
What is Cockayne syndrome
Cockayne syndrome is a rare inherited neurodevelopmental disorder characterized by an abnormally small head size (microcephaly), premature aging (progeria), a failure to gain weight and grow at the expected rate in the newborn (failure to thrive) leading to very short stature, delayed development, impaired nervous system development and moderate to severe learning delay 1. The signs and symptoms of this condition are usually apparent from infancy, and they worsen over time. Most affected individuals have an increased sensitivity to sunlight (photosensitivity), and in some cases even a small amount of sun exposure can cause a sunburn or blistering of the skin. Other signs and symptoms often include hearing loss, vision loss, severe tooth decay, bone abnormalities, hands and feet that are cold all the time, and changes in the brain that can be seen on brain scans 2.
Cockayne syndrome is very rare and is estimated to occur in 2 to 3 per million newborns in the United States and Europe 2. As of 1992, about 140 cases of Cockayne syndrome had been reported in the literature 3. Cockayne Syndrome affects males and females in equal numbers. Cockayne syndrome is caused by mutations in either the ERCC8 (CSA) or ERCC6 (CSB) genes. Inheritance is autosomal recessive 2. There are no indications of ethnic or racial partiality.
People with Cockayne syndrome have a serious reaction to an antibiotic medication called metronidazole. If affected individuals take this medication, it can cause life-threatening liver failure.
Cockayne syndrome is sometimes divided into types 1, 2 and 3 based on the severity of the disease and age of onset of symptoms. However, the differences between the types are not always clear-cut, and some researchers believe the signs and symptoms reflect a spectrum instead of distinct types. Cockayne syndrome type 2 is also known as cerebro-oculo-facio-skeletal (COFS) syndrome, and while some researchers consider it to be a separate but similar condition, others classify it as part of the Cockayne syndrome disease spectrum.
There is no cure yet for Cockayne syndrome. Treatment is supportive and may include educational programs for developmental delay, physical therapy, gastrostomy tube placement as needed; medications for spasticity and tremor as needed; use of sunscreens and sunglasses; treatment of hearing loss and cataracts; and other forms of treatment, as needed 4
Cockayne syndrome type 1
Cockayne syndrome type 1 (type A), sometimes called “classic” or “moderate” form of Cockayne syndrome, diagnosed during early childhood. Cockayne syndrome type 1 (type A) is characterized by an onset of symptoms in early childhood (usually after age 1 year).
Cockayne syndrome type 1 is characterized by a normal appearing newborn whose symptoms may not become apparent until after the first two years. Height and weight, as well as other indicators of size and growth are much within the 5th percentile. Vision, hearing, and nervous system functioning (central and peripheral) gets worse over time and seriously severe disability may result. By the time the disease has become fully manifest, height, weight, and head circumference are far below the fifth percentile. Progressive impairment of vision, hearing, and central and peripheral nervous system function leads to severe disability; death typically occurs in the first or second decade. The mean age of death is 12 years, but survival into the third decade has been reported 4.
Cockayne syndrome type 1 is suspected 4:
- In an older child when both major criteria are present and three minor criteria are present;
- In an infant or toddler when both major criteria are present, especially if there is increased cutaneous photosensitivity.
Major criteria
- Postnatal growth failure (height and weight <5th centile by age 2 years)
- Progressive microcephaly and neurologic dysfunction manifested as early developmental delay in most individuals, followed by progressive behavioral and intellectual deterioration in all individuals; brain MRI reveals leukodystrophy 5. Intracranial calcifications are seen in some individuals.
Minor criteria
- Cutaneous photosensitivity with or without thin or dry skin or hair (~75%)
- Demyelinating peripheral neuropathy diagnosed by electromyography, nerve conduction testing, and/or nerve biopsy
- Pigmentary retinopathy (~55%) and/or cataracts (~36%)
- Sensorineural hearing loss (~60%)
- Dental anomalies, including dental caries (~86%), enamel hypoplasia, anomalies of tooth number and anomalies of tooth size and shape
- A characteristic physical appearance of “cachectic dwarfism” with thinning of the skin and hair, sunken eyes, and a stooped standing posture
- Characteristic radiographic findings of thickening of the calvarium, sclerotic epiphyses, vertebral and pelvic abnormalities
Family history. The presence of a similarly affected sibling can be useful for diagnosis.
