one of these disorders, coagulation factor testing may be ordered to help determine the diagnosis and treatment.
There are nine coagulation factor proteins that can be measured clinically (see table below). These factors are referred to by a name or Roman numeral or both in some cases. For example, coagulation factor II is also known as prothrombin. When one or more of these factors are produced in too small a quantity, or are not functioning correctly, they can cause excessive bleeding.
Table 1. Coagulation factors
| Coagulation factor | other common name |
|---|---|
| I | Fibrinogen |
| II | Prothrombin |
| V | Proaccelerin or labile factor |
| VII | Proconvertin |
| VIII | Antihemophilic factor A |
| IX | Antihemophilic factor B (Christmas factor) |
| X | Stuart-Prower factor |
| XI | Plasma thromboplastin antecendent |
| XIII | Fibrin stabilizing factor |
Coagulation cascade
The coagulation pathway is a cascade of events that leads to hemostasis. The intricate pathway allows for rapid healing and prevention of spontaneous bleeding. Two paths, intrinsic and extrinsic, originate separately but converge at a specific point, leading to fibrin activation 2. The purpose is to ultimately stabilize the platelet plug with a fibrin mesh.
The mechanism by which coagulation allows for hemostasis is an intricate process that is done through a series of coagulation factors. The intrinsic pathway consists of coagulation factors I, II, IX, X, XI, and XII 2. Respectively, each one is named, fibrinogen, prothrombin, Christmas factor, Stuart-Prower factor, plasma thromboplastin, and Hageman factor. The extrinsic pathway consists of factors I, II, VII, and X. Factor VII is called stable factor. The common pathway consists of factors I, II, V, VIII, X. The factors circulate through the bloodstream as zymogens and are activated into serine proteases. These serine proteases act as a catalyst to cleave the next zymogen into more serine proteases and ultimately activate fibrinogen. The following are serine proteases: factors II, VII, IX, X, XI and XII. These are not serine proteases: factors V, VIII, XIII. The intrinsic pathway is activated through exposed endothelial collagen, and the extrinsic pathway is activated through tissue factor released by endothelial cells after external damage.
Intrinsic pathway
This pathway is the longer pathway of secondary hemostasis 2. It begins with the activation of Factor XII (a zymogen, inactivated serine protease) which becomes Factor XIIA (activated serine protease) after exposure to endothelial collagen. Endothelial collagen is only exposed when endothelial damage occurs. Factor XIIA acts as a catalyst to activate factor XI to Factor XIA. Factor XIA then goes on to activate factor IX to factor IXA. Factor IXA goes on to serve as a catalyst for turning factor X into factor Xa. This is known as a cascade. When each factor is activated, it goes on to activate many more factors in the next steps. As you move further down the cascade, the concentration of that factor increases in the blood. For example, the concentration of factor IX is more than that of factor XI. When factor II is activated by either intrinsic or extrinsic pathway, it can reinforce the intrinsic pathway by giving positive feedback to factors V, VII, VIII, XI, XIII. This makes factor XII less critical; patients can actually clot well without factor XII. The intrinsic pathway is clinically measured as the partial thromboplastin time (PTT).
Extrinsic pathway
The extrinsic pathway is the shorter pathway of secondary hemostasis 2. Once the damage to the vessel is done, the endothelial cells release tissue factor which goes on to activate factor VII to factor VIIa. Factor VIIa goes on to activate factor X into factor Xa. This is the point where both extrinsic and intrinsic pathways become one. The extrinsic pathway is clinically measured as the prothrombin time (PT).
Common Pathway
This pathway begins at factor X which is activated to factor Xa. The process of activating factor Xa is a complicated reaction. Tenase is the complex that cleaves factor X into factor Xa. Tenase has two forms: extrinsic, consisting of factor VII, factor III (tissue factor) and Ca2+, or intrinsic, made up of cofactor factor VIII, factor IXA, a phospholipid, and Ca2+. Once activated to factor Xa, it goes on to activate factor II (prothrombin) into factor IIa (thrombin). Also, factor Xa requires factor V as a cofactor to cleave prothrombin into thrombin. Factor IIa (thrombin) goes on to activate fibrinogen into fibrin. Thrombin also goes on to activate other factors in the intrinsic pathway (factor XI) as well as cofactors V and VIII and factor XIII. Fibrin subunits come together to form fibrin strands, and factor XIII acts on fibrin strands to form a fibrin mesh. This mesh helps to stabilize the platelet plug.
