Cystinuria
Cystinuria is an inborn error of metabolism resulting from poor absorption and reabsorption of the amino acid cystine in the intestines and kidneys. As urine becomes more concentrated in the kidneys, this leads to an accumulation of poorly soluble cystine in the urine and the excess cystine forms crystals resulting in the production of kidney stones (urolithiasis). Larger crystals become stones that may lodge in the kidneys or in the bladder. Sometimes cystine crystals combine with calcium molecules in the kidneys to form large stones. These crystals and stones can create blockages in the urinary tract and reduce the ability of the kidneys to eliminate waste through urine. The stones also provide sites where bacteria may cause infections. Symptoms may include acute episodes of abdominal or lower back pain, presence of blood in the urine (hematuria), and recurrent episodes of kidney stones may result in frequent urinary tract infections, which may ultimately result in renal insufficiency. The combined incidence of cystinuria has been estimated to be 1 in 7000 – 10,000. However, as with any genetic disease, the prevalence can vary greatly among communities. Prevalence is 1/100,000 in Sweden, 1/18,000 in Japan, 1/40,00 in Australia, 1/2500 in Israel, and 1/2000 in Great Britain and Spain 1.
As a genetic condition, cystinuria is present from birth and persists for life. However, symptoms including kidney stone formation, may not occur immediately. While some patients form stones at infancy, others may not be aware of their disease until decades later. Some cystinuric people will never form kidney stones, despite having high urinary cystine concentrations. The underlying cause of why some people seem to be more affected than others is currently unknown.
Cystine is an amino acid which is a fundamental building block of protein in the body. Cystine and other amino acids are typically recycled within the body as proteins are broken down and rebuilt to support the biochemical processes that enable us to live and grow. Interestingly, intestinal transport and absorption of cystine, in patients with cystinuria, tends to be impaired, but other factors offset this benefit 2. Unlike other biochemicals, amino acids are usually reclaimed from the urine during processing in the kidneys, with only small amounts ending up in the final product. However, when cystine is abundant in a person’s urine, that person is said to have cystinuria (literally, “cystine in the urine”). Cystine is the least soluble of all the essential amino acids. Cystinuria would not be a problem except for its relative insolubility in urine at physiological pH levels. Cystine solubility is highly pH-dependent because it substantially increases as the urine becomes increasingly alkaline. Cystine solubility is also affected by urinary macromolecules and ions, both of which increase cystine’s solubility.
The solubility of cystine in urine is about 250 mg/L at a pH of 6.5. This solubility increases as the urine becomes more alkaline. For example, 500 mg of cystine will dissolve in a liter of urine at a pH of 7.5. This solubility goes up to 750 mg/L at a pH of 8, but it becomes very difficult to achieve a pH above 7.5 in clinical practice, and there is also an increased risk of calcium phosphate precipitation in very alkaline urine 3.
Cystinuria is an autosomal recessive disease that affects the proximal renal tubular reabsorption of cystine, this same problem also affects lysine, ornithine, and arginine (COLA) but only cystine is clinically significant as it is the only amino acid in this group that will form stones. Some heterozygous carriers have an autosomal dominant, incomplete penetrance appearance with elevated, but typically nondisease causing, urinary cystine excretion. Cystinuria is caused by variants in genes, SLC3A1 on chromosome 2p and SLC7A9 on chromosome 19q. Initially, cystinuria was classified into subtypes 1, 2, and 3 (type 2 and 3 are also referred as nontype-1) based on the amount of urinary cystine excreted in heterozygous parental specimens. A new classification system has been proposed to distinguish the various forms of cystinuria: type A, due to variants in the SLC3A1 gene; type B, due to variants in the SLC7A9 gene; and type AB, due to 1 variant in each SLC3A1 and SLC7A9 gene 1.
Cystine stones
Cystine stones are rare and they form only in persons with cystinuria, an inherited metabolic disorder that causes high levels of cystine in the urine. While most cystine stone formers will make pure cystine stones, up to 40% may develop mixed calculi that will also contain calcium oxalate, calcium phosphate, or struvite 4.
Compared to calcium stone formers, cystine nephrolithiasis patients will tend to make larger stones, require more urological procedures and will start making stones at an earlier age. They also face a greater risk of eventual kidney damage and chronic renal failure compared to calcium nephrolithiasis patients 5.
