Familial amyloidosis
Familial amyloidosis also known as hereditary amyloidosis, refers to a group of inherited conditions in which abnormal protein deposits called amyloid is found in almost every tissue in your body where it should not be, which causes disruption of organ tissue structure and function 1. Hereditary amyloidosis is one type of the systemic amyloidosis diseases that are caused by inheriting a gene mutation. That genetic mutation then produces an amyloid protein that forms into an abnormal shape. These abnormal “misfolded” amyloid proteins can be deposited and cluster in the body’s nerves and other organs and once they build up, this may affect and harm tissue and/or organ function. In familial amyloidosis, amyloid deposits most often occur in tissues of the heart, kidneys, and nervous system 2. These protein deposits damage the tissues and interfere with how organs work. While symptoms of familial amyloidosis may appear in childhood, most individuals do not experience symptoms until adulthood 3. Even though you are born with a gene mutation, normally the harmful amyloid deposits don’t occur until adulthood. Although all the types of the familial amyloidosis can cause serious complications, there are some carriers of this genetic mutation that may not show symptoms of the disease at all. Others may have a few, more minor, health issues.
There are 2 main classifications of familial amyloidosis diseases:
- ATTR amyloidosis also known as transthyretin (ATTR) amyloidosis. ATTR amyloidosis means A for amyloid and the TTR is short for the protein “transthyretin”. Transthyretin (TTR) is a protein mainly produced in the liver with a smaller portion also coming from the choroid plexus of the brain and functions as a transporter of thyroxine (T4) and retinol (vitamin A)-binding protein 4. All the transthyretin (TTR) in the blood is produced by the liver. TTR in the brain and the eye is made separately by a structure called the choroid plexus, which is located within the brain and produces the cerebrospinal fluid that bathes the brain and spinal cord. In the past, because ATTR often involves nerve or cardiac involvement, some terms were used when the chemical variations were less defined. Examples of these outdated terms include FAP (Familial Amyloid Polyneuropathy) and FAC (Familial Amyloid Cardiomyopathy). Today, the different forms of ATTR are termed according to the “chemically based” name of the transthyretin protein variation. An example of this would be ATTRV30M (for ATTR Val30Met or p.Val50Met according to the Human Genome Variation Society recommendation), which is the most common ATTR variation. The most common TTR variants in the United States are:
- Val30Met (also the most found worldwide). What was once termed FAP (Familial amyloid polyneuropathy) is most commonly caused by Val30Met.
- Thr60Ala
- Leu58His
- Ser77Tyr
- Val122Ile — predominantly seen in the African-American population; associated with cardiomyopathy (heart conditions). What was once termed FAC (Familial amyloid cardiomyopathy) is commonly caused by Val122lle.
- Non-TTR = Non-transthyretin amyloidosis. These diseases are considered even more rare than the transthyretin amyloidosis (ATTR) variations. Examples of non-transthyretin amyloidosis (Non-TTR) include, but are not limited to, apolipoprotein AI amyloidosis (A ApoAI), gelsolin amyloidosis (A Gel), lysozyme amyloidosis (A Lys), cystatin C amyloidosis (A Cys), fibrinogen Aα-chain amyloidosis (A Fib), and apolipoprotein AII amyloidosis (A ApoAII) 5. More variations may be discovered as research continues. For example, in Europe and the United States, the most common Non-TTR proteins that cause kidney problems are Fibrinogen Aa and Apo lipoprotein AI. They are called this because of the mutations in the AFib and ApoAI genes.
Moreover, it is further complicated by the fact that there are approximately 136 different genetic variations in ATTR, and at least 60 genetic variations in Non-TTR hereditary amyloidosis diseases. It is possible that more may be discovered as research continues. Each family with a certain hereditary form of amyloidosis has its own pattern of organ involvement, approximate age of onset and associated symptoms.
Most types of familial amyloidosis or hereditary amyloidosis are inherited in an autosomal dominant manner 3.
Typically, families know when they have a hereditary form of amyloidosis because of similar symptoms and causes of illness among blood relatives, so family history is a key indicator.
The age at onset of disease-related symptoms varies between the second and ninth decades of life, with great variations across different populations. Expected age at onset is critical to determine when amyloidosis testing should be requisitioned. Portuguese and Japanese foci of patients with transthyretin familial amyloid polyneuropathy have traditionally been described as early-onset (mean age, 33 years) 6, whereas Swedish patients with transthyretin familial amyloid polyneuropathy (TTR-FAP) are characterized by a later mean age of onset (56 years) 7. Even in foci generally considered early-onset, some subgroups of patients experience a later onset. In Japan, especially, a form of later-onset transthyretin familial amyloid polyneuropathy with no genealogical relationship to the two foci of this disease now predominates 6. Patients with cardiomyopathy who have wild-type or variant transthyretin traditionally develop symptoms in their sixties.
The mean duration of disease onset to death is approximately 10 years but may vary depending on endemic region, genotype, symptoms, and other factors.
Familial amyloidosis treatment is focused on addressing symptoms of organ damage and slowing down the production of amyloid when possible through methods such as liver transplants.
How does someone get ATTR or Non-TTR familial amyloidosis?
ATTR and Non-TTR familial amyloidosis are not contagious. They are considered to be an inherited, autosomal dominant disease. This means that to get this disease, a person needs one copy of the mutant gene – in other words, it can be inherited from one parent. Each off spring of an affected parent has a 50/50 chance of inheriting the gene. If an offspring is not born with the gene mutation, then they can’t pass it onto their own offspring.
Is there a special diet that I can follow?
Eating a well-balanced, heart-healthy and nutritious diet is always recommended. Although amyloid is an abnormal protein, the amount of protein in the diet does not affect the onset of the disease. A diet low in protein and/or sodium may be necessary when the kidneys are involved. Consult with your physician on any dietary changes, and report any vitamins or other supplements that you take. You are a part of the team of people who must keep in communication with each other about your health.
Should physical activities be restricted?
If fatigue or shortness of breath occurs, it is important to rest. Patients should not push themselves or attempt strenuous activities beyond what is recommended by the doctor. Normal daily activities may be carried out as usual. Usually patients can continue working and are encouraged to do so.
Is familial amyloidosis a form of cancer?
No. At this time, none of the types of amyloidosis diseases are considered to be cancer.
How common is ATTR or Non-TTR Hereditary Amyloidosis?
ATTR and Non-TTR Amyloidosis can be found in virtually every ethnic background, but the exact incidence of familial amyloidosis is unknown. Generally, they are an uncommon condition and are considered rare diseases, with some statistics estimating 1 per 100,000 of the population in the United States 8. In Europe, the prevalence of transthyretin familial amyloid polyneuropathy is estimated to be less than one in 100,000 individuals 9. However, there is one ATTR type that alone puts the rare label in question. A variant named ATTR Val122lle is predominantly seen in the African-American population and it is associated with cardiomyopathy (heart conditions). What was once termed FAC (Familial amyloid cardiomyopathy) is commonly caused by ATTR Val122lle. Medical statistics state that ATTR Val122Ile is found in approximately 4% of the U.S. African-American population 10, with most individuals developing late-onset cardiac amyloidosis. That could mean that this gene mutation is carried by approximately 1.5 million people in the U.S. This estimate is just the ATTR Val122Ile variation of hereditary amyloidosis and it is assumed that this variant is underdiagnosed. The frequency of Val122Ile in Caucasian and Hispanic populations in the United States is 0.44% and 0%, respectively 10. The worldwide prevalence of transthyretin amyloidosis (ATTR amyloidosis) dominated by cardiomyopathy is unknown, but it is almost certainly underdiagnosed, particularly in the African American Val122Ile carrier population older than 65 years (approximately 135,000 individuals) 11. Remember, not everyone with the genetic mutation will experience symptoms, but this estimate takes the potential for carriers out of the rare category.
Val30Met is the most prevalent transthyretin familial amyloid polyneuropathy (TTR-FAP) mutation in the world, focused in Portugal, Sweden, Japan, Brazil, and Majorca, and is believed to have arisen independently in Portugal and Sweden 12. The largest cluster of individuals with transthyretin familial amyloid polyneuropathy caused by the Val30Met mutation may be found in northern Portugal (Póvoa de Varzim and Vila do Conde), where the incidence is estimated to be one in 538 individuals 13. The cardiomyopathy-related Leu111Met and Val122Ile mutations are found primarily in Danish and African American populations, respectively. However, all mutations, including Val30Met, are identified across all countries in different families without any obvious relationship.
In endemic areas of northern Sweden (Piteå and Skelleftå), the frequency of the Val30Met mutation is 4%; however, the penetrance is relatively low (11% by 50 years) 14. Conversely, in Portugal, the penetrance is high (80% by 50 years) 15. Although also endemic in some areas of Japan, the prevalence of transthyretin familial amyloid polyneuropathy is estimated to be lower than in Europe, at approximately one in 1,000,000 individuals 16.
What is a gene mutation?
Genes are pieces of DNA, and most genes contain the information for making a specific protein. Some genes tell the body how to make a protein, for example, the TTR (transthyretin) gene tells the liver how to make the TTR protein. Sometimes there are differences in the coding of the gene so the body is told to make a slightly different protein.