Negative Feedback
To prevent over-coagulation, which causes widespread thrombosis, there are certain processes to keep the coagulation cascade in check. As thrombin acts as a procoagulant, it also acts as a negative feedback by activating plasminogen to plasmin and stimulating the production of antithrombin (AT). Plasmin acts directly on the fibrin mesh and breaks it down. AT decreases the production of thrombin from prothrombin and decreases the amount of activated factor X.
Protein C and S also act to prevent coagulation, mainly by inactivating factors V and VIII.
Organs Involved
One of the organs intimately involved in the coagulation process is the liver. The liver is responsible for the formation of factors I, II, V, VII, VIII, IX, X, XI, XIII, and protein C and S. Factor VII is created by the vascular endothelium.
Pathology to the liver can cause lack of coagulation factors and lead to hemorrhage. A decrease in coagulation factors typically means severe liver damage. Factor VII has the shortest half-life, leading to elevated PT first in liver disease. International normalized ratio (INR) can be greater than 6.5 (normal is close to 1.0). Coagulopathy in liver disease is treated with fresh frozen plasma.
Coagulation defect
Thrombosis is the process of blood clot (thrombus) formation in a blood vessel. Virchow triad is an important concept that highlights the primary abnormalities in pathology that can lead to the clotting mechanism proceeding to thrombosis. The triad is composed of stasis or turbulent blood flow, endothelial injury, and hypercoagulability of the blood.
- Abnormal (stasis) or turbulent blood flow can lead to thrombosis. Normal blood flow is laminar. Turbulent blood flow leads to endothelial injury thus promoting the formation of a thrombus. An example of turbulent blood flow is in the aneurysm of weakened vessels. Another aspect of abnormal blood flow, venous stasis, such as in post-operative bed rest, long distance traveling in a car or plane, or immobility due to obesity can lead to endothelial injury thus promoting thrombosis.
- Endothelial Injury leads to platelet activation and the formation of a thrombus. This may be a result of inflammation of the endothelial surface of the vasculature. Hypercholesterolemia is an example of a chronic inflammatory condition which progresses into endothelial injury.
- Hypercoagulability (thrombophilia) is any disorder of the blood that predisposes a person to thrombosis. This may be a result of inherited clotting disorders such as a Factor V Leiden mutation or an acquired clotting disorder such as disseminated intravascular coagulation.
Hemorrhage occurs when blood escapes from its vessel walls.
Platelet dysfunction, or clotting factor dysfunction, can be further broken down into which part of the clotting mechanism physiology is affected.
Disorders of Primary Hemostasis
von Willebrand factor (vWF), Platelet defects, or Receptor Interference disorders:
- Von Willebrand Factor disease
- Bernard-Soulier disease
- Glanzmann thrombasthenia
- Medication-induced
Disorders of Secondary Hemostasis
Clotting Factor Defects
- Factor V Leiden — Factor V Leiden is a genetic mutation more prevalent in people European descent. This defect causes a state of hypercoagulability. The genetic mutation causes a defect in factor V such that protein C cannot inactivate it, allowing factor V to continuously activate downstream factors.
- Vitamin K deficiency — Vitamin K deficiency can lead to elevated prothrombin time (PT) and partial thromboplastin time (PTT). It can present as hemarthrosis, intramuscular bleeding, or gastrointestinal bleeding. Vitamin K deficiency is commonly seen in newborns due to the lack of gut colonization by bacteria. It also can be seen in malabsorption (cystic fibrosis, celiacs disease, Crohn disease).
- Hemophilia
- Hemophilia A and B are inherited in an x-linked recessive pattern. In hemophilia A there is a deficiency in factor VIII. In hemophilia B there is a deficiency in factor IX.
- Hemophilia C is an autosomal recessive mutation, where there is a deficiency in factor XI.
- Anti-phospholipid antibody syndrome
- Disseminated intravascular coagulation
- Liver disease
- Medication-induced
- Deficiencies in protein C and S also can lead to hypercoagulable states due to an inability to appropriately inhibit factors V and VIII respectively.
Defects in Small Vessels
- Trauma
- Aneurysm rupture
- Vasculitides
Clinical Significance
In addition to the pathophysiology, a few ideas to keep in mind when you have a patient with clotting mechanism disorders:
Patients with:
- Primary hemostasis defects typically present with small bleeds in the skin or mucosal membranes. This includes petechiae and/or purpura.
