What is cytokine storm
Cytokine storm also called hypercytokinemia or cytokine release syndrome, is a general term applied to massive release of cytokines and chemokines due to an uncontrolled dysregulation of the host immune defense in response to infection and other stimuli that causes loss of function of multiple organs 1. Cytokine storm is characterized by a clinical presentation of overwhelming systemic inflammation, hyperferritinemia, hemodynamic instability, and multi-organ failure, and if left untreated, it leads to death 2. Cytokine storm is an acute hyperinflammatory response that may be responsible for critical illness in many conditions including viral infections, cancer, sepsis, and multi-organ failure 3. In short, cytokine storm involves an immune response that causes collateral damage, which may be greater than the immediate benefit of the immune response. Cytokines play an important role in normal immune responses, but having a large amount of them released in the body all at once can be harmful. However, exactly what constitutes a “cytokine storm” remains ill defined, according to many experts. Given the lack of a unifying definition for cytokine storm 4 and disagreement about the distinction between cytokine storm and a physiologic inflammatory response, Fajgenbaum et al 5 propose the following three criteria for identifying cytokine storm: elevated circulating cytokine levels, acute systemic inflammatory symptoms, and either secondary organ dysfunction (often renal, hepatic, or pulmonary) due to inflammation beyond that which could be attributed to a normal response to a pathogen (if a pathogen is present), or any cytokine-driven organ dysfunction (if no pathogen is present).
The term “cytokine storm” was first used in the literature to describe the engraftment syndrome of acute graft-versus-host disease after allogeneic hematopoietic stem-cell transplantation 6. The term “cytokine release syndrome” was coined to describe a similar syndrome after infusion of muromonab-CD3 (OKT3) 7. From a historical perspective, cytokine storm was previously referred to as an influenza-like syndrome that occurred after systemic infections such as sepsis and after immunotherapies such as Coley’s toxins 8. Yersinia pestis infection (i.e., the plague) has led to major pandemics (e.g., the Black Death) and triggers alveolar macrophages to produce excessive amounts of cytokines, resulting in cytokine storm 9. An exaggerated immune response was suspected to contribute to the lethality of the 1918–1919 influenza pandemic. In fact, a reconstructed H1N1 influenza virus isolated from the 1918 pandemic, as compared with common reference strains of the influenza virus that causes influenza A, triggered marked pulmonary inflammation in mice 10. The administration of recombinant cytokines (e.g., interleukin-1, interleukin-6, interleukin-12, interleukin-18, tumor necrosis factor [TNF], and interferon-γ) in animal models and for cancer treatment in humans induces severe toxic effects or lethality consistent with the central role of cytokines as mediators of hyperinflammation in cytokine storm 11, 12. Conversely, reduction in symptoms and improvement in organ function with neutralization of specific cytokines with monoclonal antibodies also reveal that excessive levels of certain cytokines play a critical role in a number of cytokine storm disorders.
Cytokine storm and cytokine release syndrome are life-threatening systemic inflammatory syndromes involving elevated levels of circulating cytokines and immune-cell hyperactivation that can be triggered by various therapies, pathogens, cancers, autoimmune conditions, and monogenic disorders. Cytokine storm is characterized by constitutional symptoms, systemic inflammation and multiorgan dysfunction that can lead to multiorgan failure if inadequately treated (see Figure 1) 5. Cytokine storms can result in autoimmune reactions, tissue damage, or septic shock. The pathogenesis of cytokine storm is complex but includes loss of regulatory control of proinflammatory cytokines (tumor necrosis factor [TNF], interferon-γ, interleukin-1, interleukin-1β, interleukin-6 and interleukin-18) production, both at local and systemic levels 5. A wide range of clinical and laboratory abnormalities can be observed in cytokine storm leading to an increased risk of vascular hyperpermeability, multiorgan failure, and eventually death when the high cytokine concentrations are unabated over time 13. However, all cases involve elevated circulating cytokine levels, acute systemic inflammatory symptoms, and secondary organ dysfunction (often renal, hepatic, or pulmonary) 5. Cytokine storm progresses rapidly, and the mortality is high, with an all-cause mortality of approximately 40% in adults 14 and early recognition and initiation of treatment is crucial to improve patient outcomes 15.
Recognition that the immune response to the pathogen, but not the pathogen itself, can contribute to multiorgan dysfunction and that similar cytokine storm syndromes could occur with no obvious infection led to the investigation of immunomodulators and cytokine-directed therapies. One of the earliest targeted therapies for cytokine storm was the anti–interleukin-6 receptor monoclonal antibody tocilizumab, which was developed for the treatment of idiopathic multicentric Castleman’s disease in the 1990s. A host of other disorders have been described as causes of cytokine storm and targeted with immune-directed therapies, such as sepsis, primary and secondary hemophagocytic lymphohistiocytosis 16, autoinflammatory disorders, and coronavirus disease 2019 (Covid-19).
Circulating cytokine levels can be difficult to measure because cytokines have short half-lives, circulating levels may not accurately reflect local tissue levels, and measurements may not be easily obtained worldwide. Experts do not propose a specific threshold for elevations in cytokine levels above the normal range, and they do not recommend specific cytokine panels or list particular cytokines whose levels must be elevated, given the lack of available evidence 5. However, they believe that this is an important area for future research 5.
Your immune system is made up of a complex network of cells, chemicals, tissues and organs that cooperate to protect you from invasion and harm from a variety of infectious agents such as bacteria, viruses, fungi, parasites, toxins (chemicals produced by microbes) and other invaders (such as cancer cells) and also to provide you with a surveillance system to monitor the integrity of your tissues. Your immune system recognizes invaders such as bacteria, viruses and fungi as well as abnormal cells. It mounts an immune response to help the body fight the invasion. The ability to recognize and respond to foreign entities is central to the operation of the immune system. Many levels of protection are involved in this process. Therefore, a critical role of your immune system is to determine what is foreign (what immunologists often call “nonself”) from what is normally present in your body (i.e., self). As a consequence, the cells and molecules that comprise the innate immune system are preoccupied with detecting the presence of particular molecular patterns that are typically associated with infectious agents. Innate immunity involves the release of cytokines, complement, and chemokines, as well as neutrophils and macrophages to destroy the invading pathogens. When this is not enough, an antigen-specific or adaptive immune response becomes initiated, and antibodies, B cells, and T cells enter the battle 17. The generation of a specific response to an antigen is referred to as active immunity. Active immunity plays a vital role in immune responses in the event of re-exposure and your utilization of vaccines.
When harmful microbes (tiny particles) enter and invade your body, your body produces white blood cells to fight the infection. The white blood cells identify the microbe, produce antibodies to fight it, and help other immune responses to occur. They also ‘remember’ the attack. This is how vaccinations work. Vaccines expose the immune system to a dead or weakened microbe or to proteins from a microbe, so that the body is able to recognize and respond very quickly to any future exposure to the same microbe.
In addition to the immune system fundamental roles of recognition and elimination of infectious agents, it is also very useful to be able to learn from encounters with pathogens and to maintain a reserve of cells that are able to respond swiftly to a new infection with a previously encountered microbe. Forewarned is forearmed, and in this situation it may be possible to deliver a decisive blow that ends a nascent infection before it has begun. Fortunately, your immune systems have also acquired this ability, which is what your adaptive acquired immune system excels in, and this property is termed immunological memory.
Although the immune system is quite complex, its function can be boiled down to four basic roles:
- Creating a barrier to prevent pathogens from entering your body. The barrier function of the immune system acts to prevent pathogens from entering your body from the external environment. Important initial barriers to infection are physical (e.g. the skin), enhanced by substances secreted by your body, such as saliva and tears, that contain molecules that can neutralize bacteria. Chemical barriers like the acid pH of the stomach; and biological barriers like the presence of commensal organisms on the skin and in the intestinal tract, secretions like immunoglobulin A (IgA) and antimicrobial proteins in saliva and tears, and the complement system. The internal mucosal tissues (e.g. lungs & airways, and the gut) are coated with mucus that is able to trap potential infectious agents. In the airways, mobile ciliate hairs work together to transport contaminants away from vulnerable areas. Tissues such as the skin, mucosal surfaces and airways also contain populations of immune cells that can respond to infectious agents that breach these physical defences.
- Identifying pathogens if they breech a barrier. Pathogens are recognized by cells of the innate immune system, such as macrophages, monocytes and dendritic cells. This is achieved through the presence of pattern recognition receptors (PRRs) that recognize general molecular structures that are broadly shared by groups of pathogens. These structures are termed pathogen-associated molecular patterns or PAMPs. When pattern recognition receptors (PRRs) recognize pathogen-associated molecular patterns (PAMPs), the first line of host defensive responses is activated (see Figure 3 below) 18. PRRs include Toll-like receptors (TLRs). More than 10 functional TLRs have been identified in humans, each one detecting distinct MAMPs from bacteria, viruses, fungi and parasites. The best described of these are TLR4 which recognizes the lipopolysaccharides from the cell wall of Gram-negative bacteria and TLR2 which recognizes the lipoteichoic acid from the cell wall of Gram-positive bacteria 18. Several Toll-like receptors (TLRs) are expressed on the cell surface of innate immune cells because the pathogens they recognize, mainly bacteria, are extracellular. Because viruses enter host cells, it is important that there are also intracellular TLRs. Indeed, intracellular TLRs that recognize viral DNA, viral double-stranded RNA and viral single-stranded RNA exist. Among these, TLR7 and TLR8 are found in macrophages, monocytes, dendritic cells and some other cell types and are likely to be important in innate recognition of the single-stranded RNA of coronaviruses 19.
- Eliminating pathogens by a diverse repertoire of cells and molecules that act in concert to neutralize the potential threat. Extracellular bacteria can be engulfed by phagocytic cells that include macrophages and dendritic cells. After digestion of internalized bacteria, peptide fragments, termed antigens, are presented on the surface of the phagocytic cells (via Major Histocompatibility Complex 2 [MHC-II]) to antigen-specific CD4+ helper T lymphocytes. The activated helper T lymphocytes (specifically the T helper 1 phenotype [TH1]) proliferate and produce cytokines including interleukin (IL)-2 and interferon (IFN)-γ. IFN-γ promotes antigen-specific antibody production by B lymphocytes. These antibodies coat the bacteria, neutralising them and making the process of phagocytosis more efficient. In parallel with phagocytosis, innate immune cell recognition of pathogens via PRRs triggers inflammatory signalling, activation of transcription factors like nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB), inflammasome assembly, and production of classic inflammatory cytokines like tumor necrosis factor (TNF), IL-1β and IL-12. Viral infection of some cell types promotes release of type 1 IFNs (IFN-α and IFN-β) and these induce antiviral resistance, in part through activation of natural killer cells (NK cells) 20. Furthermore, virally infected cells directly activate natural killer (NK) cells which act to kill the infected cell. In addition, PRR signalling induces maturation of dendritic cells which are responsible for viral antigen processing and presentation, so initiating acquired immunity. Upregulation of MHC I on virally infected cells including both respiratory epithelial cells and dendritic cells results in presentation of viral antigens to CD8+ cytotoxic T lymphocytes. This activates them to kill virally infected cells through the release of pore forming proteins like perforin. Presentation of viral antigens via MHC II and the cytokine milieu lead to the activation of CD4+ helper T lymphocytes with switching to the T helper 1 phenotype. These cells produce IL-2, which promotes cytotoxic T lymphocyte activity, and IFN-γ, which promotes differentiation of B lymphocytes to plasma cells which produce antiviral antibodies. These antibodies can bind to free viruses neutralising them. The processes involved in antiviral immunity are summarised in figure 1.
- Generating an immunological memory. Immunological memory refers to the ability of the immune system to quickly and specifically recognize an antigen that your body has previously encountered and initiate a corresponding immune response. There are two aspects of immunological memory. First, antibodies can persist in the circulation for many months to many years, providing protection against reinfection. Second, after the cessation of an active immune response, a small number of memory T (both CD4+ and CD8+) and B lymphocytes remain; they are in a resting state but if they encounter the same antigen that triggered their formation they are able to respond immediately and lead to rapid elimination of the source of the antigen. Memory cells have a long life (up to several decades). Immunological memory is the basis of vaccination.
- Aging can be associated with a loss of immune competence, a process called immunosenescence 21. One factor linked to immunosenescence is decreased output of immune cells from bone marrow, the site of origin of all immune cells. In addition, involution of the thymus with age decreases output of naive T lymphocytes, resulting in reduced capacity to respond to new antigens. Immunosenescence means that, compared with younger adults, older people have increased susceptibility to infections including respiratory tract infections and pneumonia and poorer responses to vaccination 22. The gut mucosa is the largest site of immune tissue in humans and senescence of the gut mucosal immune system has been demonstrated in mice models, with reductions in secretory immunoglobulin A (IgA) responses, impaired oral tolerance to new antigens and impaired mucosal dendritic cell function 23. Paradoxically, ageing is also linked to an increase in blood concentrations of many inflammatory mediators, a situation termed inflammation in ageing or inflammageing 24. This state is considered to contribute to an increased risk of chronic conditions of aging like cardiovascular disease, metabolic disease (diabetes, non-alcoholic fatty liver disease), neurodegeneration and some cancers 24 and may predispose to mounting an excessive inflammatory response when infected. Although inflammation is part of the innate immune response and innate and acquired immunity should work in a coordinated and integrated way, an excessive inflammatory response can lead to impairments in acquired immunity 24.
- Obesity can be associated with a loss of immune competence 25, with impairments of the activity of helper T lymphocytes (TH), cytotoxic T lymphocytes, B lymphocytes and natural killer cells 26 and reduced antibody and IFN-γ production 27. This means that, compared with healthy weight individuals, the obese have increased susceptibility to a range of bacterial, viral and fungal infections 28 and poorer responses to vaccination 29. The impact of obesity has been well explored in relation to influenza infection and vaccination against influenza. During the 2009 H1N1 influenza A virus pandemic, obese individuals showed delayed and weakened antiviral responses to infection and showed poorer recovery from disease compared with healthy weight individuals 29. Animal studies and case studies in humans show that obesity is associated with prolonged shedding of influenza virus, indicating an impairment in viral control and killing, and the emergence of virulent minor variants 29. Green and Beck 30 note that compared with healthy weight individuals, vaccinated obese individuals have twice the risk of influenza or influenza-like illness, indicating poorer protection from vaccination in the obese. Sheridan et al 31 investigated the responses of immune cells from the blood of healthy weight, overweight and obese individuals to the influenza vaccine in vitro (test tubes). Exposure of the blood immune cells to the vaccine increased the number of activated cytotoxic T lymphocytes, the number of granzyme expressing cytotoxic T lymphocytes and the number of IFN-γ producing cytotoxic T lymphocytes. However, the responses of cells from obese individuals were blunted by 40%, almost 60% and 65%, respectively. Cells from overweight individuals showed responses intermediate between those from healthy weight and obese individuals. Similar findings for the response of blood cells to the pandemic H1N1 influenza A virus were reported by Paich et al 32. Paradoxically, obesity is also linked to an increase in blood concentrations of many inflammatory mediators, a state of chronic low-grade inflammation 33. This state is considered to contribute to an increased risk of chronic conditions of ageing 33 and may predispose to mounting an excessive inflammatory response when infected.
Immune responses against pathogens are divided roughly into three types 34.
- Type 1 cell-mediated effector immunity provides an effective response against intracellular microbes, such as bacteria, protozoa, and some viruses, and it comprises the transcription factor T-bet (also known as TBX21) and interferon-γ (IFNγ) producing helper cells (ie, CD4+ TH1 cells and ILC1s), as well as T-bet plus eomesodermin (Eomes) plus cytotoxic lymphocytes, namely CD8+ T cells (cytotoxic T lymphocytes) and natural killer (NK) cells. In type 1 cell-mediated effector immunity, pathogen clearance is mediated through effector cells including group 1 innate lymphocytes (ILC1), natural killer (NK) cells, cytotoxic T lymphocytes, and T helper 1 (TH1) cells.