This change is called a mutation in the genetic code of the DNA. DNA is isolated from a sample of blood, and then gene sequencing can be performed to see if a patient carries the genetic mutation for a particular variation of hereditary amyloidosis.
How does one gene mutation affect someone?
In simple terms, a gene is a segment of DNA that is coded to pass along a certain trait; it has a specific task (for example, determining the color of your eyes). Genes are building blocks that you inherit from your family.
The DNA sequence makes up a code that governs a particular cell’s function. A single gene on chromosome 18 is responsible for TTR genetic information. So, this gene mutation is one part of a DNA sequence and it can cause malfunctions.
Is a DNA test necessary?
One or more tissue biopsies are considered the most accurate diagnostic test available for all the amyloidosis diseases. When the tissue biopsy sample is stained with Congo red stain, it is then put under a light microscope and the amyloid deposits show up as an “apple green” birefringence (“birefringence” means double refraction) with the microscope in the lab.
Immunoassays are chemical tests done in a lab that are used to detect a specific protein or substance by using an antibody that is reactive with it. In this case, once the Congo red staining is performed and amyloid is confirmed, then additional immunoassaying of the biopsy specimen, using antiserum against TTR should also be performed.
However, this is not always enough. Since there are so many variations of hereditary amyloidosis, and more could be discovered, gene sequencing (a DNA test) is also necessary because immunoassaying may not identify a certain variant that is not within the more well known TTR types.
If there is a confirmed gene mutation, when might symptoms appear?
Most hereditary amyloidosis symptoms, if they develop, occur in adulthood. The age symptoms begin may also vary by country. In Portugal and Japan, people with familial amyloidosis usually start developing symptoms in their late 20s to 40s. In other parts of the world, people with familial amyloidosis may not have symptoms until after age 50. At that time, symptoms can range from none, to mild, to very severe. Although genetic tests can identify a particular problem gene, they cannot predict how severely that gene will affect the person who carries it.
If you are familiar with the common symptoms associated with your hereditary amyloidosis variant, you might be better able to keep a watchful eye on your health. For instance, you may not know that for some variations carpal tunnel syndrome can occur years before other symptoms develop. You could experience early signs of the clinical disease and not realize that there was a connection.
Having a genetic mutation is only a part of the story, because many illnesses develop from a mix of high-risk genes, environmental factors and/or unhealthy lifestyle (such as a smoker with a family history of familial cardiac amyloidosis). Make sure your doctor knows about the hereditary amyloidosis diseases, so they are able to monitor your health.
What is peripheral neuropathy and what are the symptoms?
Your peripheral nerves are the ones outside your brain and spinal cord. Peripheral nerves carry information to and from the brain. They also carry signals to and from the spinal cord to the rest of the body. Peripheral neuropathy means these nerves don’t work properly. Peripheral neuropathy may occur because of damage to a single nerve or a group of nerves. It may also affect nerves in the whole body. Peripheral neuropathy can be caused by inflammation of, or damage to, the nerves.
Peripheral neuropathy symptoms often start gradually, and then get worse. They include :
- Numbness
- Pain
- Burning or tingling
- Muscle weakness
- Sensitivity to touch
in any part of the body, but commonly is felt in the hands and feet.
What is autonomic neuropathy and what are the symptoms?
Autonomic neuropathy (AN) is a condition that results from damage to nerves that assist in organ and organ system functioning. Autonomic nerves control the functions of your internal organs such as the heart, stomach and intestines, as well as the glands. When your autonomic nerves are damaged, your blood pressure, heart rate, perspiration patterns, and bowel movements may be affected. In addition, you may have problems with dizziness, emptying your bladder, and/or experience gastrointestinal symptoms of diarrhea, weight loss, and poor digestion.
What if there aren’t any symptoms?
Your doctor will help you decide if you need a tissue biopsy if you have no clinical symptoms. It is important to understand that carrying the gene mutation does not always mean that you will have symptoms later on, or that this disease will seriously affect you.
If you have a parent with one of the hereditary amyloidosis types, you can be tested to see if you’ve inherited the same amyloid gene. This DNA test is done by a blood sample. If you suspect your parent has (or had) hereditary amyloidosis, but no definite diagnosis was ever made for this parent, try to speak to your parent’s doctor, or one of the doctors at the amyloid centers, in order to determine if testing is necessary.
What is the difference between DNA and RNA?
In simple terms, your DNA stores and transfers genetic information. A gene tells a cell how to make a specific protein. Proteins are formed inside your cells and it is your DNA that holds the “recipe” for making proteins. DNA and RNA work together and they both carry genetic information to make up the many different proteins you need. However, they perform different functions for this task. The RNA helps to move the DNA “code” from storage to where it can be used. RNA is converted (or “translated”) into a sequence of amino acids that makes up a protein.
In basic biological terms:
- Transcription: DNA → RNA
- Translation: RNA → protein
The collections of proteins within a cell are essential for your body’s health and function, and they work in a variety of ways, serving activity inside the cell as well as interaction outside the cell – in virtually every process within the cell.
How can the new RNAi research help with TTR amyloidosis diseases?
RNAi is short for “RNA interference.” By putting “silencing RNA” into cells that make an abnormal TTR, the translation of RNA to protein is stopped. This means that the production of the abnormal TTR that causes amyloid can be dramatically reduced. RNA interference technology is underway and shows promise. Whether it will change the course of patients with TTR amyloid is under active investigation now.
ATTR (transthyretin) amyloidosis
ATTR amyloidosis also called transthyretin (ATTR) amyloidosis, means A for Amyloid and the TTR is short for the protein “transthyretin”. Transthyretin is a protein mainly manufactured in the liver that helps carry thyroid hormone and vitamin A in the blood. ATTR amyloidosis or transthyretin amyloidosis is characterized by a slowly progressive peripheral sensorimotor and/or autonomic neuropathy as well as non-neuropathic changes of cardiomyopathy, nephropathy, vitreous opacities, and central nervous system (CNS) amyloidosis. ATTR amyloidosis usually begins in the third to fifth decade in persons from endemic foci in Portugal and Japan; onset is later in persons from other areas. Typically, sensory neuropathy starts in the lower extremities with paresthesias and hypesthesias of the feet, followed within a few years by motor neuropathy. In some persons, particularly those with early-onset disease, autonomic neuropathy is the first manifestation of the condition; findings can include: orthostatic hypotension, constipation alternating with diarrhea, attacks of nausea and vomiting, delayed gastric emptying, sexual impotence, anhidrosis, and urinary retention or incontinence. Cardiac amyloidosis is mainly characterized by progressive cardiomyopathy. Individuals with leptomeningeal amyloidosis may have the following CNS findings: dementia, psychosis, visual impairment, headache, seizures, motor paresis, ataxia, myelopathy, hydrocephalus, or intracranial hemorrhage.
Normally, transthyretin (ATTR) is made up of four identical parts. ATTR is one term that represents different kinds of mutations in a TTR gene that is inherited. That gene mutation makes the transthyretin unstable, so amyloid protein misfolding occurs. In ATTR amyloidosis, the protein becomes unstable, breaks apart, and deposits in the heart and/or the nerves. The amyloid fibrils then go out into the body and can damage nerves and/or organs, depending on the type of TTR mutation that the patient has inherited.
The 2018 International Society of Amyloidosis has defined 3 types of ATTR amyloidosis 17:
- Hereditary (familial) ATTR amyloidosis (also called ATTRv; v for variant amyloidosis). In this form, there is a change (mutation) in the DNA that is inherited and can be passed from one generation to the next. This makes the TTR protein more unstable and more likely to form amyloid fibrils. Different mutations lead to different symptoms — some may affect the nerves; some may affect the heart; and some may affect both. In the past, because ATTR often involves nerve or cardiac involvement, some terms were used when the chemical variations were less defined. Examples of these outdated terms include FAP (Familial Amyloid Polyneuropathy) and FAC (Familial Amyloid Cardiomyopathy). Today, the different forms of ATTR are termed according to the “chemically based” name of the transthyretin protein variation. An example of this would be ATTRV30M (for ATTR Val30Met), which is the most common ATTR variation.
- Wild-type ATTR amyloidosis (historically known as senile/age related amyloidosis). Unlike hereditary ATTR amyloidosis, wild-type ATTR amyloidosis (ATTRwt amyloidosis) does not involve abnormal DNA and cannot be passed on to family members. Instead, as you get older, the normal TTR protein becomes unstable, misfolds and forms amyloid fibrils. Wild-type ATTR amyloidosis (ATTRwt amyloidosis) has classically been regarded as cardiomyopathy found in elderly individuals, particularly men 18. Studies of autopsy specimens revealed that a significant proportion of the elderly population has wild-type TTR deposition in their heart, even in subjects without a history of underlying diseases 19. A recent development in diagnostic techniques revealed that cardiomyopathy resulting from wild-type ATTR amyloidosis (ATTRwt amyloidosis) is an important differential diagnosis of heart failure with preserved ejection fraction 20. Wild-type ATTR amyloidosis (ATTRwt amyloidosis) may also present features suggestive of carpal tunnel syndrome that frequently precede those of cardiomyopathy 21. Analyses of tenosynovial tissues obtained at carpal tunnel release surgery revealed that a significant proportion of patients diagnosed with idiopathic carpal tunnel syndrome had wild-type TTR amyloid deposits 22. It should be noted that both cardiomyopathy and carpal tunnel syndrome due to wild-type ATTR amyloidosis tend to affect elderly male patients 21. Other studies have also suggested an association between spinal canal stenosis and wild-type TTR deposition in ligaments 23. These findings may suggest the tendency of TTR to deposit in organs affected by shear stress 24. Moreover, a recent report described three patients with myopathy resulting from wild-type ATTR amyloidosis 25. Myopathy was the predominant feature in these patients and the initial manifestation of wild-type ATTR amyloidosis in two of these patients. Notably, serum creatine kinase levels were normal or only slightly elevated in these patients. This report further expands the spectrum of wild-type ATTR amyloidosis and increases the need for physicians’ awareness of this disease at the time of differential diagnosis of myopathy 26.