- Secondary hemostasis defects typically present with bleeds into soft tissue (muscle) or joints (hemarthrosis).
- Direct defects in small blood vessels typically present with palpable purpura and ecchymosis. These may collect and become larger to develop a hematoma.
Also, laboratory testing involving PTT or PT/INR can be divided by the physiological mechanisms:
- Disorders exclusively effecting primary hemostasis do not affect the prothrombin time (PT)/international normalized ratio (INR) or partial thromboplastin time (PTT), they only increase bleeding time
- Disorders that affect the extrinsic pathway of secondary hemostasis affect the prothrombin time (PT)/international normalized ratio (INR)
- Disorders that affect the intrinsic pathway of secondary hemostasis affect the partial thromboplastin time (PTT)
Blood coagulation test
Coagulation factor testing is performed to determine if a person has enough coagulation activity to control the blood clotting process. It is used by healthcare practitioners to determine if the level of a coagulation factor is low or absent (below the detectable limit) or if it is too high. Low coagulation factor activity is associated with reduced clot formation and excess bleeding. High coagulation factor activity can be associated with too much clot formation (thrombosis) and blockade in the circulatory system (thromboembolism).
One or more coagulation factor activity tests may be ordered to evaluate the function of specific factors. If a test result shows reduced activity, then an antigen test may be ordered as a follow-up test. This is occasionally done to determine if the low activity is due to a decreased quantity of a normal coagulation factor or due to a coagulation factor that does not function normally.
Coagulation factor testing may sometimes be ordered if a person has a family history of bleeding.
Prothrombin time (PT) and partial thromboplastin time (PTT) evaluate the time it takes for the extrinsic and intrinsic blood coagulation pathways to take effect, respectively. Mixing studies are done to determine whether a prothrombin time (PT) or partial thromboplastin time (PTT) is elevated due to a factor deficiency or a factor inhibitor (antibodies to specific factors). It is done by mixing the patient’s plasma with a control plasma. If the mixed plasma prothrombin time (PT) and partial thromboplastin time (PTT) normalize, the prothrombin time (PT) and partial thromboplastin time (PTT) prolongation is due to a factor deficiency. If they do not normalize, the prolongation is due to a factor inhibitor. An example of an inhibitor is lupus anticoagulant.
When is blood coagulation test ordered?
Coagulation factor tests are typically ordered when someone has a prolonged prothrombin time (PT) and/or partial thromboplastin time (PTT). The prothrombin time (PT) and partial thromboplastin time (PTT) tests are used as screening tools to determine if someone has a coagulation problem or as part of an investigative workup when someone has signs and symptoms of a bleeding disorder, such as unexplained bruising, bleeding gums, excess bleeding from small cuts, or frequent nose bleeds.
Factor activity may be measured when a healthcare practitioner suspects that a patient has an acquired condition that is causing bleeding, such as vitamin K deficiency or liver disease.
Factor testing may be done when a healthcare practitioner suspects that a patient has an inherited coagulation factor deficiency such as Von Willebrand disease or hemophilia A, especially when bleeding episodes begin early in life or when a close relative has an inherited factor deficiency. When an inherited deficiency is suspected, other family members may also be tested to help confirm the person’s diagnosis and to establish whether they may be carriers of the condition or have the deficiency themselves (in an asymptomatic or less severe form).
Sometimes factor testing may be done for a person with a known deficiency to monitor the factor deficiency and to evaluate the effectiveness of treatment.
Occasionally, factor testing is ordered when someone has an unexplained blood clot (thrombosis) to determine whether abnormally high factor activity is the underlying cause (e.g., elevated factor VIII activity is associated with excess clotting).
What is prothrombin time?
The prothrombin time (PT) is a test that helps evaluate a person’s ability to appropriately form blood clots. The international normalized ratio (INR) is a calculation based on results of a prothrombin time that is used to monitor individuals who are being treated with the blood-thinning medication (anticoagulant) warfarin (Coumadin®).
A prothrombin time measures the number of seconds it takes for a clot to form in a person’s sample of blood after substances (reagents) are added. The prothrombin time is often performed with a partial thromboplastin time (PTT) and together they assess the amount and function of proteins called coagulation factors that are an important part of proper blood clot formation.
