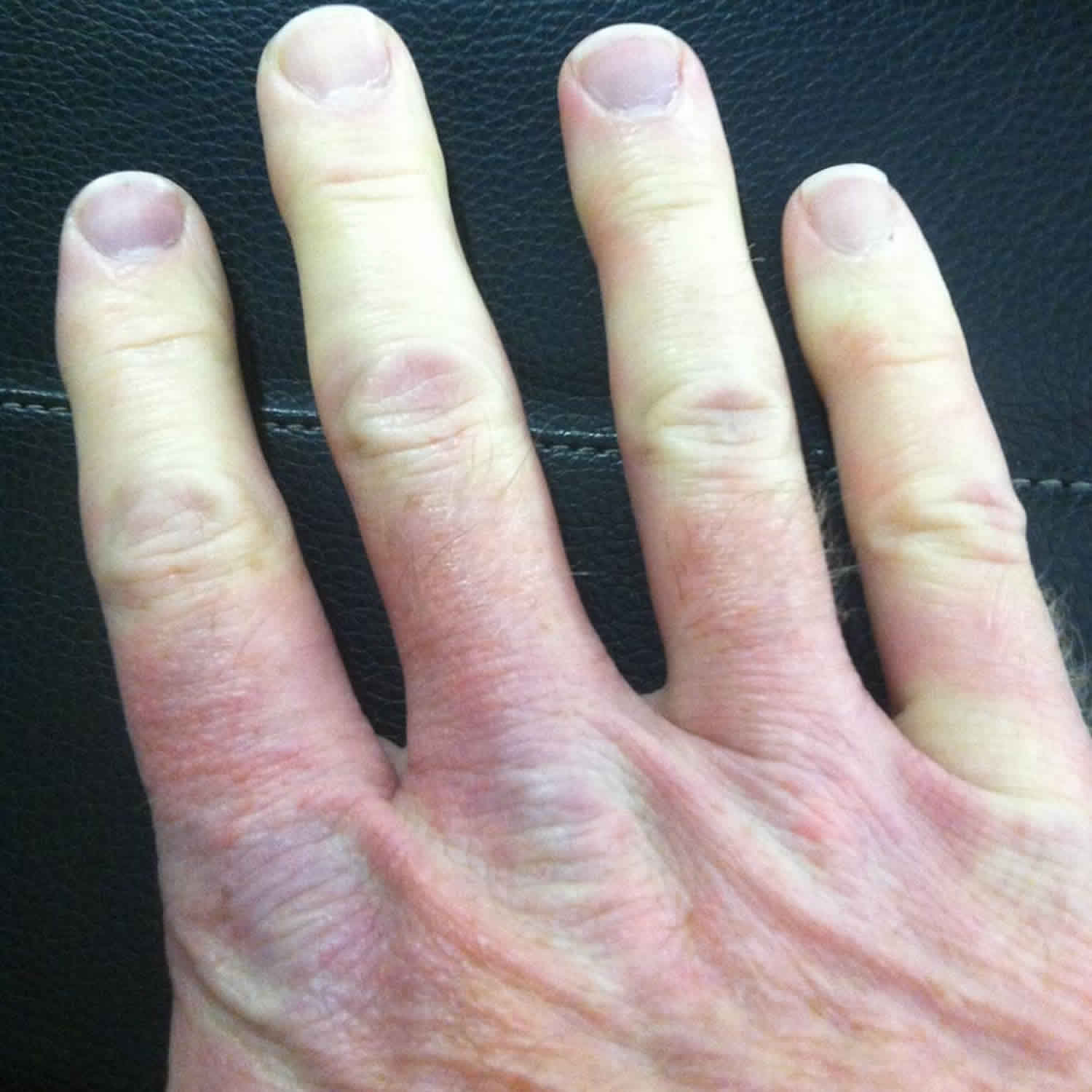
What is CREST syndrome
CREST syndrome is now called limited scleroderma, is a widespread connective tissue disease characterized by changes in the skin, blood vessels, skeletal muscles, and internal organs 1. CREST is an acronym for the clinical features that are seen in a patient with limited scleroderma or limited cutaneous systemic sclerosis. The “C” stands for calcinosis, where calcium deposits form under the skin on the fingers or other areas of the body. The “R”, stands for Raynaud’s phenomenon, spasm of blood vessels in the fingers or toes in response to cold or stress. The “E” represents esophageal dysmotility, which can cause difficulty in swallowing. The “S” is for sclerodactyly, tightening of the skin causing the fingers to bend. Finally, the letter “T” is for telangiectasia, dilated vessels on the skin of the fingers, face, or inside of the mouth. Usually only 2 of the 5 symptoms of the CREST syndrome is necessary to be diagnosed with limited scleroderma. The skin changes associated with limited scleroderma typically occur only in the lower arms and legs, below the elbows and knees, and sometimes affect the face and neck. Limited scleroderma can also affect your digestive tract, heart, lungs or kidneys.