- Acquired ATTR amyloidosis after domino liver transplantation, reported in patients who received livers from hereditary (familial) ATTR amyloidosis donors 27. As TTR is produced mainly in the liver, liver transplantation has been established as a treatment for hereditary (familial) ATTR amyloidosis 28. Because the liver removed from patients with hereditary (familial) ATTR amyloidosis (ATTRv) functions well except for the production of variant TTR, it may be transplanted into a patient with severe liver disease because of the shortage of donor livers 27. This domino liver transplantation was first performed in 1995 27. A total of 1254 domino liver transplantations were performed worldwide from 1995 to 2017 29. However, as time passes and the number of patients who have undergone domino liver transplantation increases, patients with symptoms suggesting amyloidosis have been reported 30. Analysis of a patient who received an hereditary (familial) ATTR amyloidosis (ATTRv) liver 8 years before autopsy demonstrated that systemic amyloid deposition occurs even before the appearance of symptoms associated with amyloidosis 31. The mean duration between domino liver transplantation and the first detection of amyloid deposition and symptom onset in recipients is approximately 8 years 29. Notably, the clinical features of acquired ATTR amyloidosis after domino liver transplantation are different from those of donors 29. Although liver transplantation is usually performed to treat early-onset Val30Met ATTR amyloidosis cases from endemic foci, recipients of their livers tend to complain of only sensory deficits but not autonomic symptoms, even though they are not elderly 32. These features are similar to those in patients with late-onset Val30Met ATTR amyloidosis from nonendemic areas, rather than conventional early-onset patients from endemic foci with marked autonomic dysfunctions 33. These findings may support the view that late-onset Val30Met cases prevalent throughout the world are the prototype of hereditary (familial) ATTR amyloidosis (ATTRv) 34.
The majority of hereditary amyloidosis types are TTR-related, and there are many different variations within ATTR. Most ATTR diseases have a hereditary pattern of organ involvement, approximate age of onset and associated symptoms. It is common that symptoms do not appear until a person is an adult and the degree and severity of illness depends on the individual.
If a patient has a clear family history along with clinical signs of amyloidosis, then ATTR is highly possible. However, there are times when someone is the first case to be identified in his or her family. Since many amyloidosis and other diseases can cause similar symptoms, it is vital that the patient is diagnosed properly with the type of amyloid protein clearly identified.
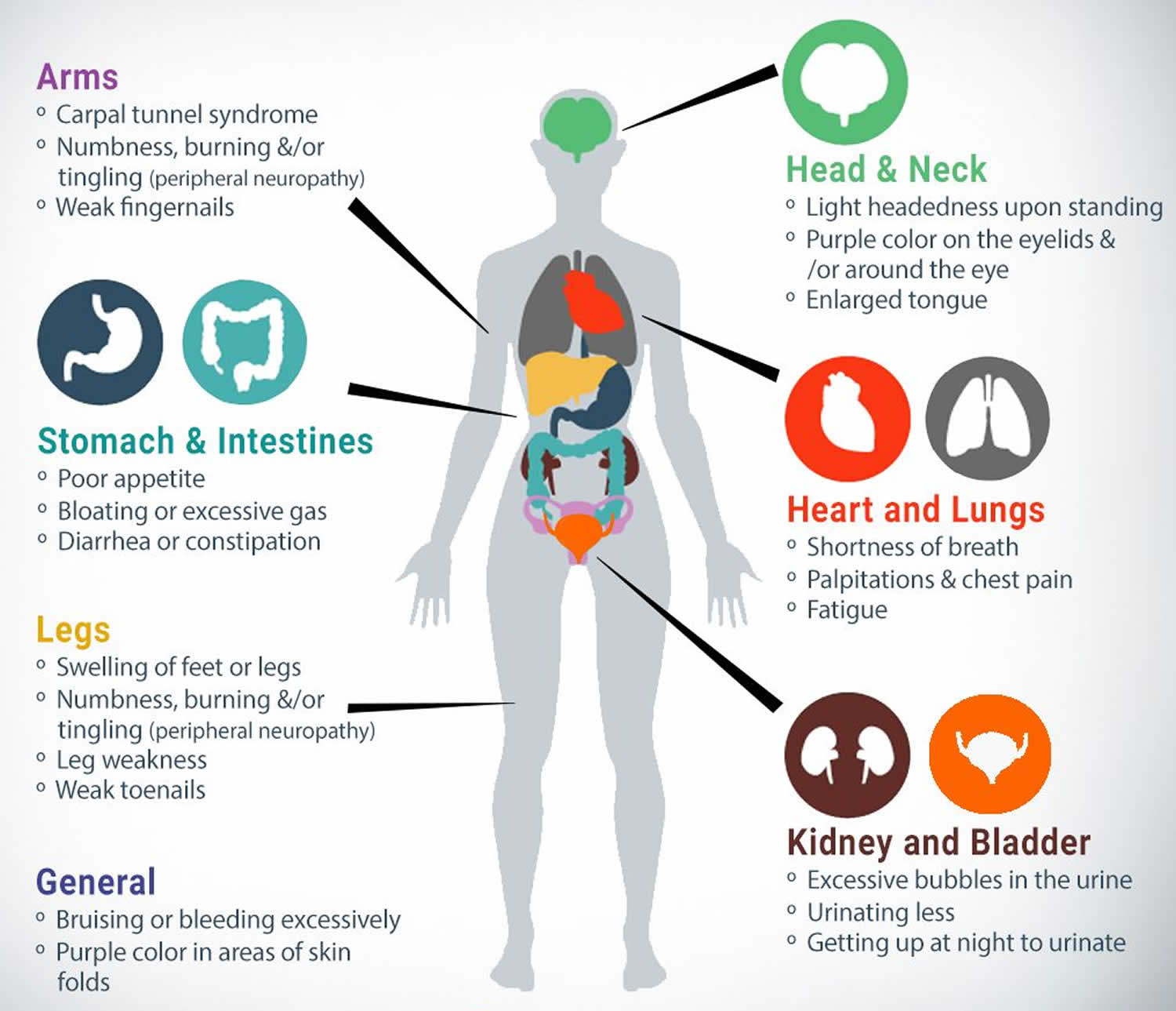
Symptoms of ATTR vary, depending on the TTR genetic variant that is involved and the organ (or multiple organs) that are affected by the amyloid deposits. It also depends on the degree that the organ function is impaired. The most common sites of amyloid deposits are associated with cardiac and/or nerve involvement (called cardiomyopathy and neuropathy) and the gastrointestinal tract. The kidneys, eyes, and carpal ligament (also known as carpal tunnel syndrome) are among other possibilities that can be affected.
ATTR amyloidosis symptoms indicating the arms are affected include:
- Carpal tunnel syndrome.
- Numbness, burning and/or tingling (peripheral neuropathy).
- Biceps tendon rupture.
Symptoms indicating the back is affected include:
- Lumbar spinal stenosis.
Symptoms indicating the legs are affected include:
- Swelling of the feet or legs.
- Numbness, burning and/or tingling (peripheral neuropathy).
- Leg weakness.
Symptoms indicating the head and neck are affected include:
- Eye floaters (vitreous opacities).
- Lightheadedness upon standing.
Symptoms indicating the heart and lungs are affected include:
- Shortness of breath.
- Palpitations.
- Chest pain.
- Fatigue.
Symptoms indicating the stomach or intestines are affected include:
- Poor appetite.
- Bloating or excessive gas.
- Diarrhea or constipation.
ATTR (transthyretin) amyloidosis causes
Mutations in the TTR gene cause transthyretin amyloidosis. The TTR gene provides instructions for producing a protein called transthyretin. Transthyretin transports vitamin A (retinol) and a hormone called thyroxine throughout the body. To transport retinol and thyroxine, four transthyretin proteins must be attached (bound) to each other to form a four-protein unit (tetramer). Transthyretin is produced primarily in the liver. A small amount of this protein is produced in an area of the brain called the choroid plexus and in the light-sensitive tissue that lines the back of the eye (the retina).
TTR gene mutations are thought to alter the structure of transthyretin, impairing its ability to bind to other transthyretin proteins and altering its normal function.
There are more than 125 different TTR mutations that have been identified in ATTR. That means that within the one ATTR type, there exist many different variations, which are then identified by the amino acid protein involvement.