- Type 2 immunity is mainly devoted to protection against helminths (parasitic worms) and venoms and is composed of GATA3 transcription factor plus lymphocytes producing IL-4, IL-5, and IL-13, namely CD4+ TH2 cells, CD8+ TC2 cells, and ILC2s. Effector molecules such as IL-4, IL-5, IL-13, and IgE that work to expel these pathogens through the concerted action of epithelial cells, mast cells, eosinophils, and basophils.
- Type 3 immunity is devoted to protection against extracellular bacteria and fungi and is composed of RORγt and lymphocytes, producing IL-17 alone or in combination with IL-22 as signature cytokines (secreted by CD4+ TH17 cells, CD8+ TC17 cells, and ILC3s) to elicit neutrophil-dependent clearance.
Relevant terms and definitions for the immune system 17:
- Immunogen: Protein or carbohydrate that is recognized and sufficiently activates an immune response
- Antigen: A molecule that is recognized by a specific antibody or T-cell receptor (TCR)
- Antibodies also known as immunoglobulins (Ig) play an important role in the immune system mechanisms of defense against extracellular microorganisms such as bacteria. Antibodies (immunoglobulins) are produced by plasma cells, which as permanently differentiated B-cells that secrete antibodies. Antibodies (immunoglobulins) fight off extracellular pathogens, for instance, bacteria and can neutralize viruses when they are in the bloodstream and other body fluids. Normal individuals have 5 classes of immunoglobulins, which are IgM, IgG, IgA, IgD and IgE and immunoglobulin subclasses including IgG1, IgG2, IgG3, IgG4, IgA1, and IgA2 35. While they have overlapping roles, IgM generally is important for complement activation; IgD is involved in activating basophils; IgG is important for neutralization, opsonization, and complement activation; IgA is essential for neutralization in the gastrointestinal tract; and IgE is necessary for activating mast cells in parasitic and allergic responses. Antibodies are expressed in two ways. The B-cell receptor (BCR), which sits on the surface of a B cell, is actually an antibody. B cells also secrete antibodies to diffuse and bind to pathogens. This dual expression is important because the initial problem, for instance a bacterium, is recognized by a unique B-cell receptor (BCR) and activates the B cell. The activated B cell responds by secreting antibodies, essentially the BCR but in soluble form. This ensures that the response is specific against the bacterium that started the whole process. Antibody deficiencies may occur due to lack of B-cells maturation, missing enzymes or failure of T-cell stimulatory signals for appropriate antibody production. In transient hypogammaglobulinemia of infancy, recurrent bacterial infections occur in children until their immune system matures 36.
- Adjuvant: Prolongs the presence of antigen in tissue and enhances the immune response to an antigen; used in acquired or artificial immunization (vaccinations)
- Dendritic cells (Antigen-Presenting cells or APCs): Facilitate activation of an antigen-specific response by the innate immune system; present antigens via major histone complexes to activate CD8+ cytotoxic T cells and CD4+ helper T cells
- CD4+ helper T cells: Facilitate cell-to-cell interactions and cytokine release to activate and control immune and inflammatory responses. Initially, CD4+ helper T cells are naive and must be activated to begin immune functions. This activation occurs by interaction with pro-Antigen-Presenting cells (“professional” antigen-presenting cells), mainly dendritic cells in lymph nodules/follicles, which leads to an intracellular pathway that up-regulates more antigen-specific TCRs on the T cell and leads to effector functions. T cells can only recognize protein-based antigens. TCRs (T cell receptors) and their co-receptors, such as CD3 and CD4 found on these cells form a complex with the major histocompatibility complex 2 (MHC-II) receptor and the antigen in question. The CD4+ cells are then activated and produce cytokines to initiate immune responses from other white blood cells/other immune cells of cell-mediated immunity. They also activate the T cell-dependent branch of humoral immunity, in which CD4+ T cells recognize protein antigens (which normally would elicit a weak or absent B cell response) and activate B cells to produce immunoglobulin in response to the antigen 37. There are three different subtypes of CD4+ helper T cells, each with a unique function: TH1 CD4+ T cells, TH2 CD4+ T cells, and TH17 CD4+ T cells.
- CD8+ cytotoxic T cells also known as killer T cells, CD8+ T cells or cytotoxic lymphocytes (CTLs), are immune cells for cell-mediated immunity. CD8+ T cells are crucial for recognizing and removing virus-infected cells and cancer cells. CD8+ cytotoxic T cells have specialized compartments, or granules, containing cytotoxins that cause apoptosis, i.e., programmed cell death. Because of its potency, the release of granules is tightly regulated by the immune system. Initially, CD8+ T cells are naive and must be activated to begin immune functions. This activation occurs by interaction with pro-Antigen-Presenting cells (“professional” antigen-presenting cells), mainly dendritic cells in lymph nodules/follicles, and leads to an intracellular pathway that up-regulates more antigen-specific T-cell receptors (TCRs) on the T cell and leads to immune functions. T cells can only recognize protein-based antigens. T-cell receptors (TCRs) and their co-receptors, such as CD3 and CD8 found on these cells form a complex with the major histocompatibility complex 1 (MHC-I) receptor and the antigen in question. Once an activated CD8+ migrates into circulation looking for antigens presented by the major histocompatibility complex 1 (MHC-I) molecules present on all nucleated cells and finds its antigenic target (expressed on a virally infected cell or cell with intracellular bacteria, for example), it utilizes its killing function 38. The killing function of CD8+ T cells is mediated by 1 of 2 mechanisms. The first mechanism involves the use of the Fas/Fas Ligand (FasL). Activated CD8+ T cells express FasL which binds to Fas (CD95), a receptor found on many cell types, leading to the activation of caspases and subsequent apoptosis of the target cell (often a cell infected with intracellular bacteria, such as Listeria species or a virally infected cell). The second method that activated CD8+ T cells can use for their killing function involves the release of granzymes and perforin (two compounds that bypass cell walls and active caspases). Activated CD8+ T cells also secrete interferon gamma (IFN-γ) (a cytokine used in the activation of macrophages/other immune processes) 39.
- Major histocompatibility complex 1 (MHC-I) also known as human leukocyte antigen (HLA): Found on all nucleated cells, play a significant role in determining “self.” Responsible for presenting intracellular antigens to CD8 T cells
- Major histocompatibility complex 2 (MHC-II): Found on antigen-presenting cells that interact with CD4 T cells; responsible for presenting exogenous antigens
- TH0 or activated T helper cell: The initial role of activated CD4+ T cells, promotes cell immunity by activating dendritic cells and stimulating lymphocyte growth; releases cytokines IL-2, IL-4, and IFN-gamma; can develop into TH1, TH2, TH17, or other TH cells
- T helper lymphocyte 1 (TH1 CD4+ T cells): TH1 CD4+ cells are important in the activation of macrophages and fighting off intracellular infections especially bacteria and fungi. TH1 CD4+ cells secrete the cytokine IFN-gamma which activates macrophages, B cells also known as B lymphocytes (to produce immunoglobulin G or IgG) and increased surface expression of the MHC 2 markers on macrophages/antigen-presenting cells. TNF-a is also secreted by these cells to activate dendritic cell migration. The activated macrophages then produced IL-12, which increases differentiation/production of TH1 T cells, further amplifying the immune response. TH1 T cells also play roles in several autoimmune reactions and disease, notably in delayed-type hypersensitivity.
- T helper lymphocyte 2 (TH2 CD4+ T cells): The response that occurs in the absence of IL-12 and IFN-gamma; promotes systemic antibody driven response. TH2 CD4+ T cells are important in combating helminthic infections. TH2 CD4+ T cells produce IL-4, IL-5, IL-10 and IL-13 cytokines, which activate and expand mast cells and eosinophils to clear the parasitic infection 40. Macrophages are also activated by TH2 CD4+ T cells to begin clear of cellular debris and inflammation caused by large parasites. TH2 cells also appear to play a role in allergic diseases/allergies.
- T helper lymphocyte 17 (TH17 CD4+ T cells): TH17 CD4+ T cells are vital in mucosal immunity and are involved in combating extracellular bacteria and and fungal infections when IL-23 is released instead of IL-12. TH17 cells produce IL17A, IL17F, IL-22, TNF-alpha, and chemokines. These cytokines activate neutrophils and monocytes as well as driving increased inflammation. It is this pro-inflammatory function that appears to play a role in the development of autoimmune inflammatory disorders (such as inflammatory bowel disease and rheumatoid arthritis) when the response is pathogenic 41.
- T regulatory cells (T regs) also known as suppressor T cells. T regulatory cells as the name suggests, monitor and inhibit the activity of other T cells. T regulatory cells or suppressor T cells modulate immune responses and inhibit autoimmune processes such as autoreactive immune cells. T regulatory cells prevent adverse immune activation and maintain tolerance or the prevention of immune responses against the body’s own cells and antigens. T regulatory cells produce inhibitory IL-10 and IL-35 cytokines to control immune responses, as well as using CTLA-4 to inhibit B7 on antigen-presenting cells to decrease immune responses. Production of T regulatory cells is spurred by cytokine IL-2 (so much so that in autoimmune disease IL-2 is often absent) and TGF-beta. T regulatory cells express the T-cell receptors (TCRs) CD3, CD4 (they likely share a common lineage with CD4+ Helper T cells), CTLA-4 and CD25. T regulatory cells also express the biomarker FOXP3 (a transcription marker important in their development and immune functions). There has been a recent interest in T regulatory cells as a method to promote wound healing after surgery and to battle cancers 42.
- B cells also known as B lymphocytes. B cells have two major functions: they present antigens to T cells, and more importantly, B cells produce antibodies to neutralize infectious microbes. Antibodies coat the surface of a pathogen and serve three major roles: neutralization, opsonization, and complement activation. Neutralization occurs when the pathogen, because it is covered in antibodies, is unable to bind and infect host cells. In opsonization, an antibody-bound pathogen serves as a red flag to alert immune cells like neutrophils and macrophages, to engulf and digest the pathogen. Complement is a process for directly destroying, or lysing, bacteria.
- Plasma cells: Permanently differentiated B-cells that secrete antibodies
- Memory B cells: Long-lasting B-cells that are responsive to one particular antigen and become activated with re-exposure to the same antigen.
- Vaccination or immunization is a way to train your immune system against a specific pathogen. Vaccination achieves immune memory without an actual infection, so the body is prepared when the virus or bacterium enters. Saving time is important to prevent a pathogen from establishing itself and infecting more cells in the body. An effective vaccine will optimally activate both the innate and adaptive immune response. An immunogen is used to activate the adaptive immune response so that specific memory cells are generated. Because B-cell receptors (BCRs) and T-cell receptors (TCRs) are unique, some memory cells are simply better at eliminating the pathogen. The goal of vaccine design is to select immunogens that will generate the most effective and efficient memory response against a particular pathogen. Adjuvants, which are important for activating innate immunity, can be added to vaccines to optimize the immune response. Innate immunity recognizes broad patterns, and without innate responses, adaptive immunity cannot be optimally achieved.
Figure 1. Cytokine storm

Abbreviations: ARDS = acute respiratory distress syndrome; CRP = C-reactive protein; VEGF = vascular endothelial growth factor.
[Source 5 ]What are cytokines?
Cytokines are a large group of signaling proteins, peptides or glycoproteins that are secreted by specific cells of immune system 43. Cytokines are a category of signaling molecules that mediate and regulate immunity, inflammation and hematopoiesis. Cytokines are produced throughout your body by cells of diverse embryological origin. Cytokine is a general name, other names are defined based on their presumed function, cell of secretion, or target of action. For example, cytokines made by lymphocytes can also be referred to as lymphokines. Many of the lymphokines are also known as interleukins (ILs), since they are not only secreted by leukocytes but also able to affect the cellular responses of leukocytes. Those cytokines secreted by monocytes or macrophages are termed monokines. And chemokines are cytokines with chemotactic activities. The advent of molecular cloning technologies led to the discovery of cytokines and chemokines involved in cytokine storm (Table 1).
Fever, a clinical hallmark of cytokine storm, can be elicited by interleukin-1, interleukin-6, or TNF through distinct mechanisms. Interleukin-1 is encoded by two genes (IL1A and IL1B), both of which bind to the same interleukin-1 receptor, activating a cascade of intracellular signaling pathways, including nuclear factor κB (NF-κB). The interleukin-1–receptor antagonist anakinra is effective as a single agent and in combination with other agents for the treatment of some forms of cytokine storm 44.
Levels of interleukin-6, an important mediator of the acute inflammatory response and pathophysiological features of cytokine storm, are highly elevated across various underlying immunopathologic disorders 45 and in mouse models of cytokine storm 46. Both tocilizumab, a monoclonal antibody directed at the interleukin-6 receptor (interleukin-6R), and siltuximab, which neutralizes interleukin-6 directly, have been shown to be effective in a number of cytokine storm disorders, including hemophagocytic lymphohistiocytosis (HLH), idiopathic multicentric Castleman’s disease, and CAR T-cell–induced cytokine storm 47.
Interleukin-6 is one of the more complex cytokines, since it is produced by and acts on immune and nonimmune cells across multiple organ systems. It can signal through two main pathways, referred to as classic cis signaling and trans signaling 47. The membrane-bound interleukin-6 receptor does not possess intracellular signaling domains but signals instead through interaction with membrane-bound gp130. In cis signaling, soluble interleukin-6 binds to membrane-bound iinterleukin-6 receptor, forming an interleukin-6–interleukin-6 receptor complex that binds to gp130, which then initiates signaling through its intracellular domain.
Downstream signal transduction is mediated by JAKs (Janus kinases) and STAT3 (signal transducer and activator of transcription 3), as well as by Akt–mTOR (mammalian target of rapamycin) and MAPK–ERK (mitogen-activated protein kinase–extracellular signal-regulated kinase) pathways. Membrane-bound gp130 is ubiquitously expressed, whereas expression of membrane-bound interleukin-6 receptor is restricted largely to immune cells. Activation of cis signaling results in pleiotropic effects on the immune system, which can contribute to cytokine storm 47. In the presence of high circulating levels of interleukin-6, which can be present in cytokine storm, trans signaling occurs through the binding of interleukin-6 to the soluble form of interleukin-6R, forming a complex with a gp130 dimer on potentially all cell surfaces. The resultant interleukin-6–soluble interleukin-6 receptor–gp130–JAK-STAT3 signaling is then activated in cells that do not express the membrane-bound interleukin-6 receptor, such as endothelial cells. This results in systemic hyperinflammation involving secretion of monocyte chemoattractant protein 1 (MCP-1), interleukin-8, and additional interleukin-6, as well as increased vascular endothelial growth factor (VEGF) and reduced E-cadherin expression on endothelial cells, which contribute to vascular hyperpermeability, leakiness, hypotension, and pulmonary dysfunction 47.
TNF is a potent, multifunctional, proinflammatory cytokine that belongs to the TNF–TNF receptor superfamily. In addition to inducing fever, augmenting systemic inflammation, and activating antimicrobial responses such as interleukin-6, TNF can induce cellular apoptosis and regulate immunity. TNF and other cytokines in the TNF–TNF receptor superfamily are potent inducers of NF-κB, leading to the expression of multiple proinflammatory genes. In mouse models of toxic shock, TNF is the cytokine driver of superantigen-driven cytokine storm 48. The effectiveness of anti-TNF therapies in certain autoinflammatory-driven cytokine storm conditions points to their potential role in the treatment of cytokine storm, but the limitations and dangers of anti-TNF therapies in patients with sepsis indicate that more work is needed.
Interleukin-18 is a member of the large interleukin-1 family that has recently been associated with cytokine storm disorders 49. Interleukin-18 and interleukin-1β are activated from precursors by inflammasomes. The inflammasome is a multimolecular cytosolic sensor that detects pathogenic microorganisms and sterile stressors and activates caspase-1 during the process of pyroptosis, which, in turn, causes the inactive precursor forms of interleukin-1β and interleukin-18 to become the active forms 50. Macrophages and dendritic cells are the primary sources of bioactive interleukin-18, which has many proinflammatory effects. Most important, it synergizes with interleukin-12 or interleukin-15 to stimulate secretion of interferon-γ from T cells and NK cells, and thus promotes Th1-type inflammatory responses. The interleukin-18 receptor is constitutively expressed on NK cells and induced on activation in most T cells. Interleukin-1β and interleukin-18 are also potent inducers of interleukin-6 secretion from macrophages 51.