The symptoms involved in CREST syndrome are associated with the generalized form of the disease systemic sclerosis (scleroderma). CREST is an acronym for the clinical features that are seen in a patient with this disease.
- (C) – Calcinosis: calcium deposits in the connective tissues
- (R) – Raynaud’s phenomenon: where the hands and feet turn white and cold and then blue, in response to cold or anxiety
- (E) – Esophageal dysfunction resulting in swallowing difficulty
- (S) – Sclerodactyly: thick and tight skin on the fingers, caused by an excess of collagen deposits within skin layers.
- (T) – Telangiectasia: small red spots on the hands and face that are caused by the swelling of tiny blood vessels.
CREST syndrome is believed to be an autoimmune disorder, where the immune system appears to stimulate the production of too much collagen which builds up in the skin and internal organs, impairing their function.
The problems caused by limited scleroderma may be minor. Sometimes, however, the disease affects the lungs or heart, with potentially serious results. Limited scleroderma has no known cure. Treatments focus on managing symptoms, preventing serious complications and improving quality of life 2.
Is CREST syndrome fatal?
There is no cure for CREST syndrome or systemic sclerosis (scleroderma). Survival is determined by the disease subset and internal organ manifestations. Interstitial lung disease and pulmonary artery hypertension account for almost two-thirds of deaths related to scleroderma (systemic sclerosis).
Early detection of CREST syndrome (limited scleroderma) can help prevent serious complications. See your doctor if you have any indications of the condition.
Proactive and routine annual screening allows early intervention with disease modifying drugs. These have led to improvement in prognosis and long term outcomes in recent years.
What is systemic sclerosis?
Systemic sclerosis also called scleroderma, is an autoimmune inflammatory condition. Systemic sclerosis results in potentially widespread fibrosis and vascular abnormalities, which can affect the skin, lungs, gastrointestinal tract, heart and kidneys 3. The skin becomes thickened and hard (sclerotic).
Systemic sclerosis has been subdivided into 2 main subtypes, according to the distribution of skin involvement.
- Diffuse cutaneous systemic sclerosis: Two-thirds of patients with systemic sclerosis have diffuse cutaneous systemic sclerosis: skin involvement is widespread and includes proximal limbs. Diffuse systemic sclerosis is distinguished from the localized variant (CREST syndrome) mainly based on the extent of cutaneous symptoms which are more extensive and prevalent in diffuse sclerosis 4. Diffuse cutaneous systemic sclerosis is often rapidly progressive, with significant internal organ involvement.
- Limited cutaneous systemic sclerosis (limited scleroderma): One-third of patients with systemic sclerosis have limited cutaneous systemic sclerosis: sclerosis is limited to the digits, distal limbs (not spreading more proximal than the elbows or knees) and face. Limited cutaneous systemic sclerosis progresses more slowly than diffuse cutaneous systemic sclerosis and with less internal organ involvement except there is a risk of pulmonary artery hypertension, especially later in the disease course.
- Limited cutaneous systemic sclerosis was previously referred to as CREST syndrome to denote key features :
- Calcinosis
- Raynaud phenomenon
- Esophageal dysmotility
- Sclerodactyly
- Telangiectases.
- Limited cutaneous systemic sclerosis was previously referred to as CREST syndrome to denote key features :
- Overlap syndrome: Up to 20% of patients with systemic sclerosis have an overlap syndrome with another connective tissue disease and develop arthritis, lupus or myositis.
- Systemic sclerosis sine scleroderma is a rare subtype without skin sclerosis. These patients have systemic sclerosis-related internal organ manifestations, Raynaud phenomenon and systemic sclerosis-specific auto-antibodies.
The general term ‘scleroderma’ is often used for both morphoea (localized scleroderma) and systemic sclerosis (systemic scleroderma). Distinguishing these two conditions is very important, as they vary greatly and require different treatment.
Systemic sclerosis is rare, with prevalence varying from 30–500 cases per million. Systemic sclerosis global prevalence is approximately 0.3 to 24 per 100,000 population. Studies indicate higher systemic sclerosis prevalence for North America, Australia, and Western Europe compared to prevalence data for Japan and Asia. Estimated incidence for the United States is 67 males and 267 females per 100,000 population annually. There has been a significant increase in reported incidence starting from the 1950s. There is an overall female predominance. This is more pronounced for CREST syndrome, with a female to male ratio of 10:1. The diffuse variant is more evenly distributed among the sexes.
All racial groups are affected. It seems there is a higher incidence in European ancestry compared to Japanese or Asian ancestry. African American and Native American ancestry seem to be associated with increased disease severity. Systemic sclerosis is rare in children 5.
- Systemic sclerosis is up to 5 times more common in females compared to males.
- All races and ethnicities may be affected, but rates appear to be slightly higher in some Native Americans and black-skinned races, and lower in those of Asian background.