In the body, when there is an injury and bleeding occurs, the clotting process called hemostasis begins. This process involves in part a series of sequential chemical reactions called the coagulation cascade, in which coagulation or “clotting” factors are activated one after another and result in the formation of a clot. There must be a sufficient quantity of each coagulation factor, and each must function properly, in order for normal clotting to occur. Too little can lead to excessive bleeding; too much may lead to excessive clotting.
In a test tube during a laboratory test, there are two “pathways” that can initiate clotting, the so-called extrinsic and intrinsic pathways. Both of these then merge into a common pathway to complete the clotting process. The prothrombin time (PT) test evaluates how well all of the coagulation factors in the extrinsic and common pathways of the coagulation cascade work together. Included are: factors I (Fibrinogen), II (Prothrombin), V, VII and X. The partial thromboplastin time (PTT) test evaluates those protein factors that are part of the intrinsic and common pathways: XII, XI, IX, VIII, X, V, II (prothrombin), and I (fibrinogen) as well as prekallikrein (PK) and high molecular weight kininogen (HK). The prothrombin time (PT) and partial thromboplastin time (PTT) evaluate the overall ability to produce a clot in a reasonable amount of time and, if any of these factors are deficient in quantity or not functioning properly, the test results will be prolonged.
The prothrombin time test is usually measured in seconds and is compared to a normal range that reflects prothrombin time values in healthy individuals. Because the reagents used to perform the prothrombin time test vary from one laboratory to another and even within the same laboratory over time, the normal ranges also will fluctuate. To standardize results across different laboratories in the U.S. and the world, a World Health Organization (WHO) committee developed and recommended the use of the Internationalized Normalized Ratio (INR), calculated based on the prothrombin time test result, for people who are receiving the anticoagulant warfarin (Coumadin®).
The Internationalized Normalized Ratio (INR) is a calculation that adjusts for changes in the prothrombin time reagents and allows for results from different laboratories to be compared. Most laboratories report both prothrombin time and Internationalized Normalized Ratio (INR) values whenever a prothrombin time test is performed. The INR should be only applicable, however, for those taking the blood-thinning medication warfarin.
A prothrombin time and Internationalized Normalized Ratio (INR) are ordered on a regular basis when a person is taking the anticoagulant drug warfarin to ensure that the prescription is working properly and that the prothrombin time/Internationalized Normalized Ratio (INR) is appropriately prolonged. There is no set frequency for doing the test. A health practitioner will order them often enough to make sure that the drug is producing the desired effect – that it is increasing the person’s clotting time to a therapeutic level without significant risk of excessive bleeding or bruising.
The prothrombin time (PT) may be ordered when a person who is not taking anticoagulant drugs has signs or symptoms of excessive bleeding or clotting, such as:
- Unexplained bleeding or easy bruising
- Nosebleeds
- Bleeding gums
- A blood clot in a vein or artery
- An acute condition such as disseminated intravascular coagulation (DIC) that may cause both bleeding and clotting as coagulation factors are used up at a rapid rate
- A chronic condition such as severe liver disease that may affect hemostasis
Prothrombin time (PT), along with partial thromboplastin time (PTT), may be ordered prior to surgery when the surgery carries an increased risk of blood loss and/or when the person has a clinical history of bleeding, such as frequent or excessive nosebleeds and easy bruising, which may indicate the presence of a bleeding disorder.
What is partial thromboplastin time?
The partial thromboplastin time (PTT) is a screening test that helps evaluate a person’s ability to appropriately form blood clots. It measures the number of seconds it takes for a clot to form in a person’s sample of blood after substances (reagents) are added. The partial thromboplastin time assesses the amount and the function of certain proteins called coagulation factors that are an important part of blood clot formation.
When body tissue(s) or blood vessel walls are injured, bleeding occurs and a process called hemostasis begins. Small cell fragments called platelets adhere to and then clump (aggregate) at the injury site. At the same time, a process called the coagulation cascade begins and coagulation factors are activated. Through the cascading reactions, threads called fibrin form and crosslink into a net that adheres to the injury site and stabilizes it. Along with the platelets adhering, this forms a stable blood clot to seal off injuries to blood vessels, prevents additional blood loss, and gives the damaged areas time to heal.