Patients with cytokine storm due to macrophage activation syndrome (MAS) have high levels of interleukin-18 in serum 52 and interleukin-18 is a biomarker of severity that correlates with hyperferritinemia, elevated aminotransferase levels, and disease flare 53. The proinflammatory effects of interleukin-18 are normally kept in check by the interleukin-18–binding protein (IL18BP), which prevents the binding of interleukin-18 to its receptor 54. The ratio of free interleukin-18 to bound interleukin-18–IL18BP complexes in serum is an important indicator of the severity of the macrophage activation syndrome 55. Tadekinig alfa is a recombinant interleukin-18–binding protein (IL18BP) currently under investigation as a treatment for hyperinflammation.
Table 1. Cytokines and chemokines involved in cytokine storm
| Mediator | Main Cell Source | Type and Function |
| Cytokines and growth factors | ||
| Interleukin-1 (IL-1) | Macrophages, epithelial cells; pyroptotic cells | Proinflammatory alarmin cytokine; pyrogenic function, macrophage and Th17 cell activation |
| Interleukin-2 (IL-2) | T cells | Effector T-cell and regulatory T-cell growth factor |
| Interleukin-6 (IL-6) | Macrophages, T cells, endothelial cells | Proinflammatory cytokine; pyrogenic function, increased antibody production, induction of acute-phase reactants |
| Interleukin-9 (IL-9) | Th9 cells | Protection from helminth infections, activation of mast cells, association with type I interferon in Covid-19 56 |
| Interleukin-10 (IL-10) | Regulatory T cells, Th9 cells | Antiinflammatory cytokine; inhibition of Th1 cells and cytokine release |
| Interleukin-12 (IL-12) | Dendritic cells, macrophages | Activation of the Th1 pathway; induction of interferon-γ from Th1 cells, cytotoxic T lymphocytes and NK cells; acting in synergy with interleukin-18 |
| Interleukin-17 (IL-17) | Th17 cells, NK cells, group 3 innate lymphoid cells | Promoting neutrophilic inflammation, protection from bacterial and fungal infections |
| Interleukin-18 (IL-18) | Monocytes, macrophages, dendritic cells | Proinflammatory alarmin cytokine; activation of Th1 pathway, acting in synergy with interleukin-12 |
| Interleukin-33 (IL-33) | Macrophages, dendritic cells, mast cells, epithelial cells | Proinflammatory alarmin cytokine; amplification of Th1 and Th2 cells, activation of NK cells, cytotoxic T lymphocytes and mast cells |
| Interferon-γ (INF-γ) | Th1 cells, cytotoxic T lymphocytes, group 1 innate lymphoid cells, and NK cells | Proinflammatory cytokine; activation of macrophages |
| Tumor necrosis factor (TNF) | Macrophages, T cells, NK cells, mast cells | Increasing vascular permeability; pyrogenic function |
| GM-CSF (granulocyte–macrophage colony-stimulating factor) | Th17 cells | Proinflammatory cytokine |
| VEGF (vascular endothelial growth factor) | Macrophages | Angiogenesis |
| Chemokines | ||
| Interleukin-8 (CXCL8) | Macrophages, epithelial cells | Recruitment of neutrophils |
| MIG (CXCL9) | Monocytes, endothelial cells, keratinocytes | Interferon-inducible chemokine; recruitment of Th1 cells, NK cells, plasmacytoid dendritic cells |
| IP-10 (CXCL10) | Monocytes, endothelial cells, keratinocytes | Interferon-inducible chemokine; recruitment of macrophages, Th1 cells, NK cells |
| MCP-1 (CCL2) | Macrophages, dendritic cells, cardiac myocytes | Recruitment of Th2 cells, monocytes, dendritic cells, basophils |
| MIP-1α (CCL3) | Monocytes, neutrophils, dendritic cells, NK cells, mast cells | Recruitment of macrophages, Th1 cells, NK cells, eosinophils, dendritic cells; pyrogenic function |
| MIP-1β (CCL4) | Macrophages, neutrophils, endothelium | Recruitment of macrophages, Th1 cells, NK cells, dendritic cells |
| BLC (CXCL13) | B cells, follicular dendritic cells | Recruitment of B cells, CD4 T cells, dendritic cells† |
| Plasma proteins | ||
| CRP (C-reactive protein) | Hepatocytes | Monomeric CRP increases interleukin-8 and MCP-1 secretion; interleukin-6 increases CRP expression |
| Complement | Hepatocytes, other cells | Complement activation contributes to tissue damage in cytokine storm; complement inhibition can reduce immunopathologic effects of cytokine storm |
| Ferritin | Ubiquitous | Primary site of iron storage in cells |
Footnote: † In idiopathic multicentric Castleman’s disease, the levels of CXCL13 are the most elevated of all the cytokines or chemokines.
Abbreviations*: BLC = B-lymphocyte chemoattractant; Covid-19 = coronavirus disease 2019; CRP = C-reactive protein; CTLs = cytotoxic T lymphocytes; CXCL = C-X-C motif chemokine ligand; GM-CSF = granulocyte–macrophage colony-stimulating factor; IP-10 = interferon-inducible protein 10; MCP-1 = monocyte chemoattractant protein 1; MIG = monokine induced by interferon-γ; MIP-1α and MIP-1β = macrophage inflammatory protein 1α and 1β, respectively; NK = natural killer; Th1, Th2, Th9, and Th17 cells types = 1, 2, 9, and 17 helper T cells, respectively; VEGF = vascular endothelial growth factor.
[Source 5 ]What are chemokines?
Chemokines are a family of cytokines that contribute to a variety of immune-cell functions, including leukocyte recruitment and trafficking. Cytokine proteins are classified as chemokines according to behavior and structural characteristics. Their name is derived from their ability to induce directed chemotaxis in nearby responsive cells; they are chemotactic cytokines. The major role of chemokines is to act as a chemoattractant to guide the migration of cells. Some chemokines are considered pro-inflammatory and can be induced during an immune response to recruit cells of the immune system to a site of infection, while others are considered homeostatic and are involved in controlling the migration of cells during normal processes of tissue maintenance or development. Cells that are attracted by chemokines follow a signal of increasing chemokine concentration towards the source of the chemokine. Some chemokines control cells of the immune system during processes of immune surveillance, such as directing lymphocytes to the lymph nodes so they can screen for invasion of pathogens by interacting with antigen-presenting cells residing in these tissues. These are known as homeostatic chemokines and are produced and secreted without any need to stimulate their source cells. Some chemokines have roles in development; they promote angiogenesis (the growth of new blood vessels), or guide cells to tissues that provide specific signals critical for cellular maturation. Other chemokines are inflammatory and are released from a wide variety of cells in response to bacterial infection, viruses and agents that cause physical damage such as silica or the urate crystals that occur in gout. Their release is often stimulated by pro-inflammatory cytokines such as interleukin 1. Inflammatory chemokines function mainly as chemoattractants for leukocytes, recruiting monocytes, neutrophils and other effector cells from the blood to sites of infection or tissue damage. Certain inflammatory chemokines activate cells to initiate an immune response or promote wound healing. They are released by many different cell types and serve to guide cells of both innate immune system and adaptive immune system. Dysregulated trafficking during inflammation may have a role in hyperinflammation.
In addition to being known for mediating chemotaxis, chemokines are all approximately 8-10 kilodaltons in mass and have four cysteine residues in conserved locations that are key to forming their 3-dimensional shape. Chemokines have been classified into four main subfamilies: CXC, CC, CX3C and C. All of these proteins exert their biological effects by interacting with G protein-linked transmembrane receptors called chemokine receptors, that are selectively found on the surfaces of their target cells 57.
Numerous regulatory cytokines such as interleukin-10 and natural cytokine antagonists such as IL1RA serve as buffers to limit systemic off-target effects. Interleukin-10 inhibits the production of TNF, interleukin-1, interleukin-6, and interleukin-12 and down-regulates antigen presentation. Furthermore, in mice lacking interleukin-10, infection leads to cytokine storm 58. Though interleukin-10 and IL1RA are often elevated in cytokine storm, this finding most likely reflects a secondary, albeit insufficient, counterregulatory response to the proinflammatory cytokines. Anakinra is a therapeutic agent that mimics the endogenous immunoregulatory effects of IL1RA.
Plasma proteins such as complement proteins and other inflammatory mediators can contribute to the pathogenesis of cytokine storm. These soluble proteins recognize pathogens, amplify cellular responses, and provide feedback on cytokine signaling. In fact, cytokines can enhance the production of complement proteins, which in turn can enhance or inhibit cytokine production. Thus, complement can be highly effective in eliminating microbes but can also cause collateral damage if excessive. Hypocomplementemia, resulting from increased consumption by immune complexes, can be observed in cytokine storm 59. Complement inhibitors are under evaluation for the treatment of cytokine storm disorders.
Cells involved in cytokine storm
The cells of the innate immune system are the first line of defense against pathogens. Neutrophils, monocytes, and macrophages recognize pathogens, produce cytokines, and engulf pathogens and cells by phagocytosis. There are many other innate immune cells, such as dendritic cells, gamma–delta T cells, and natural killer (NK) cells 60. Innate immune cells use pattern-recognition receptors, which are not specific for any particular antigen, to recognize and respond to a wide variety of microbial invaders by producing cytokines that activate cells of the adaptive immune system.
Innate cells that are most often implicated in the pathogenesis of cytokine storm include neutrophils, macrophages, and NK cells. Neutrophils can produce neutrophil extracellular traps, a network of fibers that contribute to thrombi formation and amplify cytokine production during cytokine storm. Macrophages, which are tissue-resident cells that are often derived from circulating monocytes, do not divide; they have diverse functions, from the removal of senescent cells by engulfment, to tissue repair and immunoregulation, to antigen presentation. In many forms of cytokine storm, macrophages become activated and secrete excessive amounts of cytokines, ultimately causing severe tissue damage that can lead to organ failure. Hemophagocytic macrophages are often observed in bone marrow biopsy specimens from patients with cytokine storm. Interferon-γ can induce hemophagocytosis by macrophages, and this may contribute to the cytopenias commonly observed in patients with cytokine storm 61. The cytolytic function of NK cells is diminished in some forms of cytokine storm, which can lead to prolonged antigenic stimulation and difficulty resolving inflammation 62. Excess interleukin-6 may mediate the impairment in NK-cell function by lowering perforin and granzyme production.
The adaptive immune system is composed of B cells and T cells. T cells differentiate into a number of subsets with distinct effector-cell functions potentially involved in cytokine storm (Figure 3). Type 1 helper T (Th1) cells and cytotoxic T lymphocytes (CTLs) are primarily responsible for the host defense against viral infections. Th1 cells regulate the recruitment of macrophages, whereas type 2 helper T (Th2) cells recruit eosinophils and basophils, type 9 helper T (Th9) cells recruit mast cells, and type 17 helper T (Th17) cells recruit neutrophils 63. An exaggerated Th1-type inflammatory response often occurs during cytokine storm. Th1 cells produce large quantities of interferon-γ, induce delayed hypersensitivity reactions, activate macrophages, and are essential for defense against intracellular pathogens 64. Iatrogenic causes of cytokine storm involving excessive T-cell activation, such as CAR T-cell and anti-CD28 antibody therapy, point to the ability of activated T cells to initiate cytokine storm. Impaired granule-mediated killing of infected cells or tumor cells by cytotoxic T lymphocytes is a key aspect of some forms of cytokine storm 65. Data from mouse models of hemophagocytic lymphohistiocytosis (HLH) and patients with cytokine storm indicate that the inability of cytotoxic T lymphocytes to kill efficiently leads to prolonged activation of T cells, triggering a cascade of inflammatory tissue damage 66. Th17 cells have a major role in host defense, particularly antifungal protection, and abnormal Th17-cell function can lead to autoimmunity 67. An experimental model of macrophage activation syndrome (a form of secondary hemophagocytic lymphohistiocytosis) provides evidence that Th17 cells can be drivers of a cytokine storm that is independent of interferon-γ 68.
B cells are not often associated with the pathogenesis of cytokine storm. However, the effectiveness of B-cell depletion in treating some cytokine storm disorders, such as human herpesvirus 8 (HHV-8)–associated multicentric Castleman’s disease, suggests that these cells are capable of initiating or propagating cytokine storm, particularly when virally infected.
Figure 2. Cells of the immune system
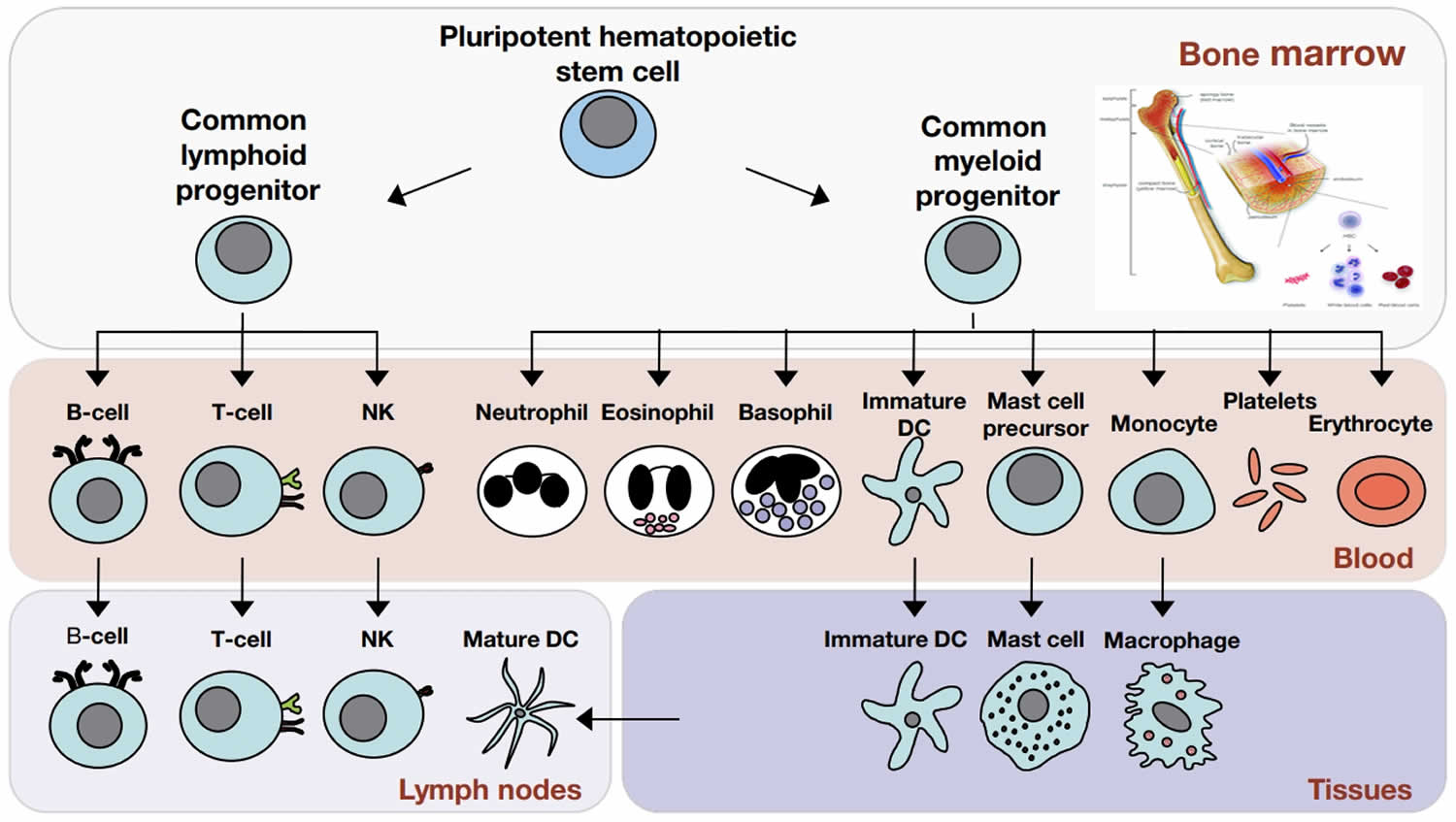
Footnote: The cells of the immune system originate in the bone marrow from pluripotent hematopoietic stem cells. Pluripotent hematopoietic stem cells give rise to a common lymphoid progenitor, which gives rise to all of the major lymphoid cell types (T‐cells, B‐cells, and Natural killer [NK] cells) or a common myeloid progenitor, which gives rise to all of the major myeloid cell types (neutrophils, eosinophils, basophils, dendritic cells (DCs), mast cells, and monocytes/macrophages) as well as the erythrocytes and megakaryocytes (which generate platelets).