- Peak age of onset of systemic sclerosis varies between approximately 35 and 55 years. Juvenile onset occurs but is rare compared to adult onset disease.
Key features of systemic sclerosis
- Skin thickening of the fingers and toes (sclerodactyly)
- Specific autoantibodies in the blood (anti-Scl70 or anti-centromere antibody and others)
- Abnormal nail fold capillaries
- Internal organ fibrosis and/or vascular damage (involving the lungs, heart, gastrointestinal tract and/or kidneys)
Systemic sclerosis causes
Systemic sclerosis is an inherited condition, and a positive family history is one of the major identifiable risk factors for systemic sclerosis 4. Autoimmunity is considered the major mechanism responsible for systemic sclerosis that is characterized by inflammation, fibrosis and vasculopathy. Although several environmental triggers are suspected, there is no identified definitive etiological agent. Some of the factors that have been implicated include Cytomegalovirus (CMV) infection (cytomegalovirus antibody has been demonstrated in a significant proportion of patients with systemic sclerosis) and industrial exposures to vinyl chloride, solvent, and silica 6.
The precise underlying mechanisms are complex and remain largely unknown. Genetic susceptibility plus a triggering event result in a cascade of innate and adaptive immunoinflammatory responses.
Genetic susceptibility
- First degree relatives of affected individuals may be at 10–16 fold increased risk of developing systemic sclerosis, compared with those with no family history of the disease.
- Studies have identified genetic loci associated with systemic sclerosis.
- Clinical subtypes map to particular genetic subsets.
- Differences in gene expression occur in fibroblasts, immune (T and B), endothelial, smooth muscle and epithelial cells.
Triggering event
Systemic sclerosis has been associated with injury, exposure to silica, vinyl chloride monomer, chlorinated solvents, trichloroethylene, welding fumes, aromatic solvents, ketones, bleomycin and possibly other drugs (vitamin K, cocaine, penicillamine, appetite suppressants and some chemotherapeutic agents).
Immune pathways
A number of pathways are likely involved in the pathogenesis of systemic sclerosis, including cytokines that injure blood vessels, growth factors that stimulate collagen production, integrin signalling, morphogen pathways, co-stimulatory pathways and more.
Paraneoplastic systemic sclerosis
Malignancy in association with systemic sclerosis is rare.
- Anti-RNAP-III antibodies are found in up to 15% of patients with paraneoplastic systemic sclerosis.
- Patients are usually older than 65 years at presentation.
- Skin involvement is atypical
- The disease tends to be treatment-resistant.
- Associated malignancies include breast, hematological and gastrointestinal cancers.
Systemic sclerosis pathophysiology
Systemic sclerosis occurs as a result of excessive activation of the process repair known as fibrosis in affected individuals 4. Fibrosis is a process of wound healing that accompanies chronic inflammation which is characterized by proliferation of collagen-producing cells known as fibroblasts.
Murine experiments have shown that systemic sclerosis arises as a result of an interplay which involves autoimmunity, obliterative vascular lesions, and chronic inflammation. The interplay leads to excessive production of fibrosis-inducing cytokines, such as Transforming growth factor Beta (TGF-ß); platelet-derived growth factor (PDGF), and interleukin-4 (IL-4).
Infectious agents such as Cytomegalovirus (CMV) have been found to contribute to the development of systemic sclerosis by triggering fibroblast activation through activation of fibroblastic cytokines.
Vasculopathy in systemic sclerosis includes a destructive vasculitis characterized by loss of small vessels and a progressive obliterative vasculopathy due to excessive fibrosis and diminished new vessel formation. These two mechanisms cause endothelial dysfunction, target organ ischemia and the other vascular complications seen in this disorder.
Other mediators of disease in systemic sclerosis are human leukocyte antigens(HLA). The following HLAs have been implicated in the causation of this disorder: HLA-B8, HLA-DR5, HLA-DR3, HLA-DQB2, and HLA- DR-52 7.
Systemic sclerosis symptoms
The signs and symptoms of systemic sclerosis are related to underlying vascular, inflammatory and fibrotic disease. Constitutional symptoms are common, such as fatigue, joint pain (arthralgia) and muscle pain (myalgia).
Cutaneous features of systemic sclerosis
Skin sclerosis
- Extent of skin fibrosis defines diffuse vs limited systemic sclerosis
- Sclerodactyly: thickening and tightness of the skin of the fingers (or toes). May be spindle-shaped
Hands
- Puffy fingers; early inflammatory phase of disease
- Raynaud phenomenon
- Abnormal nail fold capillaries
- Palmar erythema affecting thenar / hypothenar eminence
- Smaller fragile nails with ragged cuticles
- Digital pitted scars
- Digital ulcers
- Ulceration can lead to dry gangrene and eventual loss of the tips of the fingers (like frost bite).