The most common TTR variants in the United States are:
- Val30Met or p.Val50Met (also the most found worldwide). What was once termed FAP (Familial amyloid polyneuropathy) is most commonly caused by Val30Met.
- Thr60Ala
- Leu58His
- Ser77Tyr
- Val122Ile — predominantly seen in the African-American population; associated with cardiomyopathy (heart conditions). What was once termed FAC (Familial amyloid cardiomyopathy) is commonly caused by Val122lle.
ATTR (transthyretin) amyloidosis signs and symptoms
The presenting signs and symptoms in patients with ATTR are fairly nonspecific and are often attributed to more common diseases affecting both the heart and the peripheral nervous system (PNS) and autonomic nervous system. There are three main types of ATTR, identified by the organ system involved: cardiac, neuropathic, and leptomeningeal.
Familial amyloid cardiomyopathy (ATTR cardiac amyloidosis)
Patients with cardiac amyloid deposition typically present with the following typical symptoms of chronic heart failure:
- Symptoms suggestive of right-sided heart failure (ie, dyspnea on exertion, peripheral edema, hepatomegaly, ascites, elevated jugular venous pressure), diastolic dysfunction, and/or arrhythmias (ie, palpitations, lightheadedness, syncope, ECG changes)
- Heart failure with preserved ejection fraction (HFpEF) predominates in ATTR
- Patients may also present with atrial arrhythmias or conduction system disease due to amyloid fibril deposition within areas responsible for electrical impulse conduction.
- Cardiomegaly may be noted on chest imaging/echocardiogram 35.
Congestive heart failure and atrial fibrillation are the most common symptoms. The term “arrhythmia” refers to changes in the normal electrical impulses that cause the heart to beat. The result is a heart that can beat too fast, too slow or erratically. Atrial fibrillation (or a-fib for short) is one of the many forms of arrhythmia. During a-fib, the heart’s two small upper chambers cause an abnormal heart rhythm, usually rapid and irregular beating. This may result in increased heart damage, stroke or heart failure.
Familial amyloid polyneuropathy (ATTR amyloid neuropathy)
The term neuropathy means nerve damage. Neuropathic involvement in patients affected by ATTR–familial amyloid polyneuropathy (FAP) is classically a symmetric, ascending length−dependent, sensorimotor, axonal polyneuropathy subtype and may include the following:
- Peripheral neuropathy or peripheral nervous system sensimotor impairment affecting all functional classes of nerve fibers: motor, sensory and autonomic fibers (diarrhea or contipation, urinary incontinence, orthostatic hypotension, sexual impotence, glaucoma). Peripheral neuropathy can be caused by inflammation of, or damage to, the nerves. It can result in tingling, numbness and burning pain in any part of the body, but commonly is felt in the hands, feet and lower legs. Some patients may experience an increased sensitivity to pain. A loss of sensitivity to temperature may also occur. Sensorimotor impairment means the loss of a combination of sensory and motor activities. This can decrease a patient’s ability to move and feel (sensation) because of nerve damage.
- Autonomic neuropathy is a condition that results from damage to nerves that assist in organ and organ system functioning. Autonomic nerves control the functions of our internal organs such as the heart, stomach and intestines, as well as the glands. Nerves affected by ATTR amyloid deposits may cause the inability to control the muscles that expand or contract blood vessels, which affects the heart rate (irregular heart beats) and blood pressure. If a patient has a sudden drop in blood pressure such as when moving from a seated to a standing position (orthostatic hypotension), then dizziness, fainting, or lightheadedness may occur 36. Other body functions may also be affected, including poor digestion, bowel motility, constipation alternating with diarrhea, attacks of nausea and vomiting, delayed gastric emptying, erectile function. sexual impotence, anhidrosis, and urinary retention or incontinence 37. Because of sensory loss and autonomic dysfunction, trophic ulcers on the lower extremities are common. Frequently, the autonomic neuropathy produces the most significant morbidity of the disorder.
- Lower-limb neuropathy (eg, in patients with the TTR V30M mutation)
- Upper-limb neuropathy (eg, TTR I84S, TTR L58H) 35
- ATTR V30M variant: Lower extremity weakness, pain, and/or impaired sensation; autonomic dysfunction, often manifesting as sexual or urinary dysfunction 38
- Weakness and paresthesias of one or both hands, due to carpal ligament deposits (eg, in variant TTR L58H, normal-sequence TTR); symptoms of localized carpal ligament deposition sometimes precede other clinical manifestations by as long as 20 years.
Digestive system
The digestive system is also called the gastrointestinal tract (or GI tract). Amyloid deposits in the digestive system can affect the nerves that control intestinal muscle contractions causing nausea, diarrhea or constipation, and bladder control problems. Other symptoms may include weight loss, loss of appetite, or a feeling of fullness in the stomach after eating small amounts.
Kidneys
Amyloid deposits in the kidneys can affect how they filter toxins and proteins in the blood. This may result in a condition called nephrotic syndrome, where there is excess protein in the urine and the lower legs can become swollen (also called “edema”). Swelling can affect the belly, arms, and lungs as well. In some cases, the amyloid deposits will cause the kidneys to lose the ability to purify the blood, which can lead to kidney failure; also known as “renal” failure. These patients may need dialysis to replace the function of the kidneys.
Leptomeningeal amyloidosis or cerebral amyloid angiopathy
Patients with rare TTR variants that cause central nervous system (CNS) disease may present with the following features:
- Nystagmus and pyramidal signs, with spastic paraparesis 39
- Seizures, subarachnoid hemorrhages, cerebrovascular attacks (ischemic strokes), dementia, in patients with leptomeningeal/cerebrovascular deposits 39
- Hearing loss, cerebellar ataxia, in patients with isolated leptomeningeal disease (rare) 40
Other symptoms
Swelling may develop and cause other symptoms as a result of the amyloid deposits. For example, patients may have carpal tunnel syndrome. This is when amyloid deposits in the wrist area squeeze and irritate the nerve, causing tingling and numbness in the fingers and thumb.
ATTR (transthyretin) amyloidosis diagnosis
First, a patient is tested to determine if they have amyloid proteins in their body. If amyloidosis is confirmed but the type is not clearly found in these tests, it will be important to do more tests to find the exact type and also to determine the variation of ATTR.
The main diagnostic testing for any amyloidosis disease includes blood tests, urine tests and biopsies. Some tests are only done once to confirm a diagnosis, while others may be repeated to monitor the disease and response to therapy.
Blood and urine tests will be done to help your doctor determine the diagnosis of amyloidosis. These tests can also help to show which organs are involved and how much damage they may have.
In addition, a tissue biopsy will be performed. This involves the removal of a small sample of tissue for lab examination. A tissue sample is essential to confirm the diagnosis and type of amyloidosis. A “Congo red stain” is put on the biopsy tissue and if the lab examiner then sees the light wave change to an apple green color (called “birefringence”) then amyloidosis is diagnosed.
With ATTR, after amyloidosis is confirmed and it is determined that there is transthyretin amyloid protein (via biopsy and Congo red staining in the lab), the protein needs to be identified by protein sequence analysis and DNA sequencing must be performed.
When scientists examine your blood for certain genetic markers it is called genome sequencing. A simple blood sample is sent to a lab and experts examine the DNA chains. If a certain condition is in question, then sections of the DNA chain will be checked for genetic markers of the condition or defect.
Since the hereditary amyloidosis variations affect individuals differently, it is important to establish which variation you have in order to identify a treatment plan that is tailored for your type of amyloidosis. The DNA sequence analysis of TTR identifies more than 99% of disease-causing mutations.
A person should never ignore any health problem. Early detection can be important with any disease and the more tests that are done, the more accurate the diagnosis.
ATTR (transthyretin) amyloidosis treatment
Today’s treatment plans are two-fold:
- Supportive treatment – treating your symptoms and organ damage
- Source treatment – slowing down, or stopping, the overproduction of amyloid at the source of the disease.
Supportive treatment
Supportive treatment is helpful for various symptoms, including peripheral neuropathy, autonomic neuropathy, and cardiac and kidney problems, and can change the quality of life for many people. There are several medications that can be prescribed to treat peripheral neuropathy, which can cause tingling or burning in some parts of the body. These medications can help with pain relief and nerve damage. If a patient has autonomic neuropathy, symptoms can vary, with common problems affecting blood pressure, heart rate, digestion, and perspiration, depending on the location of the damage to the nerves. Other gastrointestinal dysfunctions may require treatment for symptoms that include poor nutritional health, diarrhea or constipation, and nausea or vomiting. Doctors can prescribe medications to help with these symptoms to lessen the pain and the symptom itself.
Management of heart problems, heart failure, and kidney dialysis (when needed) make a significant improvement on a patient’s quality of life. Reversing any damage to the organs and other parts of the body is difficult to achieve. If treatment begins during the early onset of clinical symptoms, the overall success rate is higher, so early detection is essential.