In cystine stone formers, the typical patient makes about one stone every 1 to 2 years, has one surgical procedure every 3 years, and has undergone 7 surgeries by the time they are middle-aged 6.
- Men are affected about twice as often as women. The peak age of presentation of the original cystine stone is 22 years of age; although, 22% of patients will start making cystine stones as children.
- The overall risk of renal injury/failure is high at up to 70%, but end-stage renal failure is relatively low in cystinuria patients at less than 5%.
- Twenty percent to 40% of cystinuria patients have other urinary chemical abnormalities such as hypocitraturia (44%), hypercalciuria (19%), and hyperuricosuria (22%).
- Infrequently, cystinuria is associated with hemophilia, muscular dystrophy, mongolism, hereditary pancreatitis, and retinitis pigmentosa.
- Recurrence rates after surgical intervention approach 45% at 3 months without prophylactic medical treatment. With treatment, the average recurrence rate drops to 25% at 3 years.
About 25% of cystine stone formers will have non-cystine chemical components in their stones. For this reason, complete stone composition analyses and 24 hour urine tests are recommended for optimal stone prophylaxis 3.
On average, individuals with untreated cystinuria experience one new stone every year and require a surgical procedure to remove the stones every 3 years. By middle age, the average cystinuria patient will have undergone seven surgical procedures 7.
The diagnosis of cystinuria is readily made by stone analysis, microscopic examination of the urine, and 24-hour urine testing. Although surgical intervention is necessary, the cornerstones of treatment are dietary and medical prevention of recurrent stone formation.
The most basic thing you can do to prevent stone formation is to drink more fluids, thereby diluting your urine. Your goal should be to urinate more than two liters per day. All fluids count toward this goal, but water is, of course, the best.
Cystine stones symptoms
Most people are diagnosed with kidney stones after the thunderclap onset of excruciating and unforgettable pain. This severe pain occurs when the kidney stone breaks loose from the place that it formed, the renal papilla, and falls into the urinary collecting system. When this happens, the stone can block the drainage of urine from the kidney, a condition known as renal colic. The pain may begin in the lower back and may move to the side or the groin. Other symptoms may include blood in the urine (hematuria), frequent or persistent urinary tract infections, urinary urgency or frequency and nausea or vomiting.
When your doctor evaluates you for a kidney stone, the first step will be a complete history and physical examination. Important information regarding current symptoms, previous stone events, medical illnesses and conditions, medications, dietary history and family history will all be collected. A physical examination will be performed to evaluate for signs of a kidney stone, such as pain in the flank, lower abdomen or groin.
Your doctor will perform a urinalysis, to look for blood or infection in the urine. A blood sample will also be collected so that kidney function and blood counts can be measured.
Even though all of these tests are necessary, a kidney stone can only be definitively diagnosed by a radiologic evaluation. In some cases, a simple X-ray, called a KUB (kidney, ureter and bladder), will be adequate to detect a stone. If your doctor requires more information, an intravenous pyelogram (IVP) or a computed tomography (CT) scan may be necessary.
Sometimes kidney stones do not cause any symptoms at all. Such painless stones can be discovered when your doctor is looking for other things on X-rays. Sometimes, although a stone does not cause any pain, it can cause other problems, such as recurring urinary tract infections or blood in the urine.
Cystine stones treatment
Surgical treatment of cystine stones is similar to that of other kidney stones except that cystine is notoriously resistant to extracorporeal shock wave lithotripsy (ESWL) unless the stones are less than 1 cm in size 1. Retrograde pyelography and the use of indwelling ureteral catheters will help to visualize stones that otherwise would be difficult to localize for ESWL (extracorporeal shock wave lithotripsy) therapy. Even then, the stones are relatively difficult to see and target reliably. They also will likely require more shocks than calcium oxalate or calcium phosphate stones. For these reasons, ureteroscopy with laser lithotripsy is preferred for most cystinuria patients with obstructing cystine stones that require surgery.
Total removal of all cystine stones and fragments has demonstrated reduced recurrence rates and better preservation of renal function. Surgery has not caused any measurable decrease in overall renal function 8.