- Granulocytes include basophils, eosinophils, and neutrophils. Basophils and eosinophils are important for host defense against parasites. They also are involved in allergic reactions.
- Neutrophils, the most numerous innate immune cell, patrol for problems by circulating in the bloodstream. Neutrophils can phagocytose, or ingest, bacteria, degrading them inside special compartments called vesicles.
- Mast cells also are important for defense against parasites. Mast cells are found in tissues and can mediate allergic reactions by releasing inflammatory chemicals like histamine.
- Monocytes, which develop into macrophages, also patrol and respond to problems. They are found in the bloodstream and in tissues. Macrophages, “big eater” in Greek, are named for their ability to ingest and degrade bacteria. Upon activation, monocytes and macrophages coordinate an immune response by notifying other immune cells of the problem. Macrophages also have important non-immune functions, such as recycling dead cells, like red blood cells, and clearing away cellular debris. These “housekeeping” functions occur without activation of an immune response.
- Dendritic cells (DC) are an important antigen-presenting cell (APC), and they also can develop from monocytes. Antigens are molecules from pathogens, host cells, and allergens that may be recognized by adaptive immune cells. APCs like DCs are responsible for processing large molecules into “readable” fragments (antigens) recognized by adaptive B or T cells. However, antigens alone cannot activate T cells. They must be presented with the appropriate major histocompatiblity complex (MHC) expressed on the APC. MHC provides a checkpoint and helps immune cells distinguish between host and foreign cells.
- Natural killer (NK) cells have features of both innate and adaptive immunity. They are important for recognizing and killing virus-infected cells or tumor cells. They contain intracellular compartments called granules, which are filled with proteins that can form holes in the target cell and also cause apoptosis, the process for programmed cell death. It is important to distinguish between apoptosis and other forms of cell death like necrosis. Apoptosis, unlike necrosis, does not release danger signals that can lead to greater immune activation and inflammation. Through apoptosis, immune cells can discreetly remove infected cells and limit bystander damage. Recently, researchers have shown in mouse models that NK cells, like adaptive cells, can be retained as memory cells and respond to subsequent infections by the same pathogen.
Figure 3. T-cells involved in cytokine storm
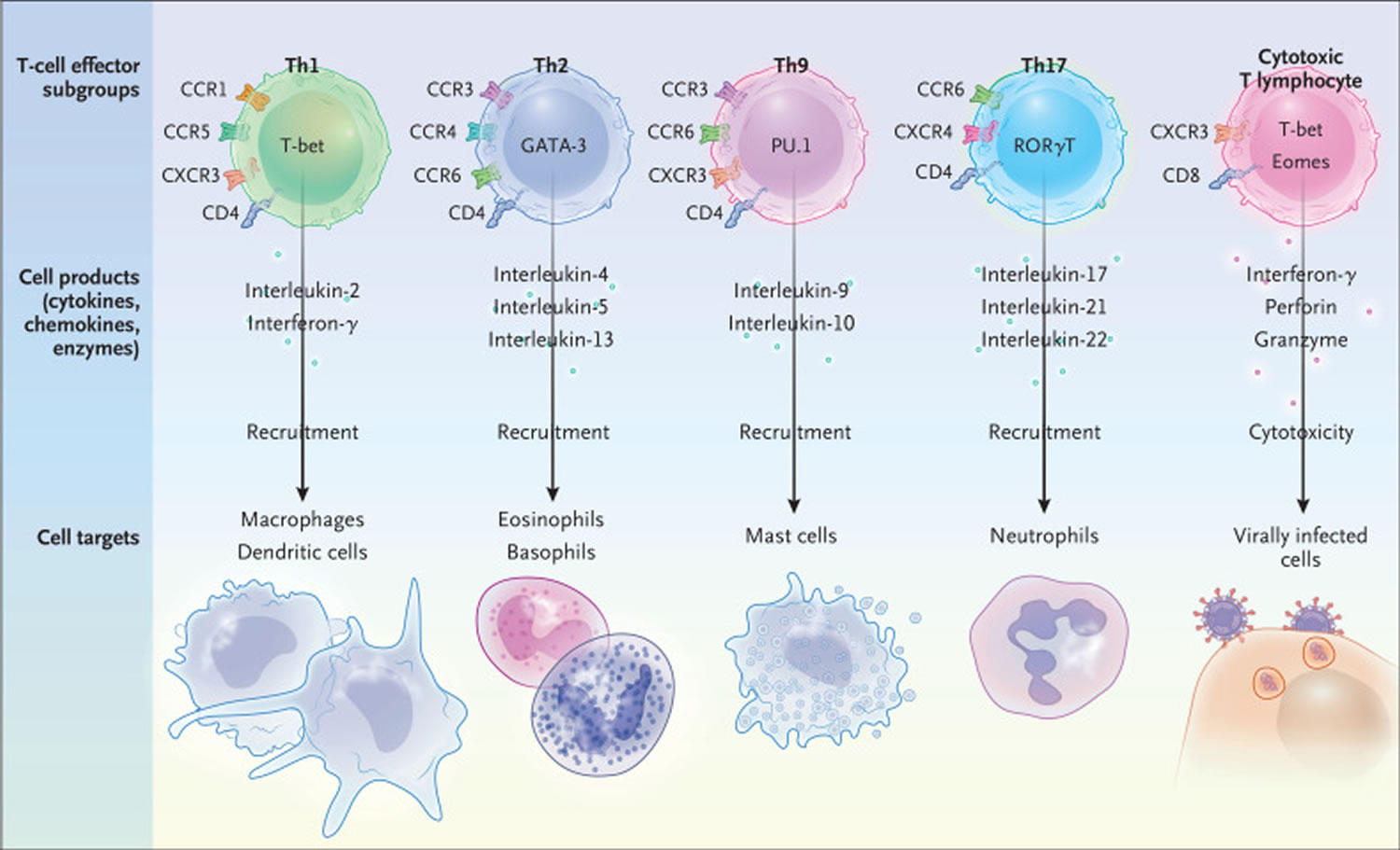
Footnote: The master transcription factors (T-bet, GATA-3, PU.1, RORγT, and eomesodermin [eomes]), effector molecules, and cell targets are shown for the following T-cell subgroups: types 1, 2, 9, and 17 helper T (Th1, Th2, Th9, and Th17, respectively) cells and cytotoxic T lymphocytes.
[Source 5 ]Causes of cytokine storm
A cytokine storm can occur as a result of an infection, autoimmune condition, or other disease. It may also occur after treatment with some types of immunotherapy. A complex, interconnected network of cell types, signaling pathways, and cytokines is involved in cytokine storm disorders 5. Interferon-γ, interleukin-1, interleukin-6, TNF, and interleukin-18 are key cytokines that often have elevated levels in cytokine storm and are thought to have central immunopathologic roles 5. The pattern of cytokine elevations varies on the basis of such factors as the microbiome, genetic features, and underlying disorders 69. The specific immune cells that secrete the various cytokines are not fully understood and most likely vary among cytokine storm disorders. Interferon-γ is primarily secreted by activated T cells and NK cells and is a potent activator of macrophages. Clinically, interferon-γ causes fever, chills, headache, dizziness, and fatigue 70. Emapalumab, a monoclonal antibody that binds interferon-γ, was recently approved for the treatment of cytokine storm in patients with primary hemophagocytic lymphohistiocytosis (HLH) 71. This agent may also be useful in other cytokine storm disorders, such as macrophage activation syndrome or CAR T-cell–associated cytokine storm, although in the latter case, it may diminish antitumor effects.
Table 2. Clinical causes of cytokine storm, pathologic drivers, and therapeutic approaches
| Type of Cytokine Storm and Trigger | Cause | Pathologic Cellular or Cytokine Driver | Common Therapeutic Approaches |
| Iatrogenic | |||
| CAR T-cell therapy | Infusion of CAR T cells | Macrophages, CAR T cells, interleukin-6, interleukin-1β | Anti–interleukin-6 antibody, glucocorticoids |
| Blinatumomab | Infusion of CD19- and CD3-specific T-cell receptor–engaging antibody | Activated T cells, macrophages, interleukin-6 | Anti–interleukin-6 antibody, glucocorticoids |
| Pathogen-induced | |||
| Bacterial sepsis | Hematogenous bacterial infection | Heterogeneous and multifactorial drivers | Intravenous antibiotics |
| EBV-associated hemophagocytic lymphohistiocytosis (HLH) | EBV infection in patient with genetic susceptibility | Interferon-γ, TNF, CD8+ T cells | B-cell–depleting therapy, glucocorticoids |
| HHV-8–associated multicentric Castleman’s disease | HHV-8 infection in patient with HIV coinfection, genetic susceptibility, or both | Viral interleukin-6, interleukin-6 | B-cell–depleting therapy |
| Covid-19 | SARS-CoV-2 infection, potentially in a susceptible person | Unknown driver | Glucocorticoids |
| Monogenic and autoimmune | |||
| Primary hemophagocytic lymphohistiocytosis (HLH) | Germline mutation in genes regulating granule-mediated cytotoxicity | CD8+ T cells, interferon-γ | T-cell inhibition or ablation, interferon-γ inhibitor, glucocorticoids |
| Secondary hemophagocytic lymphohistiocytosis (HLH) or macrophage activation syndrome | Viral cause (EBV or CMV), autoimmune disorder (rheumatoid arthritis or adult-onset Still’s disease), or neoplastic disorder in patient with genetic susceptibility (lymphoma) | CD8+ T cells, interferon-γ, interleukin-1β, myeloid-cell autoinflammation | Treatment of the underlying cause, in addition to T-cell inhibition or ablation, interleukin-1β inhibitor, JAK1 and JAK2 inhibitors, glucocorticoids |
| Autoinflammatory disorders | Germline mutations in genes regulating the innate immune system and inflammasome activation | Innate cells, TNF, interleukin-1β | Anti-TNF antibody, anti–interleukin-1 antibody |
| Idiopathic multicentric Castleman’s disease | Unknown cause | Interleukin-6, activated T cells, mTOR | Anti–interleukin-6 antibody, sirolimus, cyclosporine, cytotoxic chemotherapy, glucocorticoids |
Abbreviations: * CAR = chimeric antigen receptor; CMV = cytomegalovirus; Covid-19 = coronavirus disease 2019; EBV = Epstein–Barr virus; HHV-8 = human herpesvirus 8; HIV = human immunodeficiency virus; HLH = hemophagocytic lymphohistiocytosis; JAK1 = Janus kinase 1; JAK2 = Janus kinase 2; MAS = macrophage activation syndrome; MCD = multicentric Castleman’s disease; mTOR = mammalian target of rapamycin; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
[Source 5 ]Iatrogenic cytokine storm
Infusion of chimeric antigen receptor (CAR) T cells engineered to recognize and eliminate CD19+ lymphoma cells can induce cytokine storm, with supraphysiologic levels of interferon-γ and interleukin-6 72. The highly activated chimeric antigen receptor (CAR) T cells are clearly the initiators of the cytokine storm. Although some studies suggest that the driver cytokines are released by CAR T cells, resulting in a positive feedback loop of T-cell activation and inflammatory cytokine release 73, recent studies in mice suggest that the cytokines and factors mediating the severity of cytokine storm are produced not by the CAR T cells but by macrophages and can be reversed by interleukin-6 and interleukin-1 blockade 74. Tumor lysis most likely also contributes to the cytokine storm through the induction of pyroptosis in target cells 75. Since interleukin-6 blockade is highly effective at reversing symptoms and organ dysfunction in most patients, it is the likely cytokine driver of cytokine storm induced by CAR T-cell therapy. Glucocorticoids and interleukin-1 inhibition can also be effective in the treatment of this type of cytokine storm.
Cytokine storm can be observed with other T-cell–engaging immunotherapies as well, such as blinatumomab, a bispecific antibody that binds to CD19+ and CD3+ T cells 76. Like CAR T cells, activated T cells initiate the cytokine storm, and macrophage activation propagates blinatumomab-induced cytokine storm, which also responds to anti–interleukin-6 antibody therapy 45. The unfortunate consequences of another T-cell–activating treatment with the anti-CD28 superagonist TGN1412 show that rapid activation of large numbers of T cells can result in severe cytokine storm within minutes after infusion 77. However, cytokine storm does not develop in all patients treated with CAR T cells or blinatumomab, so additional factors, such as CAR structure and design 73, disease burden 78 and host genomic background 79, are likely to play a part. In a recent study of NK-cell CAR therapy, there were no reported cases of cytokine storm or even elevated interleukin-6 levels 80, possibly because of lower interleukin-6 production by NK cells than by T cells and different cross-talk with myeloid cells. Additional iatrogenic causes of cytokine storm include rituximab 45, gene therapies, immune checkpoint inhibitors, cardiac-bypass surgery 81 and allogeneic stem-cell transplantation, as well as bioterrorism agents such as staphylococcal enterotoxin B and Francisella tularensis.
Pathogen-induced cytokine storm
Cytokine storm can also result from naturally occurring microbial infections. Though data on relative frequencies are limited, infections are most likely the most common trigger of cytokine storm. Distinguishing between appropriate cytokine production for controlling a widespread infection and excessive cytokine production is challenging. Disseminated bacterial infections causing sepsis induce the production of many cytokines that can lead to fever, cell death, coagulopathies, and multiorgan dysfunction. The collateral damage caused by the immune response as it attempts to clear the pathogen can be more deadly than the pathogen itself. Certain bacteria, including streptococcus species and Staphylococcus aureus, can produce superantigens that cross-link the major histocompatibility complex and T-cell receptors, leading to polyclonal activation of T cells, cytokine production, and toxic shock syndrome. Superantigens are the most powerful T-cell mitogens, and bacterial superantigen concentrations of less than 0.1 pg per milliliter are sufficient to stimulate T cells in an uncontrolled manner, resulting in fever, shock, and death.
In sepsis-associated cytokine storm, it is unclear which immune cell types and cytokines may be responsible for propagating the pathologic hyperinflammation. Antibiotics are the mainstay of treatment. The administration of monoclonal antibodies directed at specific cytokines and the use of apheresis or medical devices to remove cytokines from circulation have had generally disappointing results in clinical trials 82. Although the timing of treatment in these studies may have contributed to the lack of benefit, additional host or pathogen factors may be important, beyond the specifically elevated cytokine levels. For example, reanalysis of a negative trial of interleukin-1β blockade in patients with sepsis identified a subgroup of patients with elevated ferritin levels who seemed to benefit from the treatment 83.