Source treatment
For most ATTR variations, the liver is the main source of amyloid production. However, the liver itself is not affected by the disease in most cases and the amyloid burden causes damage in other parts of the body. A liver transplant is very helpful in reducing (or stopping) the amyloid deposits. It can stabilize or improve neurological symptoms as well as gastrointestinal problems (which can correct poor nutrition and overall health). However, the statistics vary as to who can benefit from these transplants, with the more common ATTR Val30Met having the highest success rate. The outcome of liver transplantation is largely dependent on the mutation that exists in the patient. In some cases, amyloid deposition does completely stop after transplantation, so research is ongoing in this area. For those patients with cardiac symptoms, studies have shown that heart problems may continue after a liver transplant. In some situations, a combined heart and liver transplant will help a patient with an ATTR variant that produces advanced cardiac problems. A retrospective analysis of the data obtained from the Familial Amyloidotic Polyneuropathy World Transplant Registry 41 suggested an early age of onset, short disease duration, and Val30Met mutation to be better predictors for survival. However, the progression of cardiomyopathy and neuropathy resulting from wild-type TTR deposition may occur even after liver transplantation, particularly in late-onset male patients, resulting in poor prognosis after liver transplantation in these patients 42.
Several medications have been approved by the Food and Drug Administration (FDA) for treating patients with ATTR amyloidosis. Other medications continue to be investigated. In 2018, two drugs were approved by the FDA for ATTR polyneuropathy of hereditary transthyretin-mediated (hATTR) amyloidosis in adults. The first was Patisiran (Onpattro) lipid complex injection, a first of its kind RNA interference therapeutic. This drug aims to silencing the gene expression. The second drug approved in 2018 is Inotersen (Tegsedi) which reduces the production of TTR protein through a once a week subcutaneous injection. In 2019, Tafamidis (Vyndamax and Vyndaqel) were approved by the FDA for ATTR cardiomyopathy. These drugs are for oral administration taken once daily. In clinical trials, new therapies include aiming to treat the root cause of the disease, destabilized and folded TTR, using monoclonal antibodies to specifically target and clear misfolded (toxic) form a the TTR amyloid protein, and using a drug designed to reduce the production of transthyretin (TTR protein) in all types of TTR amyloidosis. Another RNA interference therapeutic is also currently in clinical trial. Advances in other treatments are likely, with new studies and clinical trials currently in view. It is possible that ATTR can cause serious health complications, so it should not be taken lightly. However, do not assume that disability or severe health issues are stamped on your future. There are treatments available and research continues.
TTR silencers
These medications act on the liver to decrease the production of TTR. Two TTR silencers approved by the FDA to treat patients with the hereditary type of TTR who also have neuropathy.
- Patisiran (Onpattro®) is an infusion that is given every three weeks.
- Inotersen (Tegsedi®) is an injection given once a week and requires weekly lab work.
Theoretically, preventing the production of TTR efficiently ameliorates systemic organ dysfunction in ATTR amyloidosis because it not only prevents amyloid fibril formation but also suppresses an increase in toxic amyloid precursors, such as TTR oligomers 43. As described earlier, TTR seems to exert harmful effects even when fibrillar structures recognized as amyloid fibrils are not formed. Given that circulating variant TTR may induce microangiopathy, which plays a role as an initial lesion of organ damage 44, a strategy that eliminates circulating TTR is more reasonable than liver transplantation and TTR stabilizers 43. This strategy became a reality with the development of gene-silencing therapeutics, including small interfering RNA (siRNA) and antisense oligonucleotide 4. In 2018, two randomized controlled trials of such gene-silencing agents (patisiran and inotersen) demonstrated an efficacy on neuropathy in patients with hereditary (familial) ATTR amyloidosis 45, 46.
Patisiran is an RNA interference therapeutic comprising siRNA formulated in a lipid nanoparticle that is predominantly delivered to the liver and reduces TTR production 4. In a phase III trial, 225 patients with hereditary (familial) ATTR amyloidosis with polyneuropathy were randomly assigned in a 2:1 ratio to receive patisiran intravenously (0.3 mg/kg of body weight) or placebo once every 3 weeks 45. The results of this study were excellent because all endpoints, including the scores related to somatic and autonomic neuropathies, quality of life score, and exploratory cardiac measures, were better in patients who received patisiran than in those who received placebo. In particular, even an improvement of primary outcome measures represented by neuropathy impairment and quality of life scores was seen in more than half of the patients receiving patisiran.
Inotersen is a second-generation antisense oligonucleotide designed to reduce the production of TTR 47. Parenterally administered antisense oligonucleotide, in general, is rapidly transferred into various organs, with the highest concentration in the liver and kidneys 48. A phase III trial involving 172 patients with hereditary (familial) ATTR amyloidosis who were randomly assigned in a 2:1 ratio to receive weekly subcutaneous injections of inotersen (300 mg) or placebo for 15 months demonstrated significantly better primary endpoints represented by neuropathy impairment and quality of life scores 46. Because glomerulonephritis and thrombocytopenia were reported as severe adverse events, close monitoring of renal function and platelet count is required in patients receiving inotersen.
TTR stabilizers
These medications stabilize the TTR protein, which in turn prevents it from breaking apart and forming amyloid fibrils.
- Tafamidis (Vyndamax®, Vyndaqel®) is approved by the FDA for patients with hereditary or wild-type ATTR that has affected their heart.
- AG10 is a medication currently being tested in a clinical trial.
- Diflunsial (Dolobid®) is a nonsteroidal anti-inflammatory drug (NSAID) that has been shown to also stabilize the TTR protein. However, this medication has not been fully studied in patients with ATTR that has affected the heart and also may not be tolerated due to side effects.
As the dissociation of TTR tetramers into monomers is the crucial step for the subsequent process of protein misfolding and amyloid fibril formation 4, an approach to stabilize the native quaternary structure of TTR tetramers using small molecules that bind to thyroxin-binding sites has been proposed as a potential approach for the treatment of not only hereditary (familial) ATTR amyloidosis but also wild-type ATTR amyloidosis (ATTRwt amyloidosis) 49. In the early 2010s, randomized controlled trials suggested the efficacy of two orally administered TTR stabilizers (i.e., tafamidis and diflunisal) for ameliorating the progression of neuropathy in patients with hereditary (familial) ATTR amyloidosis 50. As these TTR-stabilizing drugs can be administered orally, patients with late-onset hereditary (familial) ATTR amyloidosis who were not eligible for liver transplantation also became targets for disease-modifying treatment. Another recent randomized controlled trial suggested the efficacy of tafamidis even for cardiomyopathy resulting from both hereditary (familial) ATTR amyloidosis and wild-type ATTR amyloidosis (ATTRwt amyloidosis) 51.
Tafamidis is an analogue of thyroxine designed to stabilize TTR tetramers 52. A phase III clinical trial involving 128 patients with early-stage Val30Met ATTR amyloidosis who were randomly assigned in a 1:1 ratio to receive tafamidis (tafamidis meglumine) 20 mg once daily or placebo for 18 months suggested that tafamidis delayed the progression of neuropathy, although the primary endpoint could not be achieved 53. An open-label extension study for up to 6 years also demonstrated the slowing of neuropathy progression without any unexpected adverse events 54. In particular, patients who continued to receive tafamidis had less progression of neuropathy than those who switched to tafamidis following 18 months of placebo, warranting the need for early intervention 54. In addition to its efficacy on neuropathy, the efficacy of tafamidis on cardiomyopathy due to not only hereditary (familial) ATTR amyloidosis but also wild-type ATTR amyloidosis (ATTRwt amyloidosis) was suggested by a recent phase III clinical trial involving 441 patients with ATTR amyloidosis 51. This study included 335 patients with ATTRwt amyloidosis and 106 patients with hereditary (familial) ATTR amyloidosis who were randomly assigned in a 2:1:2 ratio to receive 80 mg of tafamidis, 20 mg of tafamidis, or placebo for 30 months and demonstrated reduced mortality and cardiovascular-related hospitalizations.
Diflunisal is a nonsteroidal anti-inflammatory drug that also acts to stabilize TTR 49. A study involving 130 patients with hereditary (familial) ATTR amyloidosis who were randomly assigned in a 1:1 ratio to receive 250 mg of diflunisal twice daily or placebo for 2 years suggested that diflunisal can slow the progression of neuropathy, although 67 patients (27 diflunisal patients and 40 placebo patients) discontinued the treatment before completing the 2-year protocol 50. The demographics of patients in this study were different from those in the phase III trial of tafamidis for neuropathy in patients with hereditary (familial) ATTR amyloidosis 53, because it included patients with relatively late disease onset, various disease severities, and non-Val30Met mutation.
Fibril disruptors
These medications may help break up and clear ATTR amyloid fibrils. Doxycycline (antibiotic) and green tea extract (over-the-counter supplement)have only been tested in small studies and there is limited evidence that these medications would be helpful in treating amyloidosis.
An antibody that removes TTR amylod fibrils, called PRX-004, is being tested in clinical trials.