Face
- Matt telangiectases on face, chest, palms
- Peri-oral furrowing (fat loss)
- Microstomia (limited oral aperture defined as interlabial distance < 4.5 cm)
- Beaked nose
Other
- Calcinosis affecting digits, extensor surfaces of limbs. Skin can breakdown and discharge chalky material (calcium)
- Salt and pepper dyspigmentation (hyperpigmentation and hypopigmentation)
- Pruritus
Rarer cutaneous features
- Morphoea
- Most frequently plaque, nodular or linear
- More common in limited cutaneous systemic sclerosis
- Panniculitis
Gastrointestinal symptoms
Upper gastrointestinal tract
- Gastroesophageal reflux / heart burn
- Dyspepsia
- Dysphagia
- Early satiety
- Micro-aspiration (accelerates lung disease)
Lower gastrointestinal tract
- Bloating, distention
- Nausea, vomiting
- Pain
- Alternating diarrhea / constipation
- Incontinence (anorectal sphincter dysfunction)
Cardiopulmonary symptoms
- Interstitial lung disease
- Pulmonary artery hypertension
- Cardiac scleroderma (cardiomyopathy, conductive)
- Shortness of breath
- Decreased exercise tolerance
- Chest pain
- Palpitations
Renal disease
Scleroderma renal crisis:
- Proteinuria
- High blood pressure
- Renal failure.
Other symptoms
- Fatigue
- Sicca symptoms (dry eyes, dry mouth) and Sjogren syndrome
- Musculoskeletal symptoms: friction rubs over the joints and tendons, particularly the knees; joint pain, muscle pain, weakness and limited movement resulting in contractures
- Ocular symptoms: tight eyelids, reduced tear secretion, retinopathy.
Systemic sclerosis diagnosis
The diagnosis of systemic sclerosis is confirmed when key features are present.
- Sclerodactyly
- Abnormal nail folds capillaries on capillaroscopy/dermatoscopy
- Specific autoantibodies (especially anti-Scl70 or anti-centromere antibody)
- Internal organ fibrosis and/or vascular damage
Investigations may include:
- Other blood tests: anemia, raised sedimentation rate (ESR) and C-reactive protein (CRP), positive Rheumatoid factor, increased gamma globulins (hypergammaglobulinaemia) and abnormal coagulation tests may be present.
- Skin biopsy: excessive ground substance and odd-looking endothelial cells in the dermis; and later, deposits of collagen. The epidermis is usually atrophic (thinned).
- Pulmonary function tests
- High resolution CT scan
- Echocardiogram
- Right heart catheter
- Electrocardiogram (ECG)
- Cardiac MRI
- Barium swallow, manometry
- Endoscopy with gastrointestinal biopsy.
The joint American College of Rheumatology and European League against Rheumatism classification criteria (2013) are utilized to diagnose systemic sclerosis. A score of 9 or more confirms the diagnosis.
- Skin thickening of the fingers of both hands extending proximal to metacarpal phalangeal joint: score 9
- Skin thickening of the fingers only: puffy fingers 2; sclerodactyly 4
- Finger tip lesions: digital tip ulcers 2; fingertip pitted scars 3
- Telangiectases 2
- Abnormal nailfold capillaries 2
- Pulmonary disease: pulmonary artery hypertension 2; interstitial lung disease 2
- Raynaud phenomenon 3
- Systemic sclerosis-specific autoantibodies ACA 3, Anti-scl70 3, Anti-RNA polymerase III 3 (maximum score 3)
Systemic sclerosis treatment
Treatments help with symptoms and may modify the disease outcome, especially early in the disease course. They focus on suppressing inflammation and dilating abnormal / constricted blood vessels. Some newer treatments target specific immunological pathways and signalling molecules.
General advice
- It is absolutely essential for smokers to stop smoking.
- Avoid vasoconstrictive drugs, such as decongestants, amphetamines, ergotamine.
Fatigue, weakness and generalized musculoskeletal symptoms can be debilitating.
- Hydroxychloroquine may help these constitutional symptoms.
- Gentle controlled exercise may also be of benefit.
- Simple or more complex analgesia may be required.