Disseminated viral infections can also induce profound cytokine storm. Patients with hyperinflammatory responses to microbes often have defects in pathogen detection, effector and regulatory mechanisms, or resolution of inflammation. For example, patients lacking functional perforin, which is critical for resolving infections and inflammation, have prolonged CD8+ T-cell production of interferon-γ and TNF, and HLH-associated cytokine storm develops in such patients when they are infected with EBV or cytomegalovirus 84. Experimental models suggest that cytokine storm occurs in these patients from defective perforin-mediated cytolysis that leads to prolonged engagement between lymphocytes and antigen-presenting cells and defective clearance of antigen-bearing dendritic cells, resulting in continuous activation and proliferation of T cells and macrophages, hemophagocytosis, and an autocrine loop of proinflammatory cytokines 85. Furthermore, retrospective analyses of data from persons who died from coagulopathies and hemophagocytosis during the H1N1 influenza pandemic of 2009 revealed germline mutations previously associated with hemophagocytic lymphohistiocytosis (HLH)-associated cytokine storm 69. Thus, the pathogen initiates and T-cell activation propagates cytokine storm in patients with a genetic susceptibility. Cyclosporine and anti–interleukin-6 receptor monoclonal antibody therapy can be effective in some virus-driven forms of hemophagocytic lymphohistiocytosis (HLH)-associated cytokine storm, indicating the critical role of T-cell activation and interleukin-6.
Another pathogen-induced form of cytokine storm is human herpesvirus 8 (HHV-8)–associated multicentric Castleman’s disease. In this disorder, uncontrolled infection with HHV-8 (also known as Kaposi’s sarcoma herpesvirus) leads to a cytokine storm driven primarily by excessive production of human interleukin-6 and viral interleukin-6 by human herpesvirus 8 (HHV-8)–infected plasmablasts 86. Patients with human herpesvirus 8 (HHV-8)–associated multicentric Castleman’s disease are immunocompromised as a result of human immunodeficiency virus infection or a genetic susceptibility, making it difficult to control the HHV-8 infection, which is a common, typically asymptomatic infection in the general population 87. A recent study showed that the effect of tocilizumab in patients with human herpesvirus 8 (HHV-8)–associated multicentric Castleman’s disease was minimal and short-lived, most likely because of viral interleukin-6 signaling that was independent of the neutralized interleukin-6 receptor 88. As with EBV-associated HLH,71 rituximab is highly effective in patients with HHV-8–associated multicentric Castleman’s disease, since B-cell depletion removes the primary reservoir for HHV-8 89. Many additional microbes can trigger cytokine storm, including other herpesviruses, such as herpes simplex virus, and other influenza viruses, such as H5N1.
Targeted treatment is more challenging in patients with viral infections than in patients with bacterial infections, since fewer antiviral agents are available. Intravenous immune globulin and convalescent plasma are sometimes used to help control the pathogen and provide beneficial immunomodulation. For some viral infections, treating patients with proinflammatory cytokines in the early stages of infection can help to control the virus before detrimental effects of the immune response occur 90.
Covid-19 associated cytokine storm
Covid-19 is a disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is characterized by heterogeneous symptoms ranging from mild fatigue to life-threatening pneumonia, cytokine storm, and multiorgan failure. Cytokine storm was also reported in patients with SARS and was associated with poor outcomes 91. Although the mechanisms of lung injury and multiorgan failure in Covid-19 are still under investigation,14 reports of hemophagocytosis and elevated cytokine levels — as well as beneficial effects of immunosuppressant agents — in affected patients, particularly those who are the most severely ill, suggest that cytokine storm may contribute to the pathogenesis of Covid-19 92.
Serum cytokine levels that are elevated in patients with Covid-19–associated cytokine storm include interleukin-1β, interleukin-6, IP-10, TNF, interferon-γ, macrophage inflammatory protein (MIP) 1α and 1β, and VEGF 93. Higher interleukin-6 levels are strongly associated with shorter survival 94. The relative frequencies of circulating activated CD4+ and CD8+ T cells and plasmablasts are increased in Covid-19 95. In addition to the elevated systemic cytokine levels and activated immune cells, several clinical and laboratory abnormalities, such as elevated CRP and d-dimer levels, hypoalbuminemia, renal dysfunction, and effusions, are also observed in Covid-19, as they are in cytokine storm disorders. Laboratory test results reflecting hyperinflammation and tissue damage were found to predict worsening outcomes in Covid-19 96.
Although immunologic dysregulation has been observed in severe cases of Covid-19 56, it is not known whether immune hyperactivity or a failure to resolve the inflammatory response because of ongoing viral replication or immune dysregulation underlies severe cases 5. The correlation between the nasopharyngeal viral load and cytokine levels (e.g., interferon-α, interferon-γ, and TNF), as well as a declining viral load in moderate but not severe cases, suggests that the immune response is positively associated with the viral burden 56. Alternatively, the discoveries of inborn errors of type I interferon immunity and autoantibodies against type I interferons in the most severe cases of Covid-19 suggest that an inadequate antiviral response may be contributory in some patients with Covid-19 97. Host immune responses and immune-related symptoms are extremely variable between asymptomatic patients (who have effective control of SARS-CoV-2) and patients with severe Covid-19 (who are unable to control the virus), which suggests that host immune dysregulation contributes to pathogenesis in some cases. Another hypothesized mechanism involves autoimmunity due to molecular mimicry between SARS-CoV-2 and a self-antigen. These mechanisms may be involved in subgroups of patients, such as children with postinfection multisystem inflammatory syndrome, a condition that seems to be ameliorated by immunomodulatory therapies such as intravenous immune globulin, glucocorticoids, and anti–interleukin-1 and anti–interleukin-6 therapies. Patients with multisystem inflammatory syndrome very clearly meet the definition of cytokine storm, since SARS-CoV-2 is no longer present; however, it is unclear whether the cytokine storm is a driver of Covid-19 or a secondary process. Furthermore, it is now clear that patients with SARS-CoV-2 infection can be asymptomatic or can have acute Covid-19 with heterogeneous severity, a chronic course of Covid-19, or multisystem inflammatory syndrome. A critical question concerns the factors that contribute to the severe cytokine storm–like phenotype observed in a small fraction of patients. Coexisting conditions such as hypertension, diabetes, and obesity are associated with more severe cases of Covid-19, possibly because of the preexisting chronic inflammatory state or a lower threshold for the development of organ dysfunction from the immune response.
Several important differences in therapeutic considerations should be noted between Covid-19–associated cytokine storm and many other cytokine storm disorders. First, cytokine storm triggered by infection with SARS-CoV-2 may require different therapies from those used for cytokine storm due to other causes 5. Cytokines may be both a key component of the cytokine storm and an essential factor in the antimicrobial response. Thus, blocking cytokine signaling may actually impair clearance of SARS-CoV-2, increase the risk of secondary infections, and lead to worse outcomes, as seen with influenza virus 98. Since interleukin-6 and other cytokines are potentially critical for both a healthy response to SARS-CoV-2 and a detrimental cytokine storm, it is particularly important that the right subgroups of patients with Covid-19 are selected for treatments at the right time. Despite positive anecdotal reports, two large, randomized, controlled trials of anti–interleukin-6 receptor antibody therapies did not show a survival benefit in hospitalized patients with Covid-19 99.
Second, the primary site of infection and disease most likely contributes to differences in immune responses and mechanisms underlying the cytokine storm, which have implications for treatment 5. For example, selective elimination of the primary viral reservoir is beneficial in patients with HHV-8–associated multicentric Castleman’s disease but is not possible in patients with Covid-19 5.
Third, lymphopenia is not often observed in cytokine storm disorders, but it is a hallmark of severe Covid-19 5. It is currently unclear whether the lymphopenia observed in Covid-19 is due to tissue infiltration or destruction of lymphocytes 5.
Fourth, clotting issues can occur across cytokine storm disorders, but thromboembolic events appear to be more frequent in Covid-19–associated cytokine storm 100. Finally, although cytokine panels have not been measured simultaneously on the same platform across Covid-19–associated cytokine storm and other cytokine storm disorders, preliminary results suggest that circulating levels of several cytokines, such as interleukin-6, as well as other inflammatory markers, such as ferritin, are less severely elevated in Covid-19 than in some of the other cytokine storm disorders 56. Levels of inflammatory mediators in pulmonary tissue during infection with SARS-CoV-2 remain unknown.
Despite the many unknowns, a recent randomized, controlled trial showing that dexamethasone reduces mortality among the most severe cases of Covid-19, characterized by elevated CRP levels and supplemental oxygen requirements, and potentially worsens outcomes in milder cases suggests that excessive, late-stage inflammation contributes to mortality 92. A meta-analysis of seven randomized trials showed that 28-day all-cause mortality in critically ill patients with Covid-19 was lower among those who were treated with glucocorticoids than among those who received usual care or placebo 101. An observational study suggesting that patients with Covid-19 have a good response to glucocorticoids when the CRP level is high but a poor response when the level is low is consistent with these findings 102. Further support comes from positive anecdotal reports of targeted antagonists against interleukin-1, granulocyte–macrophage colony-stimulating factor, and JAK1 and JAK2 in patients with Covid-19 103. Likewise, the observation that proinflammatory agents such as inhaled interferon-β have a positive effect if given early in the disease course is consistent with a model in which immunostimulation that enhances antiviral activity is helpful early (and probably harmful late), whereas immunosuppression is helpful late and harmful early. As with dexamethasone, the timing of treatment and selection of subgroups of patients included in studies will most likely have an effect on outcomes.
Despite unknowns regarding the role of immune dysregulation and cytokine storm in Covid-19, hundreds of immunomodulatory drugs are currently under investigation 104. Many of these treatments have been used for other cytokine storm disorders. Canakinumab, an anti–interleukin-1β monoclonal antibody, and anakinra are both being studied for Covid-19–induced ARDS. Acalabrutinib, a selective inhibitor of Bruton tyrosine kinase that regulates B-cell and macrophage signaling and activation, may have promise for dampening the hyperinflammatory response in Covid-19 105. JAK1 and JAK2 inhibitors, which are approved for the treatment of a number of autoimmune and neoplastic conditions, have the potential to inhibit signaling downstream of type I interferon, interleukin-6 (and other gp130 family receptors), interferon-γ, and interleukin-2, among other cytokines 106. Much like anti–interleukin-6 antibody therapy, inhibition of Bruton tyrosine kinase and JAK could prove to be damaging or unhelpful if given too soon, when the immune response to SARS-CoV-2 is critical in controlling viral replication and clearance.
Autoimmune cytokine storm
In rare cases, a pathogen triggers cytokine storm in patients with monogenic disorders, and in other cases, cytokine storm has autoimmune, neoplastic, or idiopathic causes. In patients with primary hemophagocytic lymphohistiocytosis (HLH), various autosomal recessive monogenic abnormalities in granule-mediated cytotoxicity lead to cytokine storm. Common pathologic mutations include those occurring in PRF1, UNC13D, STXBP1, RAB27A, STX11, SH2D1A, XIAP, and NLRC4 66. In patients with secondary hemophagocytic lymphohistiocytosis (HLH), viral, autoimmune, or neoplastic disorders trigger cytokine storm, and such patients often have heterozygous polymorphisms in the same genes that are altered in primary hemophagocytic lymphohistiocytosis (HLH) 85. Elevated levels of interferon-γ, TNF, interleukin-1, interleukin-4, interleukin-6, interleukin-8, interleukin-10, CXCL9, CXCL10, and interleukin-18 are frequently associated with hemophagocytic lymphohistiocytosis (HLH). Anti–interferon-γ antibody therapy with emapalumab has recently been approved for the treatment of primary HLH, as a bridge to allogeneic stem-cell transplantation, which is typically curative.
The beneficial effects of glucocorticoids, cyclosporine, anti–interleukin-1 antibody, JAK1 and JAK2 inhibitors, anti–interleukin-6 antibody, and cytotoxic chemotherapies in some patients with primary or secondary hemophagocytic lymphohistiocytosis (HLH) suggest that pathways targeted by these agents are key to pathogenesis. Cyclophosphamide and etoposide, which are broadly cytotoxic but particularly effective at eliminating activated CD8+ T cells, are often effective in patients with primary HLH, secondary HLH (including macrophage activation syndrome), and corresponding models 107. Etoposide also targets macrophages, including those involved in regulating inflammation, which could be harmful. Generalized T-cell and B-cell ablation with alemtuzumab and T-cell ablation with antithymocyte globulin have been reported; ablation most likely works by depleting the pathogenic CD8+ T cells, among other cell types 108. Nonablative inhibition of T cells with cyclosporine can also be helpful 109.
Autoinflammatory diseases are characterized by seemingly unprovoked inflammation and cytokine storm without signs of infection or autoimmunity. Affected patients have germline mutations in genes regulating the innate immune system and activation of the inflammasome. Several genetic disorders are associated with altered regulation of the innate immune system, including familial Mediterranean fever (MEFV), TNF receptor–associated periodic syndrome (TNFRSF1A), hyperimmunoglobulinemia D with periodic fever syndrome (MVK), familial cold autoinflammatory syndrome (NLRP3), the Muckle–Wells syndrome (NLRP3), neonatal-onset multisystem inflammatory disease (NLRP3), deficiency of ADA2 (CECR1), NLRC4 inflammasomopathies, X-linked lymphoproliferative type 2 disorder (XIAP), the Takenouchi–Kosaki syndrome (CDC42), and the Wiskott–Aldrich syndrome (CDC42). Although all patients with these disorders have periodic fevers, only a portion have cytokine storm. Given the primary genetic defects and the effective treatments that are available, innate cells are most likely the primary cell drivers involved, and TNF, interleukin-1, interleukin-18, or a combination of these cytokines probably drives pathogenesis. Patients with genetic immunodeficiency syndromes such as chronic granulomatous disease and STAT1 gain-of-function disease can, paradoxically, present with cytokine storm from overwhelming infections 110.
Idiopathic multicentric Castleman’s disease is another cytokine storm disorder that is similar to human herpesvirus 8 (HHV-8)–associated multicentric Castleman’s disease, but the cause is unknown. Patients with the thrombocytopenia, anasarca, fever, reticulin fibrosis, and organomegaly (TAFRO) subtype tend to have the most severe cytokine storm 111. Although the cause is unknown, interleukin-6 is the driver of pathogenesis in a large portion of patients. As a result, tocilizumab, which targets the interleukin-6 receptor, and siltuximab, which targets interleukin-6 directly, were developed and approved by regulatory agencies in Japan (tocilizumab) and in the United States and dozens of other countries (siltuximab) for the treatment of idiopathic multicentric Castleman’s disease. Both siltuximab and tocilizumab have been shown to resolve disease flares and sustain remission in approximately one third to one half of patients 112. However, some patients with low circulating interleukin-6 levels have a response to interleukin-6 blockade, and some patients with high systemic interleukin-6 levels do not have a response. A seven-protein panel that can predict which patients with idiopathic multicentric Castleman’s disease are most likely to benefit from siltuximab was recently identified and validated 113.
Patients with idiopathic multicentric Castleman’s disease who have progressive organ dysfunction and who do not have a response to anti–interleukin-6 therapy are often treated with combination cytotoxic chemotherapy to nonspecifically eliminate hyperinflammatory cells 114. Other elevated serum cytokines and cellular signaling pathways that could be considered for therapeutic targeting include CXCL13, CXCL10 (interferon-inducible protein 10 [IP-10]), VEGF-A 115, type I interferon 116, mTOR complex 1 (mTORC1) 117 and JAK-STAT3. These findings have led to treatment with the mTORC1 inhibitor sirolimus in patients with idiopathic multicentric Castleman’s disease who do not have a response to anti–interleukin-6 therapy 118. Sirolimus therapy is being evaluated in an ongoing clinical trial involving patients with active disease who do not yet have fulminant cytokine storm 119.
Cytokine storm symptoms
Cytokine storm or cytokine storm syndrome is a critical life-threating condition requiring intensive care admission and can present with a variety of symptoms ranging from mild, flu-like symptoms to severe life-threatening manifestations of the overshooting inflammatory response (see Figure 1 above). Cytokine storm signs and symptoms include high fever, inflammation (redness and swelling), fast heartbeat (tachycardia), fast breathing (tachypnea), low blood pressure (hypotension), malaise, generalized swelling, altered mental status, diffuse enlarged lymph nodes (lymphoadenopathy), organomegaly (particularly of the liver and spleen), and often reddish (erythematous) or purpuric rash and severe fatigue and nausea. Sometimes, a cytokine storm may be severe or life threatening and lead to multiple organ failure and death.