Medical management
Acceptable levels of urinary cystine are 250 mg/L or less at a urinary pH of 6.5 to 7. This level can often be reached through an increased fluid intake and urinary alkalinization. The use of thiol-based medications to reduce urinary cystine levels is discouraged unless hydration therapy and alkalinization treatment are insufficient to achieve the desired “optimal” cystine concentration levels (less than 250 mg/L) at an acceptable pH (6.5 to 7). A sustained urinary pH of 7.5 can be used to dissolve existing cystine stones. Some experts have recommended even lower cystine concentrations of 150 mg/L and possibly even lower at 90 mg/L as “optimal” 3.
Hydration is usually the first step in medical management. Increasing fluid intake sufficiently to reliably generate 2500 to 3000 ml or more of urine per day is often necessary. The goal is to dilute the urine sufficiently to get the urinary cystine content as close to the less than 250 mg/L recommended concentration level as possible. This frequently requires having the patient wake up in the middle of the night to void and drink extra water. Drinking 240 ml of water every hour during the day and 480 ml before bed and at least once overnight is a standard strategy for maximizing oral hydration. Hydration can be monitored by following the specific gravity which should always be 1.010 or less. Since some cystinuric patients can generate up to 1400 mg of cystine per day, hydration alone may not be sufficient, but it is always the first step in management. Up to one-third of cystine stone patients can manage their stone recurrences with fluid management. Optimal hydration will depend on the patient’s cystine excretion 3.
Alkalinization can not only prevent cystine precipitation and stone formation but may also dissolve existing cystine stones. For prophylaxis, the urinary pH should be targeted at 7.0 to 7.5, but for stone dissolution, a urinary pH higher than 7.5 needs to be maintained. At this high pH level (above 7.5), calcium phosphate stones can precipitate. In such cases, hypercalciuria needs to be controlled tightly with diet and thiazides. Mineral water and citrus juices can help increase pH levels, but potassium citrate supplementation is the mainstay of urinary alkalinization therapy. Usual daily potassium citrate dosage in cystinuria is 60 to 90 mEq total in 3 or 4 divided doses and then titrated as needed to optimize the pH. Serum potassium should also be checked periodically in patients on high dosages of potassium citrate to detect hyperkalemia 3.
Sodium bicarbonate can also be used to help with pH issues, but it tends to have a relatively short-term alkalinizing effect, and the extra sodium intake may increase urinary cystine excretion. High animal protein diets are also discouraged in cystinuric patients for the same reason 3.
Acetazolamide is a carbonic anhydrase inhibitor that increases urinary bicarbonate excretion and raises urinary pH levels. While not a first-line therapy (it can cause hypocitraturia and metabolic acidosis), it may occasionally be of some help in maintaining high urinary pH levels in addition to the other therapies mentioned. It can be particularly useful in maintaining a high overnight urinary pH without the need for multiple awakenings or additional overnight alkalinization dosing 9.
When conservative measures as outlined above are insufficient after a 3-month trial period, a thiol-based drug regimen is usually the next step in active cystine stone formers 3.
Thiol-Based Agents
Cystine is composed of 2 cysteine molecules bound together by a disulfide bond. Thiol-based drugs have sulfhydryl groups that can reduce this disulfide bond producing a mixed cysteine disulfide compound that is far more soluble than the original cystine molecule. As a general guide, most patients with a 24-hour urinary cystine excretion of 500 mg or more are likely to need a thiol medication in addition to hydration therapy and alkalinization 3.
Thiol-based treatment is thought to have the extra benefit of possibly making cystine stones more amenable to extracorporeal shock wave lithotripsy (ESWL) treatment. This may occur because of the mixing of calcium phosphate along with the cystine creating a more fragile stone that is more easily fragmented with extracorporeal shock wave lithotripsy (ESWL) therapy.
Penicillamine, a penicillin derivative, was the first thiol drug used for cystinuria. Penicillamine-cysteine disulfide is 50 times more soluble in urine than cystine. Each 250 mg penicillamine tablet can reduce urinary cystine levels by about 75 mg to 100 mg per day. The problem with penicillamine is that there is a high incidence of side effects including fever, rash, loss of taste, arthritis, leukopenia, aplastic anemia, gastrointestinal (GI) disturbances, renal membranous nephropathy with proteinuria, and pyridoxine deficiency. The incidence of significant side effects is about 50% which limits long-term compliance. Almost 70% of patients discontinued the drug due to adverse effects in one study. For these reasons, penicillamine use is limited in favor of other thiol-based drugs 10.