Familial amyloidosis causes
Familial transthyretin amyloidosis is caused by changes (mutations) in the TTR gene. The TTR gene is responsible for making a protein called transthyretin (TTR) which transports vitamin A (retinol) and a hormone called thyroxine to many parts of the body. To transport thyroxine, four transthyretin proteins must be attached (bound) to each other to form a four-protein unit (tetramer). To transport retinol, transthyretin must form a tetramer and also bind to retinol binding protein. Transthyretin is produced primarily in the liver. A small amount of this protein is produced in an area of the brain called the choroid plexus and in the light-sensitive tissue that lines the back of the eye (the retina). Mutations in TTR lead to a transthyretin protein that is not made correctly. The faulty protein then folds up to form amyloid. Amyloid builds up in various parts of the body causing nerve and tissue damage 55. Currently, >150 mutations of the TTR gene are known; most of them are amyloidogenic, producing a broad range of phenotypes associated with familial amyloid polyneuropathy 56. The original mutation described Val30Met (alternatively named pVal50Met) is the most common worldwide and is primarily associated with neuropathy 57.
Most people who have familial amyloid polyneuropathy have inherited the TTR mutation from a family member. However, a few people with familial amyloid polyneuropathy will have no family history of the disease and have a new (de novo) mutation in the TTR gene 55. Not all people who have a TTR gene mutation will develop transthyretin amyloidosis.
Familial amyloidosis inheritance pattern
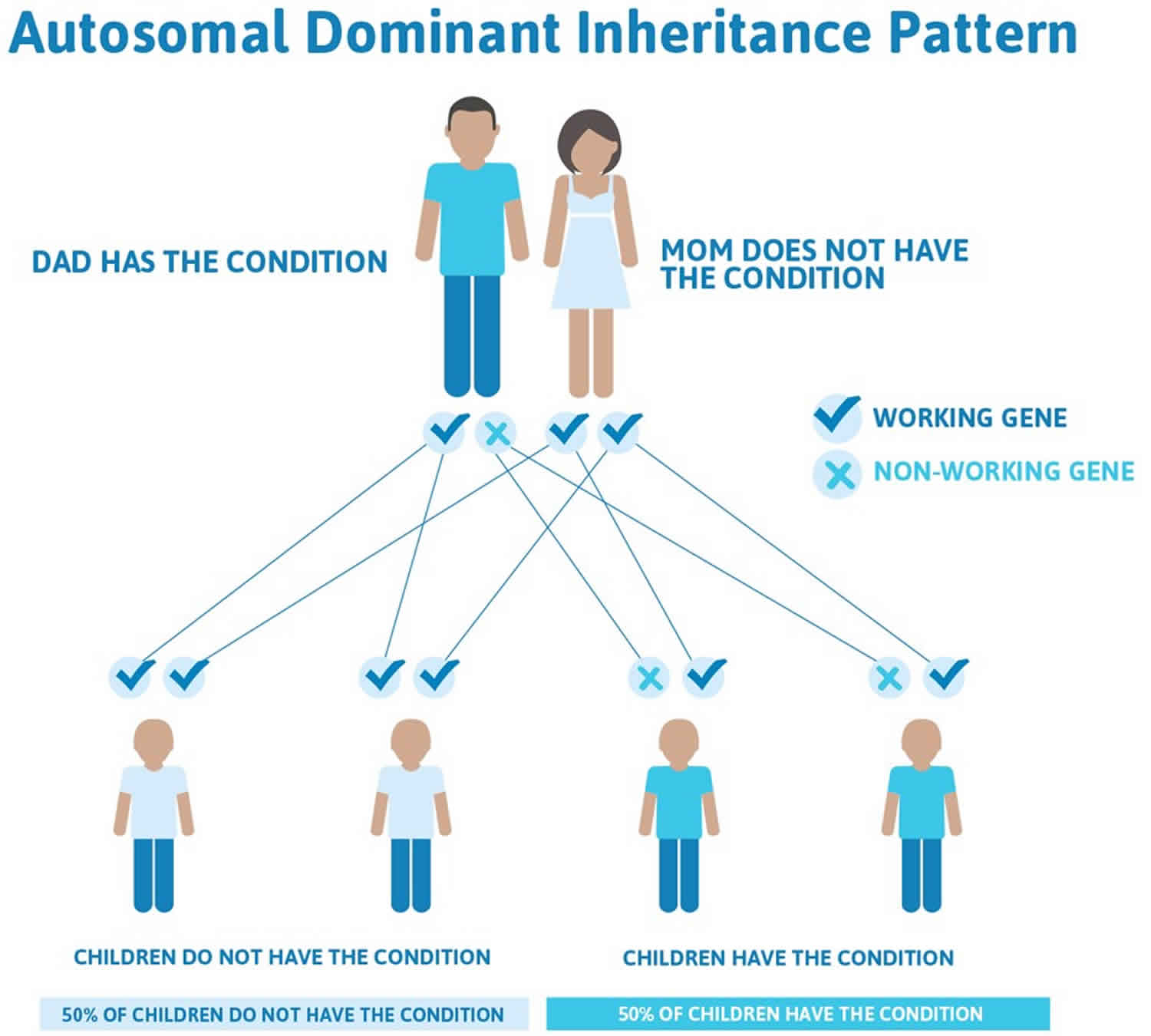
Familial amyloidosis is inherited in an autosomal dominant manner. This means that one altered copy of the disease-causing gene (called a mutation) in each cell is sufficient to cause the disease. The disease-causing mutation can be inherited from a parent or it can occur for the first time in an individual. Each child of an individual affected with familial amyloidosis has a 50% (1 in 2) risk to inherit the disease-causing mutation and a 50% chance of not inheriting the mutation. However, not all individuals with a mutation in a gene that causes familial amyloidosis will develop signs and symptoms of the disease.
Often autosomal dominant conditions can be seen in multiple generations within the family. If one looks back through their family history they notice their mother, grandfather, aunt/uncle, etc., all had the same condition. In cases where the autosomal dominant condition does run in the family, the chance for an affected person to have a child with the same condition is 50% regardless of whether it is a boy or a girl. These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
- When one parent has the abnormal gene, they will pass on either their normal gene or their abnormal gene to their child. Each of their children therefore has a 50% (1 in 2) chance of inheriting the changed gene and being affected by the condition.
- There is also a 50% (1 in 2) chance that a child will inherit the normal copy of the gene. If this happens the child will not be affected by the disorder and cannot pass it on to any of his or her children.
There are cases of autosomal dominant gene changes, or mutations, where no one in the family has it before and it appears to be a new thing in the family. This is called a de novo mutation. For the individual with the condition, the chance of their children inheriting it will be 50%. However, other family members are generally not likely to be at increased risk.
Figure 1 illustrates autosomal dominant inheritance. The example below shows what happens when dad has the condition, but the chances of having a child with the condition would be the same if mom had the condition.
Figure 1. Familial amyloidosis autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Familial amyloidosis symptoms
ATTR symptoms
Symptoms of ATTR vary, depending on the TTR genetic variant that is involved and the organ (or multiple organs) that demonstrate signs of amyloid deposition. The most common sites of amyloid deposits are associated with cardiac and/or nerve involvement (called cardiomyopathy and neuropathy) and the gastrointestinal tract. However, the most common form of familial amyloidosis affects the peripheral nervous system. The peripheral nerves send messages from the brain and spinal cord to the rest of the body. The kidneys, eyes, and carpal ligament (also known as carpal tunnel syndrome) are among other possibilities that can be affected.
For each patient, the symptoms will depend on which organs are affected by the amyloid deposits. It also depends on the degree that the organ function is impaired.
Symptoms of familial amyloidosis may include 58:
- Weakness, numbness or pain in the lower legs and feet
Carpal tunnel syndrome in both wrists
Sexual impotence
Urinary problems, protein in the urine
Diarrhea or constipation
Unexplained weight loss
Dry eyes, increased pressure in the eyes (glaucoma), seeing ‘floaters’
Abnormal heart beat, enlarged heart
Getting dizzy when moving from sitting to standing (orthostatic hypotension)
Dry eyes and mouth
Later symptoms may include muscle weakness and stiffness, difficulty with coordination, stroke, seizures, dementia, and congestive heart failure 37.
Less common symptoms include skin changes, hearing loss, shortness of breath, and anemia 59.
Heart symptoms
When amyloid deposits cause cardiomyopathy, it can result in a stiffening of the heart. Some patients may experience:
- Nausea
- Weight loss
- Inability to sleep
- Increasing fatigue
- Dizziness
- Shortness of breath
- Leg swelling (edema)
- Palpitations and abnormal heart rhythms (atrial fibrillation)
- Chest pain.
Congestive heart failure and atrial fibrillation are the most common symptoms. The term “arrhythmia” refers to changes in the normal electrical impulses that cause the heart to beat. The result is a heart that can beat too fast, too slow or erratically. Atrial fibrillation (or a-fib for short) is one of the many forms of arrhythmia. During a-fib, the heart’s two small upper chambers cause an abnormal heart rhythm, usually rapid and irregular beating. This may result in increased heart damage, stroke or heart failure.
Nervous system symptoms
The term neuropathy means nerve damage.
- Peripheral neuropathy: The following ATTR symptoms are caused by a condition known as peripheral neuropathy. Peripheral neuropathy (PN) can be caused by inflammation of, or damage to, the nerves. It can result in tingling, numbness and burning pain in any part of the body, but commonly is felt in the hands, feet and lower legs. Some patients may experience an increased sensitivity to pain. A loss of sensitivity to temperature may also occur. Sensorimotor impairment means the loss of a combination of sensory and motor activities. This can decrease a patient’s ability to move and feel (sensation) because of nerve damage. Restless leg syndrome (RLS) is also considered a sensorimotor disorder.