Mild symptoms of cytokine storm syndrome include fever, fatigue, headache, rash, joint pain (arthralgia) and muscle ache (myalgia) 120. More severe cases are characterized by hypotension as well as high fever and can progress to an uncontrolled Systemic Inflammatory Response Syndrome (SIRS) with vasopressor-requiring circulatory shock, vascular leakage, disseminated intravascular coagulation, and multi-organ system failure 120.
Respiratory symptoms are common in patients with cytokine storm syndrome. Mild cases may display cough and tachypnea but can progress to acute respiratory distress syndrome (ARDS) with dyspnea, hypoxemia, and bilateral opacities on chest X-ray. ARDS (acute respiratory distress syndrome) may sometimes require mechanical ventilation. Of note, in patients with cytokine storm syndrome the need for mechanical ventilation is oftentimes not due to respiratory distress but instead a consequence of the inability to protect the airway secondary to neurotoxicity 121. Patients with severe cytokine storm syndrome can also develop renal failure or signs of cardiac dysfunction with reduced ejection fraction on ultrasound 120. In addition, patients with severe cytokine storm syndrome frequently display vascular leakage with peripheral and pulmonary edema 120.
In severe cases cytokine storm syndrome can be accompanied by clinical signs and laboratory abnormalities that resemble hemophagocytic lymphohistiocytosis (HLH) or macrophage activation syndrome (MAS). Patients with cytokine storm syndrome-associated HLH display the typical clinical and laboratory findings of HLH/MAS such as high fevers, highly elevated ferritin levels, and hypertriglyeridemia. In a phase III study of blinatumomab in B-ALL four out of 13 cytokine storm syndrome patients showed signs of HLH 122.
Some patients develop neurotoxicity after administration of T cell-engaging therapies. Neurologic symptoms might span from mild confusion with word-finding difficulty, headaches and hallucinations to aphasia, hemiparesis, cranial nerve palsies, seizures and somnolence. In the case of CAR T cell therapy, neurotoxicity represents the second most common serious adverse event and therefore the term “CAR T cell-related encephalopathy syndrome” (CRES) has been introduced 123. The neurotoxicity of CAR T cell therapy does not seem to be directly related to cytokine storm syndrome since neurologic symptoms do not always coincide with cytokine storm syndrome onset and neurotoxicity can occur prior to cytokine storm syndrome or after cytokine storm syndrome has resolved 124. The pathophysiology of the neurologic symptoms is poorly understood, but the lack of a strict temporal association with cytokine storm syndrome indicates that it might, at least in part, be independent from cytokine storm syndrome. In addition, experience from clinical trials suggests that treatment of the neurologic symptoms is different from that of cytokine storm syndrome.
Cytokine storm diagnosis
The workup of any cytokine storm syndrome must begin with an assessment of hemodynamic stability and appropriate intensive care intervention. Next, an assessment of organ dysfunction and coagulopathy is critical, but rarely provides specific diagnostic information. Criteria for Systemic Inflammatory Response Syndrome (SIRS) were proposed in 1992 125 and have been amended a number of times to accommodate pediatric practice (Table 3) 126.
Systemic Inflammatory Response Syndrome (SIRS) in the absence of infection was defined as the presence of ≥2 of the following 127:
- Heart rate >90 beats per minute,
- Body temperature >100.4 °F (38°C) or <96.8 °F (36°C),
- White blood cells >12,000/mm or <4000/mm or >10% bands for >24 hour
- Respiratory rate >20.16 breaths per minute
Table 3. Pediatric Systemic Inflammatory Response Syndrome (SIRS) Criteria
| Presence of at least 2 of the following 4 criteria Must include abnormal temperature or leukocyte count | |
| Core Temperature | >101.3 °F (38.5°C) or <96.8 °F (36°C) |
| Abnormal Heart Rate | >2SD above normal for age#, OR |
| Unexplained persistent elevation over 0.5-to 4-hour time period, OR | |
| For children <1 year old: heart rate <10th percentile for age#, OR | |
| Unexplained persistent heart rate depression over a 0.5-hour period | |
| Respiratory Rate | >2SD above normal for age, OR |
| Acute requirement for mechanical ventilation | |
| Leukocyte Count | Elevated or depressed^ for age, OR |
| >10% immature neutrophils | |
Footnotes:
# not explained by external stimuli or drugs
^ not due to chemotherapy-induced leukopenia
[Source 126 ]Cytokine storm syndrome also have a number of common laboratory abnormalities. Blood parameters like leukocytosis or thrombocytosis can indicate the acute phase response. Alternatively, elevated cell counts can drop precipitously as a feature of nearly all cytokine storm syndrome, suggesting consumption. Clinicians can also take advantage of a host of non-specific acute phase reactants, including erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), procalcitonin, serum amyloid A, ferritin, and fibrinogen among others. Notably, and akin to acute cytopenias, an acute drop in ESR and fibrinogen are most associated with Macrophage Activation Syndrome (MAS), but can be seen in any cytokine storm syndrome and often suggest active disseminated intravascular coagulopathy (DIC) 128. Screens for coagulopathy such as fibrin split products and D-dimer are often elevated in cytokine storm syndrome even in the absence of overt DIC, suggesting subclinical endothelial activation. Likewise, hypoalbuminemia is frequently observed and likely represents systemic capillary leak. Routine testing often reflects various organs in distress including the liver, pancreas, and kidneys. Such tests are rarely capable of distinguishing direct inflammatory damage from that induced by insufficient oxygen delivery.
Researchers are working hard to understand what cytokine storm means in the context of COVID-19. Some clinicians have suggested screening patients with the disease for laboratory signs of inflammation that might indicate a cytokine storm, like elevated ferritin levels 129.
Cytokine storm treatment
The general treatment strategy for cytokine storm syndrome involves supportive care to maintain critical organ function, intensive care of hemodynamic instability, control of the underlying disease and elimination of triggers for abnormal immune system activation, and targeted immunomodulation or nonspecific immunosuppression to limit the collateral damage of the activated immune system and correction of coagulopathy 5. A number of drugs are effective across multiple disorders under the cytokine storm umbrella and still more may be effective in multiple conditions that have not yet been studied. Given the growing number of new therapeutics targeting various aspects of the immune system, further research should focus on the identification of drugs that can be used across cytokine storm disorders and precision diagnostics for selecting the right drugs for the right patients, regardless of the underlying condition 130. A study involving patients with systemic juvenile idiopathic arthritis revealed subgroups of patients with cytokine profiles in which interleukin-6 and interleukin-18 predominated, pointing toward available therapeutic approaches 131. Likewise, biomarkers were recently shown to effectively predict which patients with adult-onset Still’s disease would have a response to anakinra or tocilizumab 132. The progress made in precision oncology suggests that similar efforts across cytokine storm disorders are warranted to identify specific therapeutic targets and signatures of response to certain drugs that cross disease boundaries. JAK signaling is an interesting target in cytokine storm, because multiple cytokine–receptor pairs can be targeted simultaneously, an approach that may be effective for multiple diseases driven by different cytokines. In addition, plasma exchange and plasma filtration columns for the adsorption of cytokines are both under evaluation for cytokine storm disorders.
It is important to consider several factors in managing cytokine storm. Neutralization of a particular cytokine whose level is elevated in the circulation with an existing agent (anti–interleukin-6, anti-TNF, anti–interferon-γ, or anti–interleukin-1β antibody) will not always be effective, and blocking a cytokine with a low or normal circulating level can be effective if it is a key component of the hyperinflammatory circuit or if its level is potentially elevated in tissue. In addition, the various therapies mentioned in this review have distinctive side-effect and risk profiles. All targeted agents have target-specific risks, and combination therapy has more potential risks than single-agent therapy. Furthermore, pathologic hyperinflammation itself is an immunodeficiency that can put patients at risk for infections, and immunosuppressive agents most likely increase the risk further. In this age of cytokine profiling and individualized medicine, patients must be monitored and given appropriate prophylaxis when treated empirically, and randomized, controlled trials should always be performed to assess efficacy and safety.
References- Catanzaro M., Fagiani F., Racchi M., Corsini E., Govoni S., Lanni C. (2020). Immune response in COVID-19: addressing a pharmacological challenge by targeting pathways triggered by SARS-CoV-2. Signal Transduct. Target Ther. 5, 84. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7255975
- The COVID-19 Cytokine Storm; What We Know So Far. Front. Immunol., 16 June 2020 https://doi.org/10.3389/fimmu.2020.01446
- Cytokine Storm in COVID-19—Immunopathological Mechanisms, Clinical Considerations, and Therapeutic Approaches: The REPROGRAM Consortium Position Paper. Front. Immunol., 10 July 2020 https://www.frontiersin.org/article/10.3389/fimmu.2020.01648
- Sinha P, Matthay MA, Calfee CS. Is a “Cytokine Storm” Relevant to COVID-19? JAMA Intern Med. 2020 Sep 1;180(9):1152-1154. doi: 10.1001/jamainternmed.2020.3313
- Fajgenbaum, D. C., & June, C. H. (2020). Cytokine Storm. The New England journal of medicine, 383(23), 2255–2273. https://doi.org/10.1056/NEJMra2026131
- Ferrara JL, Abhyankar S, Gilliland DG. Cytokine storm of graft-versus-host disease: a critical effector role for interleukin-1. Transplant Proc. 1993 Feb;25(1 Pt 2):1216-7.
- Chatenoud L, Ferran C, Bach JF. The anti-CD3-induced syndrome: a consequence of massive in vivo cell activation. Curr Top Microbiol Immunol. 1991;174:121-34. doi: 10.1007/978-3-642-50998-8_9
- Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. 1893. Clin Orthop Relat Res. 1991 Jan;(262):3-11.
- Pechous, R. D., Sivaraman, V., Price, P. A., Stasulli, N. M., & Goldman, W. E. (2013). Early host cell targets of Yersinia pestis during primary pneumonic plague. PLoS pathogens, 9(10), e1003679. https://doi.org/10.1371/journal.ppat.1003679
- Kash, J. C., Tumpey, T. M., Proll, S. C., Carter, V., Perwitasari, O., Thomas, M. J., Basler, C. F., Palese, P., Taubenberger, J. K., García-Sastre, A., Swayne, D. E., & Katze, M. G. (2006). Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature, 443(7111), 578–581. https://doi.org/10.1038/nature05181
- Atkins MB. Interleukin-2: clinical applications. Semin Oncol. 2002 Jun;29(3 Suppl 7):12-7. doi: 10.1053/sonc.2002.33077
- Doherty GM, Lange JR, Langstein HN, Alexander HR, Buresh CM, Norton JA. Evidence for IFN-gamma as a mediator of the lethality of endotoxin and tumor necrosis factor-alpha. J Immunol. 1992 Sep 1;149(5):1666-70.
- Inflammatory cytokines in the BAL of patients with ARDS. Persistent elevation over time predicts poor outcome. Chest. 1995; 108: 1303-1314. https://doi.org/10.1378/chest.108.5.1303
- Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014 Apr 26;383(9927):1503-1516. doi: 10.1016/S0140-6736(13)61048-X. Epub 2013 Nov 27. Erratum in: Lancet. 2014 Apr 26;383(9927):1464.
- La Rosée P, Horne A, Hines M, von Bahr Greenwood T, Machowicz R, Berliner N, Birndt S, Gil-Herrera J, Girschikofsky M, Jordan MB, Kumar A, van Laar JAM, Lachmann G, Nichols KE, Ramanan AV, Wang Y, Wang Z, Janka G, Henter JI. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019 Jun 6;133(23):2465-2477. doi: 10.1182/blood.2018894618
- Mehta, P., Cron, R. Q., Hartwell, J., Manson, J. J., & Tattersall, R. S. (2020). Silencing the cytokine storm: the use of intravenous anakinra in haemophagocytic lymphohistiocytosis or macrophage activation syndrome. The Lancet. Rheumatology, 2(6), e358–e367. https://doi.org/10.1016/S2665-9913(20)30096-5
- Grubbs H, Kahwaji CI. Physiology, Active Immunity. [Updated 2020 Sep 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK513280
- Calder PC. Nutrition, immunity and COVID-19. BMJ Nutr Prev Health. 2020 May 20;3(1):74-92. doi: 10.1136/bmjnph-2020-000085
- Al-Qahtani AA, Lyroni K, Aznaourova M, et al. . Middle east respiratory syndrome corona virus spike glycoprotein suppresses macrophage responses via DPP4-mediated induction of IRAK-M and PPARγ. Oncotarget 2017;8:9053–66. 10.18632/oncotarget.14754
- García-Sastre A, Biron CA. Type 1 interferons and the virus-host relationship: a lesson in Detente. Science 2006;312:879–82. 10.1126/science.1125676
- Ventura MT, Casciaro M, Gangemi S, et al. . Immunosenescence in aging: between immune cells depletion and cytokines up-regulation. Clin Mol Allergy 2017;15:21 10.1186/s12948-017-0077-0
- Pera A, Campos C, López N, et al. . Immunosenescence: implications for response to infection and vaccination in older people. Maturitas 2015;82:50–5. 10.1016/j.maturitas.2015.05.004
- Ogra PL. Ageing and its possible impact on mucosal immune responses. Ageing Res Rev 2010;9:101–6. 10.1016/j.arr.2009.07.007
- Calder PC, Bosco N, Bourdet-Sicard R, et al. . Health relevance of the modification of low grade inflammation in ageing (inflammageing) and the role of nutrition. Ageing Res Rev 2017;40:95–119. 10.1016/j.arr.2017.09.001
- Andersen CJ, Murphy KE, Fernandez ML. Impact of obesity and metabolic syndrome on immunity. Adv Nutr 2016;7:66–75. 10.3945/an.115.010207
- O’Shea D, Hogan AE. Dysregulation of natural killer cells in obesity. Cancers 2019;11:E573 10.3390/cancers11040573
- Frasca D, Diaz A, Romero M, et al. . Ageing and obesity similarly impair antibody responses. Clin Exp Immunol 2017;187:64–70. 10.1111/cei.12824