Tiopronin (Thiola, alpha-mercaptopropionylglycine, or alpha-MPG) is a second-generation thiol drug that works similarly to penicillamine but is roughly 30% more effective with significantly fewer side effects. It was approved for use in the United States in 1988, so there is ample experience with the medication. The typical dose is 300 mg three times per day. Long-term compliance is about 70%. For these reasons, tiopronin is currently the thiol drug of choice for cystinuria when hydration and urinary alkalinization therapy fail to achieve optimal cystine concentration levels at an acceptable pH 11.
Captopril is an ACE inhibitor normally used for hypertension, but it is also a thiol-based drug that can form captopril-cysteine mixed disulfides that are highly soluble, in cystinuric patients. While safe with few side effects, captopril’s clinical effectiveness in cystinuric stone forming patients is uncertain as various studies provide conflicting results. It should be considered a reasonable treatment option in cystinuric patients who are also hypertensive 12.
Bucillamine is a third-generation, thiol-based drug that is currently available only in Japan and South Korea and is approved only for use in rheumatoid arthritis. As a di-thiol compound, it would theoretically be more effective than tiopronin and better tolerated since lower dosages of the drug would be needed. Experience in Asia has shown a low toxicity profile, and phase-2 studies are currently underway in the United States to determine its potential clinical usefulness in treating hyper cystinuria.
Besides Bucillamine, other new cystine binding agents or crystal growth inhibitors are being evaluated. For example, L-cystine dimethyl esters and L-cystine methyl esters have shown promising results with good therapeutic effects at relatively low concentrations which suggest better tolerability and fewer side effects than similar agents 5. Some new, investigational thiol compounds, such as thiophosphate and meso-2-3-dimercaptosuccinic acid, are undergoing testing and appear promising 13.
Cystinuria causes
Mutations in the SLC3A1 or SLC7A9 gene cause cystinuria. The SLC3A1 and SLC7A9 genes provide instructions for making the two parts (subunits) of a protein complex that is primarily found in the kidneys. Normally this protein complex controls the reabsorption of certain amino acids, including cystine, into the blood from the filtered fluid that will become urine. Mutations in either the SLC3A1 gene or SLC7A9 gene disrupt the ability of the protein complex to reabsorb amino acids, which causes the amino acids to become concentrated in the urine. As the levels of cystine in the urine increase, the crystals typical of cystinuria form. The other amino acids that are reabsorbed by the protein complex do not create crystals when they accumulate in the urine.