- Autonomic neuropathy: Autonomic neuropathy is a condition that results from damage to nerves that assist in organ and organ system functioning. Autonomic nerves control the functions of our internal organs such as the heart, stomach and intestines, as well as the glands. Nerves affected by ATTR amyloid deposits may cause the inability to control the muscles that expand or contract blood vessels, which affects the heart rate (irregular heart beats) and blood pressure. If a patient has a sudden drop in blood pressure (such as when moving from a seated to a standing position), then dizziness, fainting, or lightheadedness may occur. Other body functions may also be affected, including perspiration patterns, poor digestion, bowel motility and erectile function.
Digestive system symptoms
The digestive system is also called the gastrointestinal tract (or GI tract). Amyloid deposits in the digestive system can affect the nerves that control intestinal muscle contractions causing nausea, diarrhea or constipation, and bladder control problems. Other symptoms may include weight loss, loss of appetite, or a feeling of fullness in the stomach after eating small amounts.
Kidney symptoms
Amyloid deposits in the kidneys can affect how they filter toxins and proteins in the blood. This may result in a condition called nephrotic syndrome, where there is excess protein in the urine and the lower legs can become swollen (also called “edema”). Swelling can affect the belly, arms, and lungs as well. In some cases, the amyloid deposits will cause the kidneys to lose the ability to purify the blood, which can lead to kidney failure; also known as “renal” failure. These patients may need dialysis to replace the function of the kidneys.
Other symptoms
Swelling may develop and cause other symptoms as a result of the amyloid deposits. For example, patients may have carpal tunnel syndrome. This is when amyloid deposits in the wrist area squeeze and irritate the nerve, causing tingling and numbness in the fingers and thumb.
Non-TTR symptoms
Non-TTR symptoms can vary with each mutation, including problems with the kidneys, liver, heart or peripheral neuropathy. Some mutations have symptoms that involve the brain or the eye.
Each individual can present with different clinical symptoms. The symptoms, as well as the prognosis, depend on the tissue and organ(s) affected by the amyloid deposits. Although all the hereditary amyloidoses may cause serious complications for some individuals, there are some carriers of genetic mutations that may not show symptoms of the disease at all. Others may have a few, more minor, health issues.
Like ATTR, there are numerous variations within the protein identification.
- Apolipoprotein AI – 22 known variations with differing symptoms, including amyloid deposit involvement in the kidneys, liver, heart, peripheral neuropathy, cutaneous (skin) areas, and laryngeal (larynx).
- Fibrinogen Aa – 14 variations with symptoms that only involve the kidneys, with 1 also involving neuropathy.
- Lysozyme – 7 variations that involve kidney and/or liver symptoms, with 1 also involving the GI tract.
- Apolipoprotein AII – 5 variations that involve kidney symptoms only.
- Gelsolin – 4 variations with peripheral neuropathy symptoms, 1 also involving the eye.
- Cystatin C – 1 variation with cerebral hemorrhage complications.
Familial amyloidosis diagnosis
In the case of familial amyloidosis, the existence of a family history or similar illness is of great assistance in diagnosing the condition. However, not everyone with a mutation in a gene associated with hereditary amyloidosis will develop symptoms. Additionally, symptoms of the disease typically do not appear until older age and the condition may have been misdiagnosed in other affected family members. For these reasons, the absence of a family history may be misleading 1.
The diagnosis of amyloidosis is usually made by performing a tissue biopsy (the removal of a small piece of tissue) and staining the tissue with Congo red stain to detect the presence or absence of amyloid deposits. The biopsy may be from any affected organ, but biopsying the rectal mucosa generally results in better detection of the following familial amyloidosis: transthyretin amyloidosis, apolipoprotein AI amyloidosis, fibrinogen Aα-chain amyloidosis (A Fib), and apolipoprotein AII amyloidosis (A ApoAII) 1.
A bone scan called a technetium pyrophosphate (TcPYP) scan can detect ATTR in the heart. A positive TcPYP scan, along with blood and urine tests to rule out other forms of amyloidosis, can confirm the diagnosis without the need for a heart biopsy. When ATTR amyloidosis is confirmed, a blood test is used to find out if the ATTR is hereditary or wild-type.
Several other tests may be used to check organ function:
- Blood samples to check the kidneys, heart, and liver.
- An electrocardiogram (EKG) and echocardiogram (ultrasound of the heart) to check the heart.
- An MRI of the heart may also be done.
Additionally, when a familial amyloidosis is suspected, genetic testing may be able to confirm a diagnosis. It is important to note that genetic testing may not be available for all types of familial amyloidosis. For those individuals interested in pursuing genetic testing, it is recommended that you schedule a genetics consultation to determine whether genetic testing would be appropriate and available.
Familial amyloidosis treatment
There is no treatment available for familial transthyretin amyloidosis that reverses damage caused by amyloid deposits, but there are treatments that may prevent or delay progression 60. Treatment depends on which tissues are affected and how far the disease has progressed 59.
Liver transplantation is the “gold standard” for treatment for familial transthyretin amyloidosis, because it replaces the main source of amyloid. It may slow or halt progression of peripheral neuropathy, but the disease often still progresses in the eyes and brain. Liver transplantation ideally should be done as early as possible before there are severe neurological problems 60.
Several medications have been developed that slow the build-up of amyloid along nerves and in other parts of the body. These include tafamidis, diflunsial, and more recently inotersen and patisiran. There are other drugs that are currently under investigation for this condition 61.
Additional treatments may include heart and/or kidney transplantation, replacement of the liquid part of the eye (vitrectomy) for eye involvement, and carpal tunnel surgery 59.
Diuretics, medications that remove excess water and salt from the body, are often used to manage congestive heart failure associated with the disease. Other symptoms of FTA are treated as they arise 59.
The medication(s) listed below have been approved by the Food and Drug Administration (FDA) as orphan products for treatment of familial transthyretin amyloidosis.
- Patisiran (Brand name: Onpattro) – Manufactured by Alnylam Pharmaceuticals, Inc. Patisiran (Onpattro) was approved for the treatment of the polyneuropathy of hereditary transthyretin-mediated amyloidosis in adults.
- Inotersen (Brand name: Tegsedi) – Manufactured by Ionis Pharmaceuticals, Inc. Inotersen (Tegsedi) was approved for the treatment of the polyneuropathy of hereditary transthyretinmediated amyloidosis in adults.
ATTR (transthyretin) amyloidosis treatment
Today’s treatment plans are two-fold:
- Supportive treatment – treating your symptoms and organ damage
- Source treatment – slowing down, or stopping, the overproduction of amyloid at the source of the disease.
Supportive treatment
Supportive treatment is helpful for various symptoms, including peripheral neuropathy, autonomic neuropathy, and cardiac and kidney problems, and can change the quality of life for many people. There are several medications that can be prescribed to treat peripheral neuropathy, which can cause tingling or burning in some parts of the body. These medications can help with pain relief and nerve damage. If a patient has autonomic neuropathy, symptoms can vary, with common problems affecting blood pressure, heart rate, digestion, and perspiration, depending on the location of the damage to the nerves. Other gastrointestinal dysfunctions may require treatment for symptoms that include poor nutritional health, diarrhea or constipation, and nausea or vomiting. Doctors can prescribe medications to help with these symptoms to lessen the pain and the symptom itself.
Management of heart problems, heart failure, and kidney dialysis (when needed) make a significant improvement on a patient’s quality of life. Reversing any damage to the organs and other parts of the body is difficult to achieve. If treatment begins during the early onset of clinical symptoms, the overall success rate is higher, so early detection is essential.
Heart disease treatment
ATTR amyloid deposits in the heart cause the heart to stiffen which can lead to symptoms of heart failure. Patients can benefit from supportive treatment measures for heart failure. However many standard medications used for heart failure are not helpful for patients with cardiac amyloidosis. Careful attention to fluid balance is important.
Fluid balance
The most important principle of treatment for cardiac amyloidosis is strict fluid balance control. Specialist heart failure nurse involvement may help patients to achieve this. Many patients with ATTR cardiac amyloidosis should limit their fluid intake. This advice is extremely important, but is often overlooked.
When there is cardiac amyloidosis, the heart may be too stiff to pump the blood efficiently around the body. This can lead to fluid build- up, causing leg swelling (oedema) and breathlessness due to fluid in the lungs. This problem is exacerbated if the patient drinks too much fluid.
Fluid excess can be avoided by careful attention to the 3 Ds:
- Diet
- Fluid intake should be steady and should usually not exceed 1.5 liters per day.
- Salt intake should be limited. This includes attention not just to salt deliberately added to the food during cooking or at the table but also to ready prepared foods with high salt content such as processed foods, crisps, bacon, canned meats, sausages, canned soups and smoked fish. Apart from that, a balanced, healthy diet is always advisable. It can be very helpful to meet with a dietician for precise and personalised dietary advice.