Anti-fibrotic therapies
Topical therapies:
- Warm wax baths (hands)
- Tacrolimus ointment
- Potent corticosteroids
- Calcipotriol
Physical therapies:
- Phototherapy, especially UVA1 or PUVA
Systemic therapies:
- Mycophenolate mofetil
- Methotrexate
- Cyclophosphamide
- Corticosteroids (with caution)
- Rituximab (B-cell depletion therapy)
- Intravenous immunoglobulin
- New targeted therapies
- Anti-interleukin 6 (tocilizumab)
- Anti-cytotoxic T-cell ligand inhibitor (abatacept)
- Tyrosine kinase inhibitors (nintedanib)
- Transforming growth factor β inhibitors (fresolimumab, perfenidone)
- Anti-Interleukin 4 / 13 drugs
- Autologous stem cell transplant
Vasodilation
- Endothelin 1 antagonists (bosentan)
- Phosphodiesterase-5 inhibitors (tadalifil)
- Guanylate cyclase agonists (riociguat)
- Prostacyclin agonists (epoprostenol; selexipag)
Treatment of skin manifestations
Raynaud phenomenon
- Avoid triggers (smoking cessation, protect from cold)
- General measures; double lined gloves, exothermic hand / feet warmers
- Natural therapies (unproven)
- Vitamin C, Vitamin E
- Gamolenic acid
- Medical therapies
- Fluoxetine
- Losartan
- Diltiazem, nifedipine
- Sildenafil, tadalafil
- GTN patches
- Iloprost
- Botulinum toxin
Digital ulcers
- Ulcer prevention
- Emollients
- Avoid smoking, cold (double lined gloves), trauma
- Ulcer treatment
- Localized dressings; protect and keep moist areas of impending ulceration and ulcerated areas
- Systemic vasodilators
- Surgery
- Amputation
- Botulinum toxin injections
- Digital sympathectomy
- Prevention of ulcer complications
- Pain management
- Infection – 10% infection rate per year
Telangiectases
- Cosmetic camouflage (green tinted makeup)
- Vascular laser or intense pulsed light therapy
Calcinosis
Calcinosis is difficult to manage and there is poor evidence for therapies listed below:
- Medical therapies
- Tetracycline antibiotics (6–12 week courses)
- Diltiazem
- Bisphosphonates
- Sodium thiosulfate (systemic and localised injections are reported)
- Cinacalcet
- Physical interventions
- Surgical excision
- Curettage and cautery
- Dental drill removal
- Ablative laser
- Extracorporeal shock wave lithotripsy
Pruritus
- Emollient, soap substitutes
- Topical corticosteroids
- Antihistamines
- Montelukast
- Systemic glucocorticoids (with caution)
Facial fat loss
- Autologous fat transfer
Treatment of internal organ manifestations
Interstitial lung disease
- Antifibrotic, immunosuppressive therapies (see above)
Pulmonary artery hypertension
- Vasodilatory therapies (see above)
Renal disease
- Monitor blood pressure regularly
- Avoid corticosteroids (especially if RNAPIII +ve)
- Angiotensin converting enzyme inhibitors
Gastrointestinal tract
- Upper gastrointestinal tract: gastro-esophageal reflux, dyspepsia, dysphagia, aspiration
- Protein pump inhibitors, such as oomeprazole, ranitidine
- Treat Helicobacter pylori
- Prokinetics: domperidone, erythromycin, metoclopramide
- N-acetylcysteine
- Lower gastrointestinal tract: malabsorption, bacterial overgrowth, diarrhea, constipation, bloating, pain, anorectal dysfunction
- Nutritional supplements, advice from dietician
- Creon
- Probiotics
- Cyclical antibiotics (for example, ciprofloxacin for 10 days)
- Stool softeners
Systemic sclerosis monitoring
Monitoring of progress and treatment response is vital in systemic sclerosis.
Skin
The skin is usually monitored clinically using the modified Rodnan Skin Score (mRSS), which gives an indication of the extent and severity of the cutaneous sclerosis, which also reflects the severity and risk of internal organ involvement.
- A score of 0 (normal skin) to 3 (skin not able to be pinched) is assigned for skin tightness for 17 body sites.
- The cumulative score is calculated out of a total of 51; severe skin involvement is defined by a score of > 20.
- A high mRSS is an independent risk factor for a poorer overall outcome.
Interlabial distance (mouth opening) can also be measured
- Microstomia is defined as an inter-labial distance of less than 4.5 cm.
Internal organs
Routine annual screening for interstitial lung disease and pulmonary artery hypertension should include:
- Pulmonary function tests
- Transthoracic echocardiogram
- Electrocardiogram (ECG)
The DETECT score is a screening tool for pulmonary artery hypertension that uses pulmonary function tests (FVC and DLCO), serum urate, serum NTproBNP and other features to provide a predictive score. A right heart catheter may be indicated in some.
- High resolution CT scan may be used to characterize interstitial lung disease.
- Blood pressure and renal function monitor renal disease.
- Other organ monitoring is performed on an individualized basis, and can include cardiac MRI, esophageal manometry and gastrointestinal endoscopy.
CREST syndrome causes
The cause isn’t known, but CREST syndrome (limited scleroderma) is believed to be an autoimmune disorder, in which your immune system turns against your body. The immune system in people with CREST syndrome appears to stimulate cells called fibroblasts to produce excess amounts of collagen, a key component of connective tissue. The hallmark of scleroderma is progressive fibrosis of tissues 8. Normally, fibroblasts make collagen to help heal wounds, but in this case, the protein is produced even when it’s not needed, forming thick bands of connective tissue around the cells of the skin, blood vessels and in some cases, the internal organs. This overproduction of collagen builds up in the skin and internal organs so that they don’t function normally.