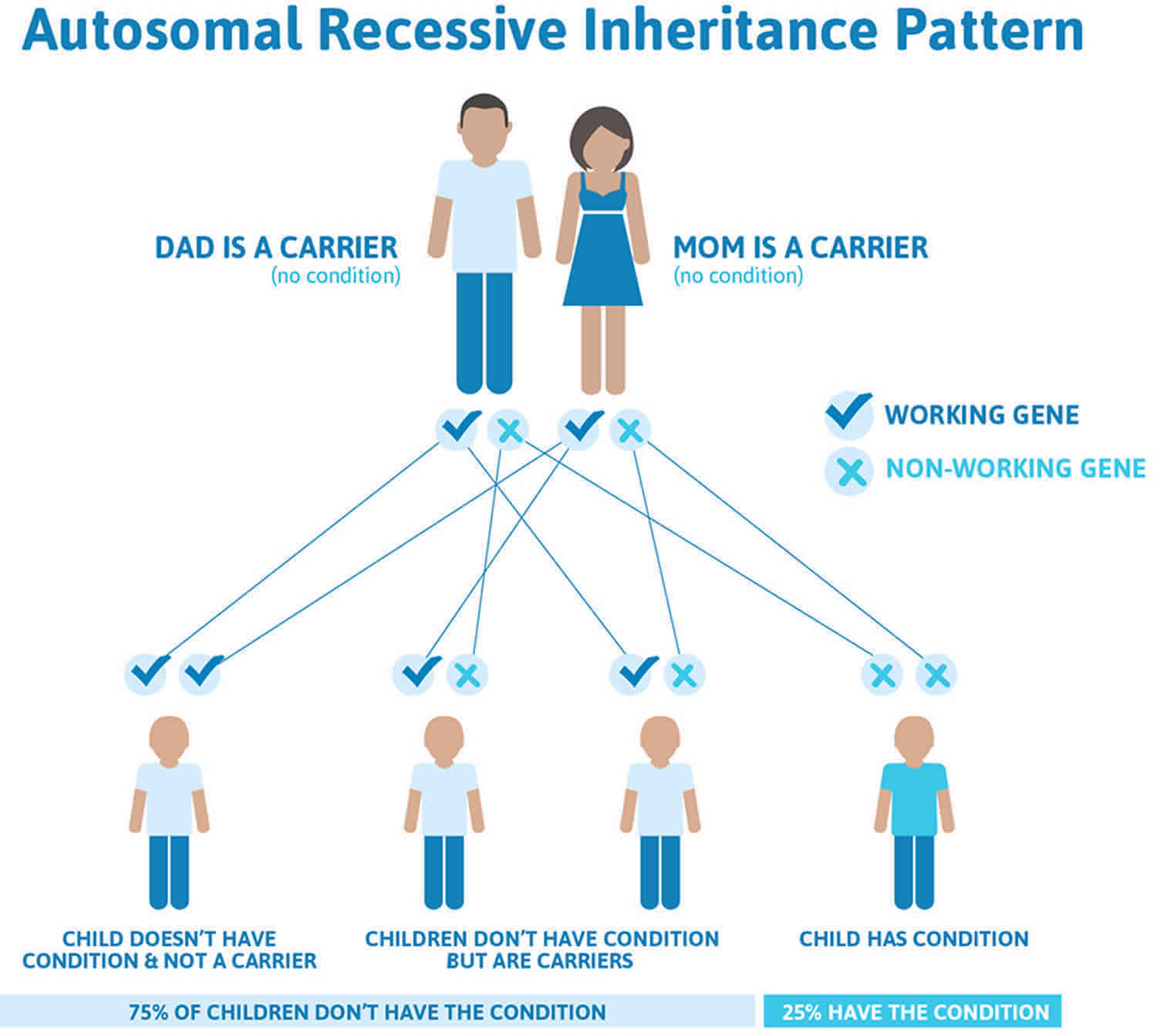
Cystinuria inheritance pattern
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 1 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 1. Cystinuria autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Cystinuria symptoms
People with cystinuria excrete abnormally high levels of cystine in the urine. The level of cystine is so high that it remains undissolved in the urine (insoluble). The amino acids lysine, arginine, and ornithine are also excreted in massive amounts by people with cystinuria. However, these amino acids dissolve more readily in the urine (more soluble) and are not associated with any particular symptoms.
Cystine crystalluria (crystals in the urine) and kidney stone formation are the only definitive symptoms of cystinuria. Kidney stones can cause their own symptoms including the presence of blood and/or signs of infection (frequent urination accompanied by a burning sensation and possible fever) in the urine due to the aggravation of tissues by the stone(s).
The initial symptom of cystinuria is usually a constant dull ache to sharp stabbing pain and severe spasms in the lower back or side of the abdomen (renal colic). Kidney stone pain, known as renal colic, is typically felt in the area between the ribs and the pelvis, wrapping from the small of the back through the side (flank) to the groin. The pain may migrate throughout this area, finally becoming more associated with the bladder and groin as a stone makes its way along the urinary tract. Men may experience pain in the testicles. The acute and chronic pains endured by people suffering with cystinuria can cause nausea and vomiting, or lead to tiredness and in some cases depression. Other symptoms may include blood in the urine (hematuria), obstruction of the urinary tract (ureters), and/or infections of the urinary tract. Frequent recurrences ultimately may lead to kidney damage.
Novice stone formers may fail to recognise a kidney stone as the cause of pain which is often difficult to define and remote from the location of the kidney. However, experienced sufferers of cystinuria are likely to become familiar with their own stone pains and able to readily recognize stone events when they occur.
People with cystinuria typically produce stones (cystine calculi) that are generally small, with a jagged crystalline surface. These stones may be accompanied by urinary “gravel,” which consists of yellowish-brown hexagonal crystals. All patients with urinary stones should be screened for cystinuria.