Each component of this hemostatic process must function properly and be present in sufficient quantity for normal blood clot formation. If there is a deficiency in one or more of these factors, or if the factors function abnormally, then a stable clot may not form and bleeding continues.
With a partial thromboplastin time, a person’s sample is compared to a normal reference interval for clotting time. When a person’s partial thromboplastin time takes longer than normal to clot, the partial thromboplastin time is considered “prolonged.” A prolonged partial thromboplastin time may be due to a condition that decreases or creates a dysfunction in one or more coagulation factors. Less often, it may be due to a condition in which the body produces certain antibodies directed against one or more coagulation factors, affecting their function.
Sometimes a partial thromboplastin time (PTT) may be prolonged because the person tested produces an autoantibody called an antiphospholipid antibody that interferes with the partial thromboplastin time (PTT) test. This type of antibody affects the results of the test because it targets substances called phospholipids that are used in the partial thromboplastin time. Though antiphospholipid antibodies can prolong the partial thromboplastin time test result, in the body they are associated with excessive clotting. A person who produces these antibodies may be at an increased risk for a blood clot. A partial thromboplastin time maybe used as part of an evaluation of a person with signs and symptoms of excessive clotting or antiphospholipid syndrome.
When a partial thromboplastin time (PTT) is used to investigate bleeding or clotting episodes, it is often ordered along with a prothrombin time (PT). A health practitioner will evaluate the results of both tests to help determine the cause of bleeding or clotting episode(s).
The partial thromboplastin time (PTT) may be ordered along with other tests such as a prothrombin time (PT) when a person has:
- Unexplained bleeding or easy bruising
- A blood clot in a vein or artery
- An acute condition such as disseminated intravascular coagulation (DIC) that may cause both bleeding and clotting as coagulation factors are used up at a rapid rate
- A chronic condition such as liver disease that may affect hemostasis
A partial thromboplastin time (PTT) may be ordered:
- When someone has had a blood clot or when a woman has had recurrent miscarriages, as part of an evaluation for lupus anticoagulant, anticardiolipin antibodies, and antiphospholipid syndrome
- On a regular basis, when a person is on standard (unfractionated) heparin therapy; when someone is switched from heparin therapy to longer-term warfarin (Coumadin®) therapy, the two are overlapped and both the partial thromboplastin time and PT are monitored until the person has stabilized.
- Prior to surgery when the surgery carries an increased risk of blood loss and/or when the person has a clinical history of bleeding, such as frequent or excessive nose bleeds and easy bruising, which may indicate the presence of a bleeding disorder
It is now understood that coagulation tests such as the prothrombin time (PT) and partial thromboplastin time (PTT) are based on what happens artificially in the test setting (in vitro) and thus do not necessarily reflect what actually happens in the body (in vivo). Nevertheless, they can be used to evaluate certain components of the hemostasis system. The partial thromboplastin time and prothrombin time (PT) tests each evaluate coagulation factors that are part of different groups of chemical reaction pathways in the cascade, called the intrinsic, extrinsic, and common pathways.
Partial thromboplastin time (PTT) results are typically reported in seconds. A partial thromboplastin time result that falls within a laboratory’s reference interval usually indicates normal clotting function. However, mild to moderate deficiencies of a single coagulation factor may be present. The partial thromboplastin time may not be prolonged until the factor levels have decreased to 30% to 40% of normal. Also lupus anticoagulant may be present but may not prolong the partial thromboplastin time result. If the lupus anticoagulant is suspected, a more sensitive lupus anticoagulant-sensitive partial thromboplastin time or a dilute Russell viper venom time can be used to test for it.
A prolonged partial thromboplastin time means that clotting is taking longer to occur than normal and may be due to a variety of causes. Often, this suggests that there may be a coagulation factor deficiency or a specific or nonspecific antibody (inhibitor) affecting the body’s clotting ability. Coagulation factor deficiencies may be acquired or inherited.
Prolonged partial thromboplastin time (PTT) tests may be due to:
- Inherited factor deficiencies:
- von Willebrand disease is the most common inherited bleeding disorder and it affects platelet function due to decreased von Willebrand factor.
- Hemophilia A and hemophilia B (Christmas disease) are two other inherited bleeding disorders resulting from a decrease in factors VIII and IX, respectively.