Cockayne syndrome type 2
Cockayne syndrome type 2 (type B), sometimes referred to as the “severe” or “early-onset” Cockayne syndrome, presenting with growth failure and developmental abnormalities at birth (congenital), with little or no postnatal neurologic development. Cockayne syndrome type 2 (type B) is the most severe and affected children usually do not survive past childhood. Type 2 Cockayne syndrome is sometimes called cerebro-oculo-facio-skeletal (COFS) syndrome or Pena-Shokeir syndrome type 2 due to the identification of a common gene defect in these patients.
The few cases of congenital Cockayne syndrome type 2 that have been reported are characterized by obvious growth failure at birth along with little or no neurological development after birth. Serious vision impairments (congenital cataracts and other structural abnormalities of the eye) are usually present at birth. Early skeletal aberrations occur as well. Affected children have early postnatal contractures of the spine (kyphosis, scoliosis) and joints. Death usually occurs by age seven years.
Cockayne syndrome type 2 is suspected:
- In infants with growth failure at birth and little postnatal increase in height, weight, or head circumference;
- When there is little or no postnatal neurologic development;
- When congenital cataracts as well as other structural defects of the eye (microphthalmos, microcornea, iris hypoplasia) are present.
Cockayne syndrome type 3
Cockayne syndrome type 3 (type C), has the mildest symptoms of the three types and appears later in childhood. Those with Cockayne syndrome type 3 (type C) live into middle adulthood.
Cockayne syndrome type 3 is rarer still and is characterized by essentially normal growth and mental development during the early years but interrupted by the late onset of the typical symptoms of Cockayne syndrome.
Xeroderma pigmentosum-Cockayne syndrome
Xeroderma pigmentosum-Cockayne syndrome is the most rare form and includes the features of both diseases. Widespread facial freckling and early skin cancers are typical of xeroderma pigmentosa and spasticity, short stature, mental retardation and sexual underdevelopment (hypogonadism) are consistent with Cockayne syndrome.
Xeroderma pigmentosum-Cockayne syndrome does not include skeletal involvement, the facial phenotype of Cockayne syndrome or central nervous system (CNS) dysmyelination and calcifications.
Cockayne syndrome causes
Cockayne syndrome can result from mutations in either the ERCC6 gene (also known as CSB) or the ERCC8 gene (also known as CSA). These genes provide instructions for making proteins that are involved in repairing damaged DNA. DNA can be damaged by ultraviolet (UV) rays from the sun and by toxic chemicals, radiation, and unstable molecules called free radicals. Cells are usually able to fix DNA damage before it causes problems. However, in people with Cockayne syndrome, DNA damage is not repaired normally. As errors build up in DNA, cells malfunction and eventually die. The faulty DNA repair underlies photosensitivity in affected individuals, and researchers suspect that it also contributes to the other features of Cockayne syndrome. It is unclear how ERCC6 or ERCC8 gene mutations cause all of the varied features of this condition.