Cystinuria diagnosis
Since cystine contains sulfur, the urine of hyper-cystinuric individuals may have a rotten egg odor. Typical hexagonal cystine crystals can sometimes be seen on urinalysis in affected patients. When these hexagonal crystals are seen on urinalysis, it suggests supersaturation of the urine with cystine 14.
The sodium cyanide-nitroprusside test is often the initial laboratory screening test for cystinuria as it is fast, simple and provides a reliable, qualitative assessment of urinary cystine levels. The cyanide converts cystine to cysteine which then binds to the nitroprusside creating an intense purple color in just a few minutes. The test typically turns positive at cystine levels above 75 mg/gm creatinine 15.
The definitive diagnosis of obstructing calculi will require imaging which includes CT scans, KUB (kidney, ureter and bladder) x-rays and/or ultrasound. CT scans without contrast remain the “gold standard” for the diagnosis of urolithiasis and will demonstrate cystine stones clearly as will an ultrasound for renal calculi, but they cannot distinguish cystine from other stone chemical constituents. Plain x-rays of the abdomen will not show cystine stones well as they are only faintly radiopaque and will tend to have a ground glass appearance 3.
Cystinuria treatment
No curative treatment of cystinuria exists, and patients will have a life-long risk of stone formation, repeated surgery, impaired renal function and quality of life. The primary objective of treatment for cystinuria is to reduce the cystine concentration in the urine. Drinking of large amounts of fluid both day and night maintains a high volume of urine (generate 2500 to 3000 mL of urine daily or more) and reduces cystine concentration in your urine. Making the urine more alkaline (alkalization) helps cystine to dissolve more readily in the urine and may also prevent the formation of stones. Drugs that may be prescribed to make the urine more alkaline include potassium citrate, and acetazolamide. This treatment is accompanied by dietary salt restriction. The goal is to achieve a cystine concentration of 250 mg/L or less at a pH of at least 6.5. “Optimal” cystine level is generally considered to be 150 mg/L or less, but some reports suggest that a cystine concentration goal of 90 mg/L is “optimal.”
Another approach to the treatment of cystinuria is administration of d- penicillamine, although there are some risks of side effects with this drug. D-penicillamine promotes the formation of cystine in a different chemical form (mixed disulfide), which is more soluble in the urine and is excreted.
The orphan drug alpha-mercaptopropionyl glycine, also known as tiopronin (Thiola) has been approved as a treatment for cystinuria. This drug is manufactured by Mission Pharmacal. Thiola has been shown to lower the level of cystine in the urine of patients with cystinuria. Another medicine that is used at times is captopril.
Kidney and/or bladder surgery sometimes becomes necessary, but stones (calculi) commonly recur. Small stones may pass spontaneously on their own with high fluid intake and, if needed, pain medications. If spontaneous stone passage is unsuccessful, stones may be removed by a special procedure. The surgeon can view the stones through an illuminated optic instrument inserted in the urethra and passed up into the upper urinary tract.The stones are then removed with special instruments (endoscopic basket extraction). Laser techniques and ultrasound have also been used to dissolve stones in the bladder and/or kidneys that are caused by cystinuria.
Genetic counseling is recommended for patients and their families. Other treatment is symptomatic and supportive.
Cystinuria prognosis
No curative treatment of cystinuria exists, and patients will have a life-long risk of stone formation, repeated surgery, impaired renal function and quality of life. One study reported 1.22 stone episodes per year. Recurrence rates after surgical intervention approach 45% at 3 months without medical management. The recurrence rate with medical management improves to approximately 25% at 3 years after surgery but is still inferior to rates for other types of calculi. The probability of a recurrence-free survival at 1- and 5-year follow-up is 0.73 and 0.27, respectively 16. Barbey et al 7 reported one new stone formation per patient per y ear and an average of one surgical procedure every 3 years, with 7 surgical procedures for nephrolithiasis by middle age.
Urinary calculi are generally the only manifestation of cystinuria, although 10% of cases are complicated by hypertension, and one study found a weak association with short stature. Patients with cystinuria who form stones are at higher risk for anatomical renal loss (nephrectomy) than those who form calcium oxalate stones. The risk of renal impairment is high; up to 70% of patients may be affected depending on the length of follow-up and medical therapy. However, according to Lindell et al 17, end-stage renal disease occurs in less than 5% of patients with cystinuria. Other complications include chronic pyelonephritis, hypertension, mental illness and mental retardation 18.