- Deficiencies of other coagulation factors, like factors XII and XI
- Acquired blood coagulation factor deficiencies:
- An example of an acquired deficiency is one due to lack of vitamin K. Vitamin K is essential for the formation of coagulation factors. Vitamin K deficiencies are rare but can occur due to an extremely poor diet, malabsorption disorders, or prolonged use of certain antibiotics, for example.
- Most coagulation factors are produced by the liver, thus liver disease may cause prolonged prothrombin time (PT) and partial thromboplastin time (PTT). With liver disease and vitamin K deficiency, prothrombin time (PT) is more likely to be prolonged than is partial thromboplastin time (PTT).
- A nonspecific inhibitor such as the lupus anticoagulant—the presence of these inhibitors is usually associated with inappropriate clotting (thrombosis), but can prolong the partial thromboplastin time (PTT).
- A specific inhibitor—although relatively rare, these are antibodies that specifically target certain coagulation factors, such as antibodies that target factor VIII. They may develop in someone with a bleeding disorder who is receiving factor replacements (such as factor VIII, which is used to treat hemophilia A) or spontaneously as an autoantibody. Factor-specific inhibitors can cause severe bleeding.
- Heparin—is an anticoagulant and will prolong a partial thromboplastin time (PTT), either as a contaminant of the sample or as part of anticoagulation therapy. For anticoagulant therapy, the target partial thromboplastin time is often about 1.5 to 2.5 times longer than a person’s pretreatment level.
- Warfarin (Coumadin®) anticoagulation therapy—the partial thromboplastin time is not used to monitor warfarin therapy, but it may be affected by it. Typically, the prothrombin time (PT) is used to monitor warfarin therapy.
- Other anticoagulants—anticoagulation therapy with direct thrombin inhibitor (e.g., argatroban, dabigatran) or direct factor Xa inhibitor (e.g., rivaroxaban)
- Prolonged partial thromboplastin time (PTT) levels may also be seen with leukemia, excessive bleeding in pregnant women prior to or after giving birth, or recurrent miscarriages.
Results of the partial thromboplastin time (PTT) are often interpreted with that of the prothrombin time (PT) in determining what condition may be present.
Table 2. Abnormal partial thromboplastin time (PTT) and prothrombin time (PT) causes
| Prothrombin time (PT) result | Partial thromboplastin time (PTT) result | Common condition present |
|---|---|---|
| Prolonged | Normal | Liver disease, decreased vitamin K, decreased or defective factor VII |
| Normal | Prolonged | Hemophilia A or B (decreased or defective factor VIII or IX) or factor XI deficiency, von Willebrand disease, factor XII deficiency, or lupus anticoagulant present |
| Prolonged | Prolonged | Decreased or defective factor I (fibrinogen), II (prothrombin), V or X, severe liver disease, disseminated intravascular coagulation (DIC) |
| Normal | Normal or slightly prolonged | May indicate normal hemostasis; however PT and PTT can be normal in conditions such as mild deficiencies in other factors and mild form of von Willebrand disease. Further testing may be required to diagnose these conditions. |
Shortened partial thromboplastin time (PTT) tests may be due to:
- Disseminated intravascular coagulation (DIC) —in the early stages of DIC, there are circulating procoagulants that shorten the partial thromboplastin time (PTT).
- Extensive cancer (ovarian, pancreatic, colon), except when the liver is involved
- An acute-phase reaction: this is a condition causing pronounced tissue inflammation or trauma that elevates factor VIII levels. It is usually a temporary change that is not monitored with a partial thromboplastin time (PTT) test. When the condition causing the acute phase reaction is resolved, the PTT will return to normal.
Several factors can affect results of a partial thromboplastin time (PTT) and the interpretation of test results:
- People with high hematocrit levels may have prolonged partial thromboplastin times (in vitro artifact).
- Heparin contamination – this is the most common problem, especially when blood is collected from intravenous lines that are being kept “open” with heparin washes.
- Drugs such as antihistamines, vitamin C (ascorbic acid), aspirin, and chlorpromazine
- In some cases, heparin can unintentionally decrease a person’s platelet count in a complication called heparin-induced thrombocytopenia. When this occurs, substitute anticoagulants such as a direct thrombin inhibitor (e.g., argatroban or bivalirudin) may be given. The partial thromboplastin time test is also used to monitor these therapies. It does not directly measure the anticoagulants used but measures their effect on blood clotting.
Is the partial thromboplastin time always used to monitor heparin therapy?
In a few situations, it is not.