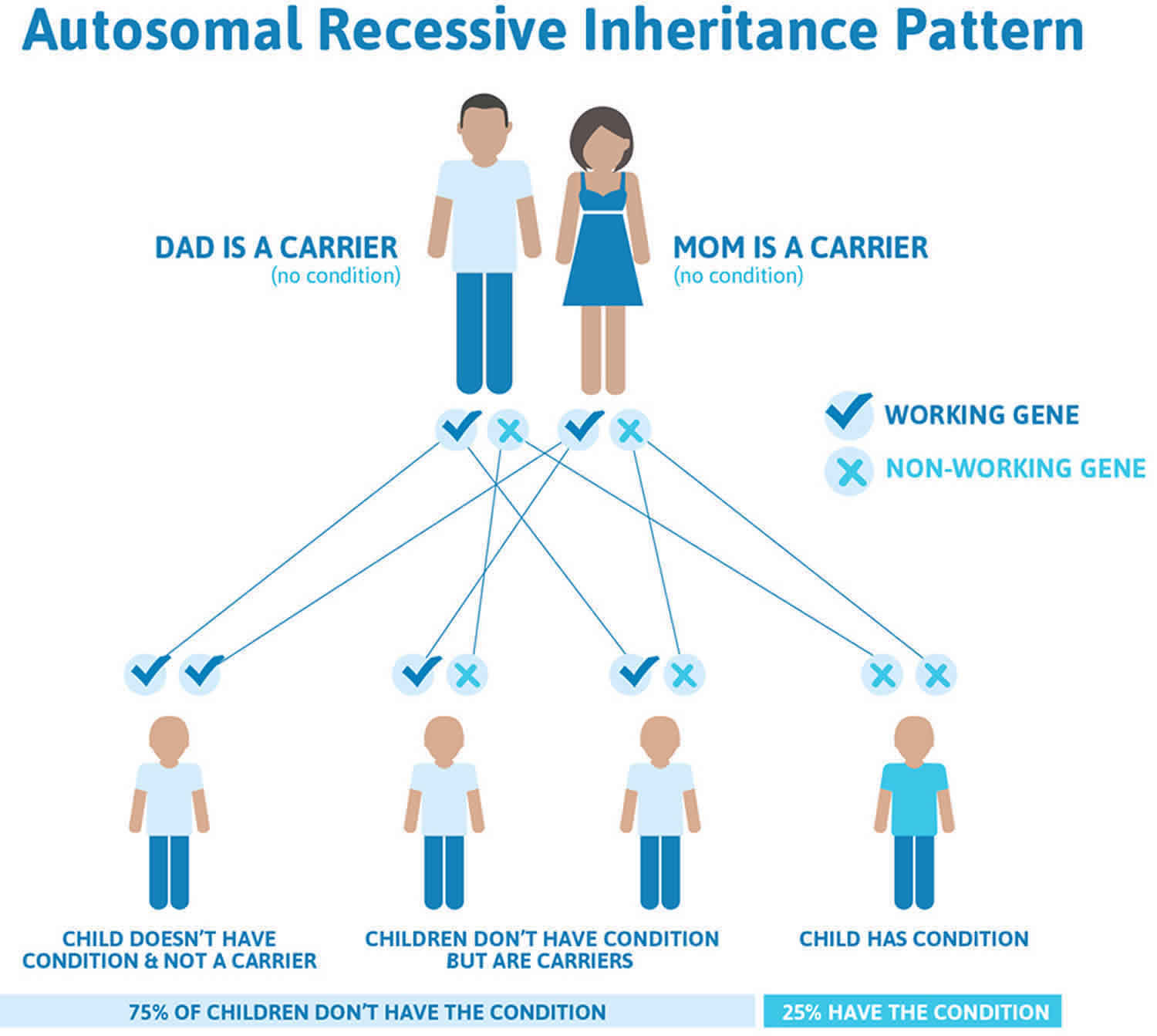
Cockayne syndrome inheritance pattern
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 1 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 1. Cockayne syndrome autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Cockayne syndrome symptoms
The symptoms of all forms of Cockayne Syndrome are similar. The different types of the disease are defined by the age of the onset. The following is a list of the most common characteristics noted in reported cases of Cockayne syndrome. No child will necessarily have all the characteristics listed, and some of these findings are progressive.
- Social, jovial personalities
- Sunburns easily
- Progeria (premature aging)
- Shortened life span
- Microcephaly
- Neurodevelopment delay
- Short stature (height <5th percentile)
- Contractures
- Retinopathy and/or cataracts
- Hearing loss
- Poor circulation (cold hands and feet
- Low body temperature
- Mircopenis
- Feeding problems
- Sleeping with eyes open
- Tremors
- White matter abnormalities
- Basal ganglia calcifications
- Liver abnormalities; elevated liver enzymes
- Hypertension
- Severe itchiness
- Unsteady gait
- Spasticity
- Rounded back
- Deep set eyes, small slender straight nose
- Dental caries (cavities)
The major characteristics of Cockayne Syndrome include the stunting of normal growth (dwarfism) during late infancy, extreme sensitivity to light (photosensitivity), and a prematurely aged appearance (progeroid). The skin appears wrinkled and aged, especially on the face, arms, and legs, due to the loss of fat under the skin (subcutaneous adipose tissue). Children with this disorder may scar easily and have an increased amount of color (pigmentation) in the skin.
Children with Cockayne Syndrome have unusual physical features including an abnormally small head (microcephaly), an unusually thin nose, a “hollow” or sunken appearance to the eyes, large misshapen ears, poor eyelid closure, and/or the abnormal forward projection of both the upper and lower jaws (prognathism). There may be an unusual amount of dental decay due to the abnormal placement of the teeth. Affected individuals typically have large hands and feet, and unusually long arms and legs in proportion to the size of their body. Joints may also be abnormally large and remain in a fixed position (flexed), and the spine may be curved outward when viewed from the side (kyphosis). Other features of Cockayne Syndrome may include a decrease in the amount of sweating (hypohidrosis), lack of proper tearing in the eyes, and/or the premature graying of the hair.