Risk factors for CREST syndrome
Although an abnormal immune system response and the resulting production of excess collagen appears to be the main cause of limited scleroderma, researchers suspect that other factors increase the risk to have the disease, including:
- Your sex. Women are far more likely to develop limited scleroderma than men are.
- Age. Limited scleroderma is more common between the ages of 30 and 50.
- Race. In the United States, limited scleroderma tends to be more severe in blacks and Native Americans than in whites.
- Genetic factors. If someone in your family has an autoimmune disease — such as lupus, rheumatoid arthritis or Hashimoto’s disease — you have an increased risk of developing limited scleroderma.
- Exposure to toxins. Certain toxic substances — such as polyvinyl chloride, benzene, silica and trichloroethylene — might trigger scleroderma in people with a genetic predisposition to the disease.
CREST syndrome symptoms
While some varieties of scleroderma occur rapidly, signs and symptoms of limited scleroderma formerly known as CREST syndrome (Calcinosis, Raynaud’s phenomenon, Esophageal dysmotility, Sclerodactyly, and Telangiectasia) usually develop gradually. Multiple organ systems including the lungs and skin are affected. However, the progression of fibrosis is slower in the CREST syndrome compared to the diffuse form of the disease. Pulmonary hypertension and pulmonary fibrosis are leading causes of death in scleroderma.
CREST syndrome symptoms and signs include:
- Tight, hardened skin. In limited scleroderma, skin changes typically affect only the lower arms and legs, including fingers and toes, and sometimes the face and neck. Skin can look shiny from being pulled taut over underlying bone. It may become difficult to bend your fingers or to open your mouth.
- Raynaud’s phenomena. This condition occurs when small blood vessels in your fingers and toes spasm in response to cold or emotional stress, blocking the flow of blood. In most people, the skin turns white before becoming blue, cold and numb. When circulation improves, the skin usually reddens and might throb or tingle. Raynaud’s phenomena is often the first sign of limited scleroderma, but many people who have Raynaud’s never develop scleroderma.
- Red spots or lines on skin. The swelling of tiny blood vessels near the skin’s surface cause these small red spots or lines (telangiectasias). Not painful, they occur primarily on the hands and face.
- Bumps under the skin. Limited scleroderma can cause tiny calcium deposits (calcinosis) to develop under your skin, mainly on your elbows, knees and fingers. You can see and feel these deposits, which sometimes are tender or become infected.
- Swallowing difficulties. Limited scleroderma commonly causes problems with the tube that connects the mouth and stomach (esophagus). Poor functioning of the muscles in the upper and lower esophagus can make swallowing difficult and allow stomach acids to back up into the esophagus, leading to heartburn, inflammation and scarring of esophageal tissues.
CREST syndrome complications
The visible signs of limited scleroderma — tight, thick skin on your fingers, hands and face — can change your appearance; make everyday tasks, such as opening a jar or shaving, more difficult; and affect your speech. But the most serious complications tend to occur beneath your skin.
- Gastrointestinal problems. Changes in the functioning of esophageal muscles can cause difficulty swallowing and chronic heartburn. When limited scleroderma affects your intestine, it can cause constipation, diarrhea, bloating after meals, unintended weight loss and malnutrition.
- Ulcers on fingers and toes. Severe Raynaud’s phenomena can obstruct blood flow to your fingers and toes, causing ulcers that can be difficult to heal. Also, abnormal or narrowed blood vessels combined with severe Raynaud’s phenomena can lead to gangrene of fingers or toes, which might require amputation.
- Lung damage. Limited scleroderma can cause a variety of problems with your lungs. In some cases, excess collagen collects in the tissue between the lungs’ air sacs, making the lung tissue stiffer and less able to work properly. Increased blood pressure in the arteries between your heart and lungs makes the heart work harder and eventually weakens it.
- Heart problems. Scarring of heart tissue can lead to abnormal heart rhythms (arrhythmias) and, in rare cases, to an inflamed heart muscle (myocarditis).
- Kidney problems. Although kidney damage is more common in other forms of scleroderma, it can occur in limited scleroderma. The first indication might be high blood pressure. Restricted blood flow to the kidneys can result in renal crises, which, if untreated, can lead to kidney failure.
- Dental problems. Severe tightening of facial skin can make it difficult to open your mouth wide enough to brush your teeth. Acid reflux can destroy tooth enamel, and changes in gum tissue may cause your teeth to become loose or even fall out.
- Dry eyes and mouth. Limited scleroderma can cause very dry eyes and mouth.