- When very high doses of heparin are used, as may occur during open-heart surgery, the partial thromboplastin time (PTT) loses its sensitivity; it will not clot. At this intense level of anticoagulation, the activated clotting time (ACT) can be used as a monitoring tool.
- Some doctors and laboratories now monitor standard (unfractionated) heparin therapy using the chromogenic anti-factor Xa test.
- Low molecular weight heparin (LMWH) is a fast-acting form of heparin often used in the treatment of conditions such as deep vein thrombosis (DVT) prevention. Though generally not requiring monitoring, it must be monitored using the anti-factor Xa test.
- For people with lupus anticoagulant and clotting and who are being treated with heparin, the partial thromboplastin time (PTT) is not reliable; thus the anti-factor Xa assay must be used to monitor their heparin therapy.
What does abnormal blood coagulation test result mean?
Normal coagulation factor activity usually means normal clotting function. Low activity of one or more coagulation factors usually means impaired clotting ability. Each coagulation factor must be present in sufficient quantity in order for normal clotting to occur, but the level required is different for each factor. Results are frequently reported as a percentage with 100% being normal. For example, a factor VIII that is 30% would be considered abnormally low.
Deficiencies in coagulation factors can develop in several different ways. They may be acquired as part of other diseases or inherited. Deficiencies vary in severity and may be permanent or temporary.
If more than one clotting factor is decreased, it is usually due to an acquired condition.
Acquired deficiencies are rare and may be caused by chronic or acute conditions, including:
- Excess blood clotting that uses up coagulation factor (e.g., disseminated intravascular coagulation)
- Liver disease (i.e., cirrhosis)
- Some cancers
- Exposure to snake venom
- Fat malabsorption
- Vitamin K deficiency
- Anticoagulation therapy (warfarin)
- Massive blood transfusions (e.g., transfuse only red blood cell units)
Inherited coagulation factor deficiencies are rare. They tend to involve only one factor, which may be reduced or absent (below the detectable limit).
Hemophilia A and B are the most common examples of inherited disorders. They are X-linked deficiencies of factors VIII and IX that occur almost exclusively in men. Women are more likely to be asymptomatic carriers of these genetic traits or have mild bleeding. Other inherited factor deficiencies, not associated with the X chromosome, are found equally in both men and women.
The severity of symptoms associated with an inherited factor deficiency depends on the factor involved, its functionality, and the amount of factor available. Symptoms may vary from episode to episode, ranging from excessive bleeding after dental procedures to severe recurrent bleeding into joints or muscles.
Individuals with a modest reduction in coagulation factor activity may have few symptoms and may discover their deficiency in adulthood, after a surgical procedure or trauma or during screening that includes a prothrombin time (PT) or partial thromboplastin time (PTT) test.
Individuals with severe factor deficiencies may have their first bleeding episode very early; for example, a male infant with a deficiency of Factor VIII, IX, or XIII may bleed excessively after circumcision.
For both inherited and acquired factor deficiencies, the missing factor(s), once identified, can be replaced as needed. This may be done with a transfusion of fresh frozen plasma (FFP), which contains various coagulation factors, with a concentrated cryoprecipitate, or with replacement factors (some are available commercially, such as recombinant factor VIII). These treatments may be used during a bleeding episode or as a preventive measure to prevent excessive bleeding during an upcoming surgery or other invasive procedure.
Elevated levels of several coagulation factors are seen in situations of acute illness, stress, or inflammation. Some people have persistent elevations of factor VIII that may be associated with an increased risk of venous thrombosis.
Why are some inherited bleeding disorders more severe than others?
The severity of bleeding depends on the individual, the degree of abnormality of the factor involved, as well as which factor is deficient. Those who have a severely deficient factor or one with significant factor dysfunction will have more severe manifestations of the disease. People with one normal gene copy and one altered gene copy (heterozygous) will tend to have less severe bleeding than those with two altered copies (homozygous).
It should be pointed out that people with a deficiency of factor XII are usually asymptomatic. This rare factor deficiency causes abnormal partial thromboplastin time (PTT) results but is not associated with increased bleeding risk.
References- Garmo C, Burns B. Physiology, Clotting Mechanism. [Updated 2018 Jun 21]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK507795
- Chaudhry R, Babiker HM. Physiology, Coagulation Pathways. [Updated 2017 Dec 31]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482253





{kind=link}