Other symptoms of Cockayne syndrome may include an abnormal blue tint to the skin (cyanosis) on the arms and legs, which may also feel cold to the touch. Neurological symptoms may include rhythmic, quivering movements (tremors), an unsteady gait (ataxia), and/or the inability to coordinate movement. Affected children may experience varying degrees of mental retardation, partial loss of hearing, and/or the progressive loss of previously acquired intellectual abilities.
The symptoms of Cockayne syndrome that affect the eyes (ocular) may include progressive clouding of the lens of the eyes (cataracts), loss of vision because of the wasting of the nerve fibers within the eyes (optic atrophy), degeneration of the retina, and/or the abnormal accumulation of retinal coloration (pigmentation).
Some people with Cockayne syndrome may also have abnormally high blood pressure (hypertension), an enlarged liver (hepatomegaly), and/or the premature accumulation of fatty plaques on the walls of the arteries around the heart (arteriosclerotic disease). Adults with this disorder may be sexually underdeveloped.
Cockayne syndrome type 1 symptoms
Prenatal growth is typically normal. Birth length, weight, and head circumference are normal. Within the first two years, however, growth and development fall below normal. By the time the disease has become fully manifest, height, weight, and head circumference are far below the fifth percentile. Progressive impairment of vision, hearing, and central and peripheral nervous system function leads to severe disability. Severe dental caries occur in up to 86% of individuals. Photosensitivity can be severe, but individuals are not predisposed to skin cancers.
Additional clinical abnormalities occurring in 10% or more of individuals include the following:
- Neurologic. Increased tone/spasticity, hyper- or hyporeflexia, abnormal gait or inability to walk, ataxia, incontinence, tremor, abnormal or absent speech, seizures, weak cry/poor feeding (as an infant), muscle atrophy, and behavioral abnormality
- Dermatologic. Anhidrosis and malar rash
- Ophthalmologic. Enophthalmos, pigmentary retinopathy (60%-100%), abnormal electroretinogram, cataracts of various types (15%-36%), optic atrophy, miotic pupils, farsightedness, decreased or absent tears, strabismus, nystagmus, photophobia, narrowed retinal arterioles, and microphthalmia
- Dental. Absent or hypoplastic teeth, delayed eruption of deciduous teeth, and malocclusion
- Renal. Abnormal renal function and pathologic abnormalities; noted in case reports, but usually not clinically significant
- Endocrine. Undescended testes, delayed/absent sexual maturation. No individuals with classic or severe Cockayne syndrome (types 1 or 2) have been known to reproduce. A successful (but very difficult) pregnancy has been reported in a young woman with mild Cockayne syndrome (type 3) 6
- Gastrointestinal. Elevated liver function tests, enlargement of liver or spleen
Death typically occurs in the first or second decade. The mean age of death is 12 years, but survival into the third decade has been reported.
Cockayne syndrome type 2 symptoms
Children with severe Cockayne syndrome have evidence of growth failure at birth, with little or no postnatal neurologic development. Congenital cataracts or other structural anomalies of the eye are present in 30%. Affected individuals have arthrogryposis or early postnatal contractures of the spine (kyphosis, scoliosis) and joints. Affected children typically die by age seven years. Cockayne syndrome type 2 overlaps clinically with the cerebrooculofacioskeletal syndrome (COFS), which is also referred to as Pena-Shokeir syndrome type 2.
Cockayne syndrome type 3 symptoms
Recently, DNA sequencing has confirmed the diagnosis of Cockayne syndrome type 3 in some individuals who have clinical features associated with Cockayne syndrome but whose growth and/or cognition exceeds the expectations for Cockayne syndrome type 1.
Xeroderma pigmentosum-Cockayne syndrome symptoms
Since the discovery of the genes in which pathogenic variants underlie Cockayne syndrome, it has become evident that the distinctions between genotype, cellular phenotype, and clinical phenotype are not absolute. Xeroderma pigmentosum, a related DNA repair disorder, includes facial freckling and early skin cancers — features not found in Cockayne syndrome. The DeSanctis-Cacchione variant of xeroderma pigmentosum includes some features of Cockayne syndrome (e.g., intellectual disability, spasticity, short stature, and hypogonadism) but does not include skeletal dysplasia, the facial phenotype of Cockayne syndrome, or CNS dysmyelination and calcifications. Individuals with an XP clinical phenotype have been seen in association with a Cockayne syndrome cellular phenotype and with a pathogenic variant in ERCC6 7. Conversely, individuals with clinical features of Cockayne syndrome but with xeroderma pigmentosum-like skin cancers have been assigned to the XPB, XPD, and XPG complementation groups based on their biochemical characteristics (the ability to restore normal function to various DNA repair-deficient cell lines) 8. Individuals with other features of Cockayne syndrome, but lacking sun sensitivity, have been reported. Mallery et al 9 has reported a poor correlation between genotype and phenotype for this group of diseases.