- Frasca D, Blomberg BB. The Impact of Obesity and Metabolic Syndrome on Vaccination Success. Interdiscip Top Gerontol Geriatr. 2020;43:86-97. doi: 10.1159/000504440
- Honce R, Schultz-Cherry S. Impact of obesity on influenza A virus pathogenesis, immune response, and evolution. Front Immunol 2019;10:1071 10.3389/fimmu.2019.01071
- Green WD, Beck MA. Obesity impairs the adaptive immune response to influenza virus. Ann Am Thorac Soc 2017;14:S406–9. 10.1513/AnnalsATS.201706-447AW
- Sheridan PA, Paich HA, Handy J, et al. . Obesity is associated with impaired immune response to influenza vaccination in humans. Int J Obes 2012;36:1072–7. 10.1038/ijo.2011.208
- Paich HA, Sheridan PA, Handy J, et al. . Overweight and obese adult humans have a defective cellular immune response to pandemic H1N1 influenza A virus. Obesity 2013;21:2377–86. 10.1002/oby.20383
- Calder PC, Ahluwalia N, Brouns F, et al. . Dietary factors and low-grade inflammation in relation to overweight and obesity. Br J Nutr 2011;106:S5–78. 10.1017/S0007114511005460
- Annunziato, F., Romagnani, C. & Romagnani, S. The 3 major types of innate and adaptive cell-mediated effector immunity. J. Allergy Clin. Immunol. 135, 626–635 (2015). https://doi.org/10.1016/j.jaci.2014.11.001
- Justiz Vaillant AA, Jamal Z, Ramphul K. Immunoglobulin. [Updated 2021 Mar 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK513460
- Justiz Vaillant AA, Qurie A. Immunodeficiency. [Updated 2020 Dec 30]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK500027
- Bourne N, Perry CL, Banasik BN, Miller AL, White M, Pyles RB, Schäfer H, Milligan GN. Increased Frequency of Virus Shedding by Herpes Simplex Virus 2-Infected Guinea Pigs in the Absence of CD4+ T Lymphocytes. J Virol. 2019 Feb 5;93(4):e01721-18. doi: 10.1128/JVI.01721-18
- Lyu F, Ozawa T, Hamana H, Kobayashi E, Muraguchi A, Kishi H. A novel and simple method to produce large amounts of recombinant soluble peptide/major histocompatibility complex monomers for analysis of antigen-specific human T cell receptors. N Biotechnol. 2019 Mar 25;49:169-177. doi: 10.1016/j.nbt.2018.11.005
- Nakiboneka R, Mugaba S, Auma BO, Kintu C, Lindan C, Nanteza MB, Kaleebu P, Serwanga J. Interferon gamma (IFN-γ) negative CD4+ and CD8+ T-cells can produce immune mediators in response to viral antigens. Vaccine. 2019 Jan 3;37(1):113-122. doi: 10.1016/j.vaccine.2018.11.024
- Hirata H, Yukawa T, Tanaka A, Miyao T, Fukuda T, Fukushima Y, Kurasawa K, Arima M. Th2 cell differentiation from naive CD4+ T cells is enhanced by autocrine CC chemokines in atopic diseases. Clin Exp Allergy. 2019 Apr;49(4):474-483. doi: 10.1111/cea.13313
- Osborne LM, Brar A, Klein SL. The role of Th17 cells in the pathophysiology of pregnancy and perinatal mood and anxiety disorders. Brain Behav Immun. 2019 Feb;76:7-16. doi: 10.1016/j.bbi.2018.11.015
- Batista-Duharte A, Téllez-Martínez D, Roberto de Andrade C, Portuondo DL, Jellmayer JA, Polesi MC, Carlos IZ. Sporothrix brasiliensis induces a more severe disease associated with sustained Th17 and regulatory T cells responses than Sporothrix schenckii sensu stricto in mice. Fungal Biol. 2018 Dec;122(12):1163-1170. doi: 10.1016/j.funbio.2018.08.004
- What are Cytokines. https://www.sinobiological.com/resource/cytokines/what-are-cytokines
- Eloseily EM, Weiser P, Crayne CB, Haines H, Mannion ML, Stoll ML, Beukelman T, Atkinson TP, Cron RQ. Benefit of Anakinra in Treating Pediatric Secondary Hemophagocytic Lymphohistiocytosis. Arthritis Rheumatol. 2020 Feb;72(2):326-334. doi: 10.1002/art.41103
- Teachey, D. T., Rheingold, S. R., Maude, S. L., Zugmaier, G., Barrett, D. M., Seif, A. E., Nichols, K. E., Suppa, E. K., Kalos, M., Berg, R. A., Fitzgerald, J. C., Aplenc, R., Gore, L., & Grupp, S. A. (2013). Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood, 121(26), 5154–5157. https://doi.org/10.1182/blood-2013-02-485623
- van der Stegen SJ, Davies DM, Wilkie S, Foster J, Sosabowski JK, Burnet J, Whilding LM, Petrovic RM, Ghaem-Maghami S, Mather S, Jeannon JP, Parente-Pereira AC, Maher J. Preclinical in vivo modeling of cytokine release syndrome induced by ErbB-retargeted human T cells: identifying a window of therapeutic opportunity? J Immunol. 2013 Nov 1;191(9):4589-98. doi: 10.4049/jimmunol.1301523
- Kang S, Tanaka T, Narazaki M, Kishimoto T. Targeting Interleukin-6 Signaling in Clinic. Immunity. 2019 Apr 16;50(4):1007-1023. doi: 10.1016/j.immuni.2019.03.026
- Faulkner L, Cooper A, Fantino C, Altmann DM, Sriskandan S. The mechanism of superantigen-mediated toxic shock: not a simple Th1 cytokine storm. J Immunol. 2005 Nov 15;175(10):6870-7. doi: 10.4049/jimmunol.175.10.6870
- Garlanda, C., Dinarello, C. A., & Mantovani, A. (2013). The interleukin-1 family: back to the future. Immunity, 39(6), 1003–1018. https://doi.org/10.1016/j.immuni.2013.11.010
- Frank, D., & Vince, J. E. (2019). Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell death and differentiation, 26(1), 99–114. https://doi.org/10.1038/s41418-018-0212-6
- Netea MG, Kullberg BJ, Verschueren I, Van Der Meer JW. Interleukin-18 induces production of proinflammatory cytokines in mice: no intermediate role for the cytokines of the tumor necrosis factor family and interleukin-1beta. Eur J Immunol. 2000 Oct;30(10):3057-60. doi: 10.1002/1521-4141(200010)30:10<3057::AID-IMMU3057>3.0.CO;2-P
- Mazodier, K., Marin, V., Novick, D., Farnarier, C., Robitail, S., Schleinitz, N., Veit, V., Paul, P., Rubinstein, M., Dinarello, C. A., Harlé, J. R., & Kaplanski, G. (2005). Severe imbalance of IL-18/IL-18BP in patients with secondary hemophagocytic syndrome. Blood, 106(10), 3483–3489. https://doi.org/10.1182/blood-2005-05-1980
- Shimizu M, Yokoyama T, Yamada K, Kaneda H, Wada H, Wada T, Toma T, Ohta K, Kasahara Y, Yachie A. Distinct cytokine profiles of systemic-onset juvenile idiopathic arthritis-associated macrophage activation syndrome with particular emphasis on the role of interleukin-18 in its pathogenesis. Rheumatology (Oxford). 2010 Sep;49(9):1645-53. doi: 10.1093/rheumatology/keq133
- Dinarello, C. A., Novick, D., Kim, S., & Kaplanski, G. (2013). Interleukin-18 and IL-18 binding protein. Frontiers in immunology, 4, 289. https://doi.org/10.3389/fimmu.2013.00289
- Novick D, Kim S, Kaplanski G, Dinarello CA. Interleukin-18, more than a Th1 cytokine. Semin Immunol. 2013 Dec 15;25(6):439-48. doi: 10.1016/j.smim.2013.10.014
- Lucas, C., Wong, P., Klein, J., Castro, T., Silva, J., Sundaram, M., Ellingson, M. K., Mao, T., Oh, J. E., Israelow, B., Takahashi, T., Tokuyama, M., Lu, P., Venkataraman, A., Park, A., Mohanty, S., Wang, H., Wyllie, A. L., Vogels, C., Earnest, R., … Iwasaki, A. (2020). Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature, 584(7821), 463–469. https://doi.org/10.1038/s41586-020-2588-y
- Mélik-Parsadaniantz S, Rostène W. Chemokines and neuromodulation. J Neuroimmunol. 2008 Jul 31;198(1-2):62-8. doi: 10.1016/j.jneuroim.2008.04.022
- Behrens, E. M., Canna, S. W., Slade, K., Rao, S., Kreiger, P. A., Paessler, M., Kambayashi, T., & Koretzky, G. A. (2011). Repeated TLR9 stimulation results in macrophage activation syndrome-like disease in mice. The Journal of clinical investigation, 121(6), 2264–2277. https://doi.org/10.1172/JCI43157
- Gorelik, M., Torok, K. S., Kietz, D. A., & Hirsch, R. (2011). Hypocomplementemia associated with macrophage activation syndrome in systemic juvenile idiopathic arthritis and adult onset still’s disease: 3 cases. The Journal of rheumatology, 38(2), 396–397. https://doi.org/10.3899/jrheum.100833
- Hashimoto, D., Chow, A., Noizat, C., Teo, P., Beasley, M. B., Leboeuf, M., Becker, C. D., See, P., Price, J., Lucas, D., Greter, M., Mortha, A., Boyer, S. W., Forsberg, E. C., Tanaka, M., van Rooijen, N., García-Sastre, A., Stanley, E. R., Ginhoux, F., Frenette, P. S., … Merad, M. (2013). Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity, 38(4), 792–804. https://doi.org/10.1016/j.immuni.2013.04.004
- Zoller, E. E., Lykens, J. E., Terrell, C. E., Aliberti, J., Filipovich, A. H., Henson, P. M., & Jordan, M. B. (2011). Hemophagocytosis causes a consumptive anemia of inflammation. The Journal of experimental medicine, 208(6), 1203–1214. https://doi.org/10.1084/jem.20102538
- Perez N, Virelizier JL, Arenzana-Seisdedos F, Fischer A, Griscelli C. Impaired natural killer activity in lymphohistiocytosis syndrome. J Pediatr. 1984 Apr;104(4):569-73. doi: 10.1016/s0022-3476(84)80549-1
- Sallusto F. Heterogeneity of Human CD4(+) T Cells Against Microbes. Annu Rev Immunol. 2016 May 20;34:317-34. doi: 10.1146/annurev-immunol-032414-112056
- Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145-73. doi: 10.1146/annurev.iy.07.040189.001045
- Crayne, C. B., Albeituni, S., Nichols, K. E., & Cron, R. Q. (2019). The Immunology of Macrophage Activation Syndrome. Frontiers in immunology, 10, 119. https://doi.org/10.3389/fimmu.2019.00119
- Schulert GS, Cron RQ. The genetics of macrophage activation syndrome. Genes Immun. 2020 May;21(3):169-181. doi: 10.1038/s41435-020-0098-4
- Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485-517. doi: 10.1146/annurev.immunol.021908.132710
- Avau A, Mitera T, Put S, Put K, Brisse E, Filtjens J, Uyttenhove C, Van Snick J, Liston A, Leclercq G, Billiau AD, Wouters CH, Matthys P. Systemic juvenile idiopathic arthritis-like syndrome in mice following stimulation of the immune system with Freund’s complete adjuvant: regulation by interferon-γ. Arthritis Rheumatol. 2014 May;66(5):1340-51. doi: 10.1002/art.38359
- Schulert, G. S., Zhang, M., Fall, N., Husami, A., Kissell, D., Hanosh, A., Zhang, K., Davis, K., Jentzen, J. M., Napolitano, L., Siddiqui, J., Smith, L. B., Harms, P. W., Grom, A. A., & Cron, R. Q. (2016). Whole-Exome Sequencing Reveals Mutations in Genes Linked to Hemophagocytic Lymphohistiocytosis and Macrophage Activation Syndrome in Fatal Cases of H1N1 Influenza. The Journal of infectious diseases, 213(7), 1180–1188. https://doi.org/10.1093/infdis/jiv550
- Vadhan-Raj S, Nathan CF, Sherwin SA, Oettgen HF, Krown SE. Phase I trial of recombinant interferon gamma by 1-hour i.v. infusion. Cancer Treat Rep. 1986 May;70(5):609-14.
- Locatelli F, Jordan MB, Allen C, Cesaro S, Rizzari C, Rao A, Degar B, Garrington TP, Sevilla J, Putti MC, Fagioli F, Ahlmann M, Dapena Diaz JL, Henry M, De Benedetti F, Grom A, Lapeyre G, Jacqmin P, Ballabio M, de Min C. Emapalumab in Children with Primary Hemophagocytic Lymphohistiocytosis. N Engl J Med. 2020 May 7;382(19):1811-1822. doi: 10.1056/NEJMoa1911326
- Porter, D. L., Hwang, W. T., Frey, N. V., Lacey, S. F., Shaw, P. A., Loren, A. W., Bagg, A., Marcucci, K. T., Shen, A., Gonzalez, V., Ambrose, D., Grupp, S. A., Chew, A., Zheng, Z., Milone, M. C., Levine, B. L., Melenhorst, J. J., & June, C. H. (2015). Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Science translational medicine, 7(303), 303ra139. https://doi.org/10.1126/scitranslmed.aac5415
- Xu XJ, Tang YM. Cytokine release syndrome in cancer immunotherapy with chimeric antigen receptor engineered T cells. Cancer Lett. 2014 Feb 28;343(2):172-8. doi: 10.1016/j.canlet.2013.10.004
- Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, Sanvito F, Ponzoni M, Doglioni C, Cristofori P, Traversari C, Bordignon C, Ciceri F, Ostuni R, Bonini C, Casucci M, Bondanza A. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018 Jun;24(6):739-748. doi: 10.1038/s41591-018-0036-4
- Liu Y, Fang Y, Chen X, Wang Z, Liang X, Zhang T, Liu M, Zhou N, Lv J, Tang K, Xie J, Gao Y, Cheng F, Zhou Y, Zhang Z, Hu Y, Zhang X, Gao Q, Zhang Y, Huang B. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci Immunol. 2020 Jan 17;5(43):eaax7969. doi: 10.1126/sciimmunol.aax7969
- Topp MS, Gökbuget N, Stein AS, Zugmaier G, O’Brien S, Bargou RC, Dombret H, Fielding AK, Heffner L, Larson RA, Neumann S, Foà R, Litzow M, Ribera JM, Rambaldi A, Schiller G, Brüggemann M, Horst HA, Holland C, Jia C, Maniar T, Huber B, Nagorsen D, Forman SJ, Kantarjian HM. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015 Jan;16(1):57-66. doi: 10.1016/S1470-2045(14)71170-2. Epub 2014 Dec 16. Erratum in: Lancet Oncol. 2015 Apr;16(4):e158.
- Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006 Sep 7;355(10):1018-28. doi: 10.1056/NEJMoa063842
- Frey NV, Porter DL. Cytokine release syndrome with novel therapeutics for acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program 2016;2016:567-572.
- Dahmer MK, Randolph A, Vitali S, Quasney MW. Genetic polymorphisms in sepsis. Pediatr Crit Care Med 2005;6:Suppl:S61-S73.
- Liu E, Marin D, Banerjee P, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med 2020;382:545-553.
- Nebelsiek T, Beiras-Fernandez A, Kilger E, Möhnle P, Weis F. Routine use of corticosteroids to prevent inflammation response in cardiac surgery. Recent Pat Cardiovasc Drug Discov 2012;7:170-174.
- Fisher CJ Jr, Dhainaut JF, Opal SM, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome: results from a randomized, double-blind, placebo-controlled trial. JAMA 1994;271:1836-1843.
- Shakoory B, Carcillo JA, Chatham WW, et al. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: reanalysis of a prior phase III trial. Crit Care Med 2016;44:275-281.
- Lykens JE, Terrell CE, Zoller EE, Risma K, Jordan MB. Perforin is a critical physiologic regulator of T-cell activation. Blood 2011;118:618-626.
- Zhang, M., Bracaglia, C., Prencipe, G., Bemrich-Stolz, C. J., Beukelman, T., Dimmitt, R. A., Chatham, W. W., Zhang, K., Li, H., Walter, M. R., De Benedetti, F., Grom, A. A., & Cron, R. Q. (2016). A Heterozygous RAB27A Mutation Associated with Delayed Cytolytic Granule Polarization and Hemophagocytic Lymphohistiocytosis. Journal of immunology (Baltimore, Md. : 1950), 196(6), 2492–2503. https://doi.org/10.4049/jimmunol.1501284
- Polizzotto MN, Uldrick TS, Wang V, et al. Human and viral interleukin-6 and other cytokines in Kaposi sarcoma herpesvirus-associated multicentric Castleman disease. Blood 2013;122:4189-4198.
- Dispenzieri A, Fajgenbaum DC. Overview of Castleman disease. Blood 2020;135:1353-1364.
- Ramaswami R, Lurain K, Peer CJ, et al. Tocilizumab in patients with symptomatic Kaposi sarcoma herpesvirus-associated multicentric Castleman disease. Blood 2020;135:2316-2319.
- Dalla Pria A, Pinato D, Roe J, Naresh K, Nelson M, Bower M. Relapse of HHV8-positive multicentric Castleman disease following rituximab-based therapy in HIV-positive patients. Blood 2017;129:2143-2147.
- Grajales-Reyes GE, Colonna M. Interferon responses in viral pneumonias. Science 2020;369:626-627.