References- Leslie SW, Nazzal L. Renal Calculi (Cystinuria, Cystine Stones) [Updated 2019 May 6]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK470527
- Shen L, Cong X, Zhang X, Wang N, Zhou P, Xu Y, Zhu Q, Gu X. Clinical and genetic characterization of Chinese pediatric cystine stone patients. J Pediatr Urol. 2017 Dec;13(6):629.e1-629.e5
- Andreassen KH, Pedersen KV, Osther SS, Jung HU, Lildal SK, Osther PJ. How should patients with cystine stone disease be evaluated and treated in the twenty-first century? Urolithiasis. 2016 Feb;44(1):65-76.
- Reinstatler L, Stern K, Batter H, Scotland KB, Ardekani GS, Rivera M, Chew BH, Eisner B, Krambeck AE, Monga M, Pais VM. Conversion from Cystine to Noncystine Stones: Incidence and Associated Factors. J. Urol. 2018 Dec;200(6):1285-1289.
- Yang Y, Albanyan H, Lee S, Aloysius H, Liang JJ, Kholodovych V, Sahota A, Hu L. Design, synthesis, and evaluation of l-cystine diamides as l-cystine crystallization inhibitors for cystinuria. Bioorg. Med. Chem. Lett. 2018 May 01;28(8):1303-1308.
- Streeper NM, Wertheim ML, Nakada SY, Penniston KL. Cystine Stone Formers Have Impaired Health-Related Quality of Life Compared with Noncystine Stone Formers: A Case-Referent Study Piloting the Wisconsin Stone Quality of Life Questionnaire Among Patients with Cystine Stones. J. Endourol. 2017 Apr;31(S1):S48-S53.
- Barbey F, Joly D, Rieu P, et al. Medical treatment of cystinuria: critical reappraisal of long-term results. J Urol. 2000 May. 163(5):1419-23.
- Moore SL, Somani BK, Cook P. Journey of a cystinuric patient with a long-term follow-up from a medical stone clinic: necessity to be SaFER (stone and fragments entirely removed). Urolithiasis. 2019 Apr;47(2):165-170.
- Sterrett SP, Penniston KL, Wolf JS, Nakada SY. Acetazolamide is an effective adjunct for urinary alkalization in patients with uric acid and cystine stone formation recalcitrant to potassium citrate. Urology. 2008 Aug;72(2):278-81.
- Joly D, Rieu P, Méjean A, Gagnadoux MF, Daudon M, Jungers P. Treatment of cystinuria. Pediatr. Nephrol. 1999 Nov;13(9):945-50.
- Fattah H, Hambaroush Y, Goldfarb DS. Cystine nephrolithiasis. Transl Androl Urol. 2014 Sep 01;3(3):228-233.
- Cohen TD, Streem SB, Hall P. Clinical effect of captopril on the formation and growth of cystine calculi. J. Urol. 1995 Jul;154(1):164-6.
- Sahota A, Tischfield JA, Goldfarb DS, Ward MD, Hu L. Cystinuria: genetic aspects, mouse models, and a new approach to therapy. Urolithiasis. 2019 Feb;47(1):57-66.
- Courbebaisse M, Prot-Bertoye C, Bertocchio JP, Baron S, Maruani G, Briand S, Daudon M, Houillier P. [Nephrolithiasis of adult: From mechanisms to preventive medical treatment]. Rev Med Interne. 2017 Jan;38(1):44-52.
- Nakagawa Y, Coe FL. A modified cyanide-nitroprusside method for quantifying urinary cystine concentration that corrects for creatinine interference. Clin. Chim. Acta. 1999 Nov;289(1-2):57-68.
- Chow GK, Streem SB. Contemporary urological intervention for cystinuric patients: immediate and long-term impact and implications. J Urol. 1998 Aug. 160(2):341-4; discussion 344-5.
- Lindell A, Denneberg T, Granerus G. Studies on renal function in patients with cystinuria. Nephron. 1997. 77(1):76-85.
- Prot-Bertoye C, Lebbah S, Daudon M, et al. CKD and Its Risk Factors among Patients with Cystinuria. Clin J Am Soc Nephrol. 2015 May 7. 10 (5):842-51.





{kind=link}