CREST syndrome diagnosis
Like other unusual and complex autoimmune disorders, CREST syndrome (limited scleroderma) can be difficult to diagnose. Signs and symptoms vary widely and often resemble those other connective tissue and autoimmune diseases. Further complicating matters is that limited scleroderma sometimes occurs with other autoimmune conditions — such as polymyositis, lupus and rheumatoid arthritis.
The diagnosis of CREST syndrome (limited scleroderma) is generally made based on your signs and symptoms (calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly and telangiectasia). During the physical exam, your doctor will look for changes in the texture, color and appearance of your skin. People with CREST syndrome usually present at least three of these five main clinical features, but the diagnosis can be made when only two these five features are present. Tests that might aid in the diagnosis include:
- Lab tests. A sample of your blood can be tested for antibodies (ANA and anticentromere) that are frequently found in the blood of people with limited scleroderma. But this isn’t a definitive test because not everyone with CREST syndrome (limited scleroderma) has these antibodies.
- Skin biopsy. Sometimes doctors take a small sample of skin that’s examined under a microscope in a laboratory. Although biopsies can be helpful, they can’t definitively diagnose CREST syndrome (limited scleroderma).
- Your doctor might recommend additional tests to identify lung, heart, kidney or gastrointestinal complications. Along with a blood test and skin biopsy, additional tests to identify lung, heart or gastrointestinal complications may also be conducted. The diagnosis of calcinosis (suspected based on finding the nodules) is confirmed with imaging studies. Multiple imaging studies are available to evaluate esophageal motility. The least invasive evaluation involves radiologic barium studies.
CREST syndrome treatment
CREST syndrome or limited scleroderma has no known cure. Treatment focuses on relieving signs and symptoms and preventing complications.
Calcinosis: No treatment seems to be available to prevent or eliminate calcinosis. Large or painful calcium deposits sometimes need to be surgically removed, and amputation of fingertips may be necessary if the skin ulcers progress to gangrene. One or a combination of the following treatments may be tried on a case-by-case basis; however, larger studies are needed to prove efficacy.
- Corticosteroids oral or injected inside the lesion (intralesional)
- Probenecid
- Diltiazem
- Warfarin
- Aluminum hydroxide
- Bisphosphonate
- Minocycline
- Colchicine
- Intravenous immunoglobulin therapy
Raynaud phenomenon: The following treatment is suggested for the treatment of people with Raynaud phenomenon:
- Reduce and remove risk factors and possible triggers such as smoking, avoid a medication known as beta-blockers, and avoid any other remediable underlying cause (eg, use of vibratory equipment)
- Keep hands and body warm
- Take a medication that are long-acting formulations of calcium channel blockers
- Use topical nitroglycerin paste if needed.
Esophageal dysmotility: The treatment of esophageal dysmotility and gastroesophageal reflux in people with scleroderma is the same as in patients without scleroderma. Treatment involves behavior changes, medication (H2 blockers), and a surgical procedure (esophageal dilatation) in more severe cases. Esophageal dilatation can help when severe feeding difficulty or regurgitation occur due to an esophageal narrowing.
Sclerodactyly: Treatments include medication (corticosteroids, nonsteroidal anti-inflammatory drugs, D-penicillamine, IFN-gamma, cyclosporine, and cytostatic drugs). Skin involvement in CREST syndrome typically is not severe; therefore, treatment is not need in many cases.
Telangiectasia: Pulsed-dye laser treatment seems effective for the treatment the telangiectasia in the face, but this has not been specifically studied in CREST patients. Treatment may include estrogen-progesterone or desmopressin, laser ablation, or sclerotherapy.
Other treatment may include stretching exercises for the finger joints, and facial exercises that may help keep the face and mouth flexible, under a physical therapist supervision. If CREST syndrome is making it difficult to do daily tasks, an occupational therapist can help individuals learn new ways of doing things. For example, special toothbrushes and flossing devices can make it easier to care for the teeth. Surgery may be necessary for some affected individuals.
About 45% of people with systemic sclerosis may have depression and 64% also develop anxiety, so it is recommended to have an early assessment and treatment.
For pain management, studies have shown that oxycodone is effective and safe for pain due to severe skin ulcers, while topical lidocaine helps reduce pain of the finger’s ulcers in people with systemic scleroderma 9.
There are also some lifestyle changes and home remedies that may be helpful for some people with CREST syndrome. For example:
- Wearing gloves and mittens is the weather is cool and/or dressing in layers and wear a hat, thermal socks and boots to maintain the body’s core temperature and to reduce Raynaud’s symptoms
- Quit smoking because nicotine constricts the blood vessels, making Raynaud’s phenomenon worse
- Eating soft, moist foods and chewing food well when having swallowing difficulties
- Eating small, frequent meals; avoiding spicy or fatty foods, chocolate, caffeine, and alcohol; and avoid exercising immediately before or after eating to improve symptoms of acid reflux.
- Use of skin moisturizers and using an humidifier at home to improving moisture levels in the home to ease skin and breathing symptoms.