- Huang, K. J., Su, I. J., Theron, M., Wu, Y. C., Lai, S. K., Liu, C. C., & Lei, H. Y. (2005). An interferon-gamma-related cytokine storm in SARS patients. Journal of medical virology, 75(2), 185–194. https://doi.org/10.1002/jmv.20255
- RECOVERY Collaborative Group, Horby, P., Lim, W. S., Emberson, J. R., Mafham, M., Bell, J. L., Linsell, L., Staplin, N., Brightling, C., Ustianowski, A., Elmahi, E., Prudon, B., Green, C., Felton, T., Chadwick, D., Rege, K., Fegan, C., Chappell, L. C., Faust, S. N., Jaki, T., … Landray, M. J. (2021). Dexamethasone in Hospitalized Patients with Covid-19. The New England journal of medicine, 384(8), 693–704. https://doi.org/10.1056/NEJMoa2021436
- Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., Zhang, L., Fan, G., Xu, J., Gu, X., Cheng, Z., Yu, T., Xia, J., Wei, Y., Wu, W., Xie, X., Yin, W., Li, H., Liu, M., Xiao, Y., … Cao, B. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet (London, England), 395(10223), 497–506. https://doi.org/10.1016/S0140-6736(20)30183-5
- Del Valle DM, Kim-Schulze S, Huang H-H, et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med 2020;26:1636-1643. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7869028
- Mathew, D., Giles, J. R., Baxter, A. E., Oldridge, D. A., Greenplate, A. R., Wu, J. E., Alanio, C., Kuri-Cervantes, L., Pampena, M. B., D’Andrea, K., Manne, S., Chen, Z., Huang, Y. J., Reilly, J. P., Weisman, A. R., Ittner, C., Kuthuru, O., Dougherty, J., Nzingha, K., Han, N., … Wherry, E. J. (2020). Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science (New York, N.Y.), 369(6508), eabc8511. https://doi.org/10.1126/science.abc8511
- Caricchio R, Gallucci M, Dass C, Zhang X, Gallucci S, Fleece D, Bromberg M, Criner GJ; Temple University COVID-19 Research Group. Preliminary predictive criteria for COVID-19 cytokine storm. Ann Rheum Dis. 2021 Jan;80(1):88-95. doi: 10.1136/annrheumdis-2020-218323
- Bastard, P., Rosen, L. B., Zhang, Q., Michailidis, E., Hoffmann, H. H., Zhang, Y., Dorgham, K., Philippot, Q., Rosain, J., Béziat, V., Manry, J., Shaw, E., Haljasmägi, L., Peterson, P., Lorenzo, L., Bizien, L., Trouillet-Assant, S., Dobbs, K., de Jesus, A. A., Belot, A., … Casanova, J. L. (2020). Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science (New York, N.Y.), 370(6515), eabd4585. https://doi.org/10.1126/science.abd4585
- Lauder, S. N., Jones, E., Smart, K., Bloom, A., Williams, A. S., Hindley, J. P., Ondondo, B., Taylor, P. R., Clement, M., Fielding, C., Godkin, A. J., Jones, S. A., & Gallimore, A. M. (2013). Interleukin-6 limits influenza-induced inflammation and protects against fatal lung pathology. European journal of immunology, 43(10), 2613–2625. https://doi.org/10.1002/eji.201243018
- Stone, J. H., Frigault, M. J., Serling-Boyd, N. J., Fernandes, A. D., Harvey, L., Foulkes, A. S., Horick, N. K., Healy, B. C., Shah, R., Bensaci, A. M., Woolley, A. E., Nikiforow, S., Lin, N., Sagar, M., Schrager, H., Huckins, D. S., Axelrod, M., Pincus, M. D., Fleisher, J., Sacks, C. A., … BACC Bay Tocilizumab Trial Investigators (2020). Efficacy of Tocilizumab in Patients Hospitalized with Covid-19. The New England journal of medicine, 383(24), 2333–2344. https://doi.org/10.1056/NEJMoa2028836
- Klok, F. A., Kruip, M., van der Meer, N., Arbous, M. S., Gommers, D., Kant, K. M., Kaptein, F., van Paassen, J., Stals, M., Huisman, M. V., & Endeman, H. (2020). Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: An updated analysis. Thrombosis research, 191, 148–150. https://doi.org/10.1016/j.thromres.2020.04.041
- WHO Rapid Evidence Appraisal for COVID-19 Therapies (REACT) Working Group, Sterne, J., Murthy, S., Diaz, J. V., Slutsky, A. S., Villar, J., Angus, D. C., Annane, D., Azevedo, L., Berwanger, O., Cavalcanti, A. B., Dequin, P. F., Du, B., Emberson, J., Fisher, D., Giraudeau, B., Gordon, A. C., Granholm, A., Green, C., Haynes, R., … Marshall, J. C. (2020). Association Between Administration of Systemic Corticosteroids and Mortality Among Critically Ill Patients With COVID-19: A Meta-analysis. JAMA, 324(13), 1330–1341. https://doi.org/10.1001/jama.2020.17023
- Keller MJ, Kitsis EA, Arora S, et al. Effect of systemic glucocorticoids on mortality or mechanical ventilation in patients with COVID-19. J Hosp Med 2020;15:489-493. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7518134
- Bronte, V., Ugel, S., Tinazzi, E., Vella, A., De Sanctis, F., Canè, S., Batani, V., Trovato, R., Fiore, A., Petrova, V., Hofer, F., Barouni, R. M., Musiu, C., Caligola, S., Pinton, L., Torroni, L., Polati, E., Donadello, K., Friso, S., Pizzolo, F., … Olivieri, O. (2020). Baricitinib restrains the immune dysregulation in patients with severe COVID-19. The Journal of clinical investigation, 130(12), 6409–6416. https://doi.org/10.1172/JCI141772
- Fajgenbaum, D. C., Khor, J. S., Gorzewski, A., Tamakloe, M. A., Powers, V., Kakkis, J. J., Repasky, M., Taylor, A., Beschloss, A., Hernandez-Miyares, L., Go, B., Nimgaonkar, V., McCarthy, M. S., Kim, C. J., Pai, R. L., Frankl, S., Angelides, P., Jiang, J., Rasheed, R., Napier, E., … Pierson, S. K. (2020). Treatments Administered to the First 9152 Reported Cases of COVID-19: A Systematic Review. Infectious diseases and therapy, 9(3), 435–449. https://doi.org/10.1007/s40121-020-00303-8
- Roschewski, M., Lionakis, M. S., Sharman, J. P., Roswarski, J., Goy, A., Monticelli, M. A., Roshon, M., Wrzesinski, S. H., Desai, J. V., Zarakas, M. A., Collen, J., Rose, K., Hamdy, A., Izumi, R., Wright, G. W., Chung, K. K., Baselga, J., Staudt, L. M., & Wilson, W. H. (2020). Inhibition of Bruton tyrosine kinase in patients with severe COVID-19. Science immunology, 5(48), eabd0110. https://doi.org/10.1126/sciimmunol.abd0110
- Zhang, W., Zhao, Y., Zhang, F., Wang, Q., Li, T., Liu, Z., Wang, J., Qin, Y., Zhang, X., Yan, X., Zeng, X., & Zhang, S. (2020). The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The Perspectives of clinical immunologists from China. Clinical immunology (Orlando, Fla.), 214, 108393. https://doi.org/10.1016/j.clim.2020.108393
- Johnson, T. S., Terrell, C. E., Millen, S. H., Katz, J. D., Hildeman, D. A., & Jordan, M. B. (2014). Etoposide selectively ablates activated T cells to control the immunoregulatory disorder hemophagocytic lymphohistiocytosis. Journal of immunology (Baltimore, Md. : 1950), 192(1), 84–91. https://doi.org/10.4049/jimmunol.1302282
- Marsh, R. A., Allen, C. E., McClain, K. L., Weinstein, J. L., Kanter, J., Skiles, J., Lee, N. D., Khan, S. P., Lawrence, J., Mo, J. Q., Bleesing, J. J., Filipovich, A. H., & Jordan, M. B. (2013). Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatric blood & cancer, 60(1), 101–109. https://doi.org/10.1002/pbc.24188
- Mouy R, Stephan JL, Pillet P, Haddad E, Hubert P, Prieur AM. Efficacy of cyclosporine A in the treatment of macrophage activation syndrome in juvenile arthritis: report of five cases. J Pediatr. 1996 Nov;129(5):750-4. doi: 10.1016/s0022-3476(96)70160-9
- Faitelson Y, Grunebaum E. Hemophagocytic lymphohistiocytosis and primary immune deficiency disorders. Clin Immunol. 2014 Nov;155(1):118-125. doi: 10.1016/j.clim.2014.09.008
- Iwaki N, Fajgenbaum DC, Nabel CS, Gion Y, Kondo E, Kawano M, Masunari T, Yoshida I, Moro H, Nikkuni K, Takai K, Matsue K, Kurosawa M, Hagihara M, Saito A, Okamoto M, Yokota K, Hiraiwa S, Nakamura N, Nakao S, Yoshino T, Sato Y. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease. Am J Hematol. 2016 Feb;91(2):220-6. doi: 10.1002/ajh.24242
- Nishimoto N, Kanakura Y, Aozasa K, Johkoh T, Nakamura M, Nakano S, Nakano N, Ikeda Y, Sasaki T, Nishioka K, Hara M, Taguchi H, Kimura Y, Kato Y, Asaoku H, Kumagai S, Kodama F, Nakahara H, Hagihara K, Yoshizaki K, Kishimoto T. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood. 2005 Oct 15;106(8):2627-32. doi: 10.1182/blood-2004-12-4602
- Dustin Shilling, Jason R Ruth, Christopher Sheild Nabel, Sheila K Pierson, Mary Guilfoyle, Craig Tendler, Manjula P Reddy, Michael Weinblatt, Nancy Shadick, Mark Bower, Alessia Dalla Pria, Yasufumi Masaki, Philip Beineke, Sofija Miljovska, Laura Katz, Sushila Shenoy, Ana B Oromendia, Jason Mezey, Jeff Wiser, Jef Benbanaste, David Lee, Alexander Fosså, Frits van Rhee, David C Fajgenbaum; Serum Proteomics Reveals Distinct Subtypes Associated with Treatment Response in Idiopathic Multicentric Castleman Disease. Blood 2018; 132 (Supplement 1): 3716. doi: https://doi.org/10.1182/blood-2018-99-119135
- van Rhee, F., Voorhees, P., Dispenzieri, A., Fosså, A., Srkalovic, G., Ide, M., Munshi, N., Schey, S., Streetly, M., Pierson, S. K., Partridge, H. L., Mukherjee, S., Shilling, D., Stone, K., Greenway, A., Ruth, J., Lechowicz, M. J., Chandrakasan, S., Jayanthan, R., Jaffe, E. S., … Fajgenbaum, D. C. (2018). International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood, 132(20), 2115–2124. https://doi.org/10.1182/blood-2018-07-862334
- Pierson SK, Stonestrom AJ, Shilling D, Ruth J, Nabel CS, Singh A, Ren Y, Stone K, Li H, van Rhee F, Fajgenbaum DC. Plasma proteomics identifies a ‘chemokine storm’ in idiopathic multicentric Castleman disease. Am J Hematol. 2018 Jul;93(7):902-912. doi: 10.1002/ajh.25123
- Pai, R. L., Japp, A. S., Gonzalez, M., Rasheed, R. F., Okumura, M., Arenas, D., Pierson, S. K., Powers, V., Layman, A., Kao, C., Hakonarson, H., van Rhee, F., Betts, M. R., Kambayashi, T., & Fajgenbaum, D. C. (2020). Type I IFN response associated with mTOR activation in the TAFRO subtype of idiopathic multicentric Castleman disease. JCI insight, 5(9), e135031. https://doi.org/10.1172/jci.insight.135031
- Arenas, D. J., Floess, K., Kobrin, D., Pai, R. L., Srkalovic, M. B., Tamakloe, M. A., Rasheed, R., Ziglar, J., Khor, J., Parente, S., Pierson, S. K., Martinez, D., Wertheim, G. B., Kambayashi, T., Baur, J., Teachey, D. T., & Fajgenbaum, D. C. (2020). Increased mTOR activation in idiopathic multicentric Castleman disease. Blood, 135(19), 1673–1684. https://doi.org/10.1182/blood.2019002792
- Fajgenbaum, D. C., Langan, R. A., Japp, A. S., Partridge, H. L., Pierson, S. K., Singh, A., Arenas, D. J., Ruth, J. R., Nabel, C. S., Stone, K., Okumura, M., Schwarer, A., Jose, F. F., Hamerschlak, N., Wertheim, G. B., Jordan, M. B., Cohen, A. D., Krymskaya, V., Rubenstein, A., Betts, M. R., … Uldrick, T. S. (2019). Identifying and targeting pathogenic PI3K/AKT/mTOR signaling in IL-6-blockade-refractory idiopathic multicentric Castleman disease. The Journal of clinical investigation, 129(10), 4451–4463. https://doi.org/10.1172/JCI126091
- Sirolimus in Previously Treated Idiopathic Multicentric Castleman Disease. https://clinicaltrials.gov/ct2/show/NCT03933904
- Shimabukuro-Vornhagen A, Gödel P, Subklewe M, et al. Cytokine release syndrome. Journal for ImmunoTherapy of Cancer 2018;6:56. doi: 10.1186/s40425-018-0343-9
- Hay KA, Hanafi L-A, Li D, Gust J, Liles WC, Wurfel MM, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T cell therapy. Blood. 2017.
- Kantarjian H , Stein A , Gökbuget N , Fielding AK , Schuh AC , Ribera J-M , et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376:836–847. doi:10.1056/NEJMoa1609783 pmcid:5881572
- Neelapu SS , Tummala S , Kebriaei P , Wierda W , Gutierrez C , Locke FL , et al. Chimeric antigen receptor T-cell therapy – assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15:47–62. doi:10.1038/nrclinonc.2017.148
- Lee DW , Gardner R , Porter DL , Louis CU , Ahmed N , Jensen M , et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–195. doi:10.1182/blood-2014-05-552729 pmcid:4093680
- American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference: definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med. 1992 Jun;20(6):864-74.
- Goldstein B, Giroir B, Randolph A; International Consensus Conference on Pediatric Sepsis. International pediatric sepsis consensus conference: definitions for sepsis and organ dysfunction in pediatrics. Pediatr Crit Care Med. 2005 Jan;6(1):2-8. doi: 10.1097/01.PCC.0000149131.72248.E6
- Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992 Jun;101(6):1644-55. doi: 10.1378/chest.101.6.1644
- Canna, S. W., & Behrens, E. M. (2012). Making sense of the cytokine storm: a conceptual framework for understanding, diagnosing, and treating hemophagocytic syndromes. Pediatric clinics of North America, 59(2), 329–344. https://doi.org/10.1016/j.pcl.2012.03.002
- COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395(10229):1033-1034. https://doi.org/10.1016/S0140-6736(20)30628-0
- de Jesus, A. A., Hou, Y., Brooks, S., Malle, L., Biancotto, A., Huang, Y., Calvo, K. R., Marrero, B., Moir, S., Oler, A. J., Deng, Z., Montealegre Sanchez, G. A., Ahmed, A., Allenspach, E., Arabshahi, B., Behrens, E., Benseler, S., Bezrodnik, L., Bout-Tabaku, S., Brescia, A. C., … Goldbach-Mansky, R. (2020). Distinct interferon signatures and cytokine patterns define additional systemic autoinflammatory diseases. The Journal of clinical investigation, 130(4), 1669–1682. https://doi.org/10.1172/JCI129301
- Shimizu M, Nakagishi Y, Yachie A. Distinct subsets of patients with systemic juvenile idiopathic arthritis based on their cytokine profiles. Cytokine. 2013 Feb;61(2):345-8. doi: 10.1016/j.cyto.2012.11.025
- Vercruysse, F., Barnetche, T., Lazaro, E., Shipley, E., Lifermann, F., Balageas, A., Delbrel, X., Fautrel, B., Richez, C., Schaeverbeke, T., & Truchetet, M. E. (2019). Adult-onset Still’s disease biological treatment strategy may depend on the phenotypic dichotomy. Arthritis research & therapy, 21(1), 53. https://doi.org/10.1186/s13075-019-1838-6





{kind=link}