Neuropathology. A characteristic “tigroid” pattern of demyelination in the subcortical white matter of the brain and multifocal calcium deposition, with relative preservation of neurons and without senile plaques, amyloid, ubiquitin, or tau deposition, is observed 10.
Cockayne syndrome diagnosis
Classic Cockayne syndrome is diagnosed by clinical findings including postnatal growth failure and progressive neurologic dysfunction along with other minor criteria. Molecular genetic testing or a specific DNA repair assay on fibroblasts can confirm the diagnosis. The two genes in which pathogenic variants are known to cause Cockayne syndrome are ERCC6 (65% of individuals) and ERCC8 (35% of individuals).
Cockayne syndrome treatment
The treatment of Cockayne syndrome is symptomatic and supportive. Specialized imaging testing (MRI) may demonstrate the loss of the fatty covering (demyelination) on some nerve fibers in the brain.
A supportive team approach may be of benefit for children with Cockayne syndrome and may include special education, physical therapy, and other medical, social, and/or vocational services. Genetic counseling may benefit family members.
Individualized educational programs for developmental delay; physical therapy to maintain ambulation; gastrostomy tube placement as needed; medications for spasticity and tremor as needed; use of sunscreens and sunglasses for cutaneous photosensitivity and lens/retina protection, respectively; treatment of hearing loss, cataracts, and other ophthalmologic complications as in the general population.
Prevention of secondary complications: Physical therapy to prevent joint contractures; aggressive dental care to minimize dental caries; home safety assessment to prevent falls.
Surveillance: Yearly assessment for complications such as hypertension, renal or hepatic dysfunction, and declining vision and hearing.
Agents/circumstances to avoid: Excessive sun exposure.
Cockayne syndrome prognosis
Patients with Cockayne syndrome 1 have progressive, unremitting, neurologic deterioration usually leading to death by the second or third decade of life. Patients with Cockayne syndrome 2 typically have a worse prognosis, with death occurring earlier, typically by age 6 or 7 years.
References- What is Cockayne Syndrome? http://cockaynesyndrome.org/about-cs
- Cockayne syndrome. https://ghr.nlm.nih.gov/condition/cockayne-syndrome
- Cockayne syndrome. https://rarediseases.org/rare-diseases/cockayne-syndrome/
- Laugel V. Cockayne Syndrome. 2000 Dec 28 [Updated 2012 Jun 14]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1342
- Sugita K, Takanashi J, Ishii M, Niimi H. Comparison of MRI white matter changes with neuropsychologic impairment in Cockayne syndrome. Pediatr Neurol. 1992;8:295–8.
- Lahiri S, Davies N. Cockayne’s Syndrome: case report of a successful pregnancy. BJOG. 2003;110:871–2.
- Colella S, Nardo T, Botta E, Lehmann AR, Stefanini M. Identical mutations in the CSB gene associated with either Cockayne syndrome or the DeSanctis-cacchione variant of xeroderma pigmentosum. Hum Mol Genet. 2000;9:1171–5.
- van Hoffen A, Kalle WH, de Jong-Versteeg A, Lehmann AR, van Zeeland AA, Mullenders LH. Cells from XP-D and XP-D-CS patients exhibit equally inefficient repair of UV-induced damage in transcribed genes but different capacity to recover UV-inhibited transcription. Nucleic Acids Res. 1999;27:2898–904.
- Mallery DL, Tanganelli B, Colella S, Steingrimsdottir H, van Gool AJ, Troelstra C, Stefanini M, Lehmann AR. Molecular analysis of mutations in the CSB (ERCC6) gene in patients with Cockayne syndrome. Am J Hum Genet. 1998;62:77–85.
- Itoh M, Hayashi M, Shioda K, Minagawa M, Isa F, Tamagawa K, Morimatsu Y, Oda M. Neurodegeneration in hereditary nucleotide repair disorders. Brain Dev. 1999;21:326–33.





{kind=link}