Medications
Several types of medications can help ease the signs and symptoms of limited scleroderma, including:
- Topical antibiotics. If skin ulcers become infected, you might need to apply topical antibiotics and bandage the area. If topical treatment doesn’t work, you might need oral or intravenous antibiotics.
- Antacid drugs. For heartburn, your doctor might suggest drugs that reduce the production of stomach acid.
- Blood pressure lowering drugs. Medications that open small blood vessels and increase circulation might help relieve Raynaud’s symptoms and reduce increased pressure in the arteries between the heart, lungs and kidney.
- Drugs to suppress the immune system. These types of medications have shown promise in preventing a condition in which excess collagen collects in the tissue between the lungs’ air sacs.
Therapy
Stiff, painful joints and skin are common problems in limited scleroderma. Physical or occupational therapy can teach exercises to help you maintain your flexibility and strength.
- Physical therapy. Stretching exercises are important to help prevent loss of mobility in your finger joints. A physical therapist can also show you facial exercises that can help keep your face and mouth flexible.
- Occupational therapy. If needed, an occupational therapist can help you learn new ways of performing daily tasks. For example, special toothbrushes and flossing devices can make it easier for you to care for your teeth.
Surgery
Surgery might be necessary for certain problems, such as:
- Calcium deposits. Large or painful calcium deposits are sometimes surgically removed.
- Red spots or lines. Laser surgery can reduce the appearance of red spots or lines caused by swollen blood vessels near the surface of the skin.
CREST syndrome life expectancy
In most cases, localized scleroderma carries a good prognosis and patients have a normal life span. Most patients with limited CREST syndrome can also expect a favorable outlook. Many internal organs may become affected over time, however disease progression tends to be slow. In a large 2003 US study by Mayes et al 10, the survival rate from time of diagnosis was computed to be 77.9% at 5 years, 55.1% at 10 years, 37.4% at 15 years, and 26.8% at 20 years. The extent of skin involvement is a good predictor of survival in patients with scleroderma.
Limited cutaneous disease is associated with a better survival rate than diffuse disease 11. Similarly, patients with sclerodactyly alone have better survival rates than patients with truncal skin involvement 12. Calcinosis cutis may be problematic, even in patients with limited cutaneous disease 13.
Renal involvement is responsible for half of all scleroderma-related deaths in patients with widespread skin changes, while patients with sclerodactyly alone do not tend to develop any type of renal disease.
The mortality in patients with limited skin involvement results from cardiac, pulmonary, and gastrointestinal causes.
References- Scleroderma. https://www.niams.nih.gov/health-topics/scleroderma
- Limited scleroderma. https://www.mayoclinic.org/diseases-conditions/crest-syndrome/symptoms-causes/syc-20355535
- Hunzelmann N. [Current treatment of systemic scleroderma]. Hautarzt. 2018 Nov;69(11):901-907
- Adigun R, Hariz A. Systemic Sclerosis (CREST syndrome) [Updated 2019 Jan 11]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK430875
- Silman AJ. Epidemiology of scleroderma. Ann. Rheum. Dis. 1991 Nov;50 Suppl 4:846-53.
- Sharif K, Watad A, Coplan L, Lichtbroun B, Krosser A, Lichtbroun M, Bragazzi NL, Amital H, Afek A, Shoenfeld Y. The role of stress in the mosaic of autoimmunity: An overlooked association. Autoimmun Rev. 2018 Oct;17(10):967-983.
- Xu Y, Mo N, Jiang Z, Fu S, Wei X, Zhao D, Xie Z, Jia W, Liu J, Wang X, Shi D, Jiao Y, Liu C, Yang X. Human leukocyte antigen (HLA)-DRB1 allele polymorphisms and systemic sclerosis. Mod Rheumatol. 2018 Sep 02;:1-20.
- CREST Syndrome. https://emedicine.medscape.com/article/1064663-overview
- Giuggioli D, Manfredi A, Colaci M, Ferri C. Oxycodone in the long-term treatment of chronic pain related to scleroderma skin ulcers. Pain Medicine. October 2010; 11(10):1500-1503
- Mayes MD, Lacey JV Jr, Beebe-Dimmer J, Gillespie BW, Cooper B, Laing TJ, et al. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 2003 Aug. 48(8):2246-55.
- Mayes MD. Scleroderma epidemiology. Rheum Dis Clin North Am. 1996 Nov. 22(4):751-64.
- Barnett AJ, Miller MH, Littlejohn GO. A survival study of patients with scleroderma diagnosed over 30 years (1953-1983): the value of a simple cutaneous classification in the early stages of the disease. J Rheumatol. 1988 Feb. 15(2):276-83.
- Düzgün N. Cutaneous calcinosis in a patient with limited scleroderma: CREST Syndrome. Eur J Rheumatol. 2017 Dec. 4 (4):305-306.





{kind=link}