- Diuretics (water tablets)
- Doctors will often prescribe diuretics (water tablets) which increase the amount of urine produced and help the body to lose excess salt and water in the urine. This can help to reduce ankle swelling and breathlessness. Diuretics prescribed may include furosemide and spironolactone. Taking these drugs is not a substitute for avoidance of excessive dietary salt and water. Patients should follow their doctor’s advice carefully regarding the dose of diuretic and the time of day when the tablet should be taken.
- Daily weights
- Some patients benefit from recording their weight regularly, usually daily or weekly. It is important that weight should be measured consistently – using the same scales, at the same time of day. This is usually best done first thing in the morning after passing urine, just wearing underclothes. Several litres of fluid can accumulate in the body without it being very noticeable. An increase in weight can be an early sign of fluid overload. The doctor or nurse can then recommend appropriate measures such as increased diuretic dose, before the patient even feels unwell because of the fluid overload.
Heart transplantation
For hereditary ATTR amyloidosis, combined heart and liver transplant has been performed in a few dozen cases around the world. This operation is only an option for a very small minority of patients, and it carries significant risks.
Treatment of peripheral neuropathy symptoms
Medications that may help to alleviate neuropathic pain include gabapentin, pregabalin and duloxetine. Medical staff can give advice regarding appropriate foot care and footwear. This is important in order to prevent painless ulcers at pressure points and to protect areas of the foot that lack sensation.
Treatment of autonomic neuropathy symptoms
If there is orthostatic hypotension (drops in blood pressure and faintness on standing up from sitting or lying positions), elastic stockings may be recommended. Patients may benefit from instruction in how to change position carefully from lying to sitting, sitting to standing and standing to walking. Drug treatment with midodrine or fludrocortisone may also be helpful to maintain blood pressure and allow higher diuretic doses. Care should be taken to avoid dehydration if there is vomiting and diarrhoea. Intravenous fluids and anti‑nausea drugs may be necessary, but it is important to avoid fluid overload if there is heart disease. There are drugs that can help to control diarrhoea and constipation, and others that can help to combat erectile dysfunction.
Dealing with postural hypotension:
- After sitting down for more than 1/2 hour always stand for about 30 seconds before moving off.
- If feeling light headed or you have ringing in your ears when walking, stand still for a few moments until it passes. Sometimes you can walk through it after some practice!
- If it gets really bad always ask for help or sit down.
- When you get a cold or virus the postural hypotension can get worse. It may help by taking the standard doses of acetaminophen (paracetamol).
- When getting up from bed in the morning, always sit on the side for a few moments before walking off.
- Never run and always pace yourself.
- Always allow more time than you think you need.
Source treatment
For most ATTR variations, the liver is the main source of amyloid production. However, the liver itself is not affected by the disease in most cases and the amyloid burden causes damage in other parts of the body. A liver transplant is very helpful in reducing (or stopping) the amyloid deposits. It can stabilize or improve neurological symptoms as well as gastrointestinal problems (which can correct poor nutrition and overall health). However, the statistics vary as to who can benefit from these transplants, with the more common ATTR Val30Met having the highest success rate. The outcome of liver transplantation is largely dependent on the mutation that exists in the patient. In some cases, amyloid deposition does completely stop after transplantation, so research is ongoing in this area. For those patients with cardiac symptoms, studies have shown that heart problems may continue after a liver transplant. In some situations, a combined heart and liver transplant will help a patient with an ATTR variant that produces advanced cardiac problems.
Several medications have been approved by the Food and Drug Administration (FDA) for treating patients with ATTR amyloidosis. Other medications continue to be investigated. In 2018, two drugs were approved by the FDA for ATTR polyneuropathy of hereditary transthyretin-mediated (hATTR) amyloidosis in adults. The first was Patisiran (Onpattro) lipid complex injection, a first of its kind RNA interference therapeutic. This drug aims to silencing the gene expression. The second drug approved in 2018 is Inotersen (Tegsedi) which reduces the production of TTR protein through a once a week subcutaneous injection. In 2019, Tafamidis (Vyndamax and Vyndaqel) were approved by the FDA for ATTR cardiomyopathy. These drugs are for oral administration taken once daily. In clinical trials, new therapies include aiming to treat the root cause of the disease, destabilized and folded TTR, using monoclonal antibodies to specifically target and clear misfolded (toxic) form a the TTR amyloid protein, and using a drug designed to reduce the production of transthyretin (TTR protein) in all types of TTR amyloidosis. Another RNA interference therapeutic is also currently in clinical trial. Advances in other treatments are likely, with new studies and clinical trials currently in view. It is possible that ATTR can cause serious health complications, so it should not be taken lightly. However, do not assume that disability or severe health issues are stamped on your future. There are treatments available and research continues.
ATTR silencers
These medications act on the liver to decrease the production of TTR. Two ATTR silencers approved by the FDA to treat patients with the hereditary type of ATTR who also have neuropathy.
- Patisiran (Onpattro®) is an infusion that is given every three weeks.
- Inotersen (Tegsedi®) is an injection given once a week and requires weekly lab work.
ATTR stabilizers
These medications stabilize the TTR protein, which in turn prevents it from breaking apart and forming amyloid fibrils.
- Tafamidis (Vyndamax®, Vyndaqel®) is approved by the FDA for patients with hereditary or wild-type ATTR that has affected their heart.
- AG10 is a medication currently being tested in a clinical trial.
- Diflunsial (Dolobid®) is a nonsteroidal anti-inflammatory drug (NSAID) that has been shown to also stabilize the TTR protein. However, this medication has not been fully studied in patients with ATTR that has affected the heart and also may not be tolerated due to side effects.
Fibril disruptors
These medications may help break up and clear ATTR amyloid fibrils. Doxycycline (antibiotic) and green tea extract (over-the-counter supplement)have only been tested in small studies and there is limited evidence that these medications would be helpful in treating amyloidosis.
An antibody that removes TTR amylod fibrils, called PRX-004, is being tested in clinical trials.
Non-ATTR amyloidosis treatment
Non-ATTR amyloidosis treatment include:
- Supportive treatment – treating your symptoms and organ damage; and,
- Source treatment – slowing down, or stopping, the overproduction of amyloid at the source of the disease.
Supportive treatment
Supportive treatment can vary with each mutation of the non-TTR diseases, however, many of them present with kidney or heart damage, so organ transplantation has been used in these cases with success. Although not a cure, and even if production of the variant amyloid protein continues, an organ transplant can slow the progression of the disease, improve quality of life and prolong survival significantly.
Source treatment
Liver transplants are less of a source treatment option for most of the Non-TTR amyloidosis diseases. However, Fibrinogen is one variation that has amyloid production occurring solely in the liver, so a liver transplant can slow, or stop, amyloid production at the source. Often, a kidney transplant is also performed because kidney damage is the main target of this disease. So, transplants of both these organs can provide a successful supportive (kidney), and source (liver), treatment in this Fibrinogen variation.
Lysozyme, Apo lipoprotein A-I and AII, along with the other Non-TTR variations, are so varied that you should consult with a specialist at an amyloid center to recommend a treatment for your individual needs. Source treatments for the hereditary Non-TTR amyloidosis variants are not as advanced as they are for ATTR. These types are more rare and seem to progress slowly.
Current organ transplants and other treatments include:
- Apolipoprotein A1
- Supportive Treatment Possibilities (affected organ) – kidney and/or heart transplant, depending on organ damage
- Source Treatment (to reduce amyloid production) – liver transplant
- Fibrinogen Aa
- Supportive Treatment Possibilities (affected organ) – kidney transplant
- Source Treatment (to reduce amyloid production) – liver transplant
- Lysozyme
- Supportive Treatment Possibilities (affected organ) – kidney and/or liver transplant, depending on organ damage
- Source Treatment (to reduce amyloid production) – none at this time
- Apolipoprotein A2
- Supportive Treatment Possibilities (affected organ) – kidney transplant
- Source Treatment (to reduce amyloid production) – none at this time
- Gelsolin
- Supportive Treatment Possibilities (affected organ) – cornea transplant
- Source Treatment (to reduce amyloid production) – none at this time
- Cystatin C
- Supportive Treatment Possibilities – avoid fever
- Source Treatment (to reduce amyloid production) – none at this time
Familial amyloidosis prognosis
The prognosis and life expectancy for each person with familial transthyretin amyloidosis (hereditary transthyretin amyloidosis) varies and depends on the TTR gene mutation present, organ(s) involved, and how early a person is diagnosed and treated. Some people whose symptoms begin at a younger age may live for only a few years after diagnosis, while older patients with slowly progressive disease can live for decades after the onset of symptoms and may never develop life-threatening disease 62. Life expectancy on average of people with familial amyloid polyneuropathy typically live for 7-12 years after they are first diagnosed 63. Death is most often due to cardiac dysfunction, infection, or cachexia 64.
The long-term outlook after liver transplant is also influenced by many factors, including the type of amyloid present, nutritional status, age, and how much the brain and heart are involved 56. Newer medications are now available that help slow the build-up of amyloid and delay symptoms and it is not yet clear how these medications will affect the long-term outlook for people with familial amyloid polyneuropathy. People with questions about their personal outlook should speak with their healthcare providers.
Familial amyloid polyneuropathy prognosis
The natural course of familial amyloid polyneuropathy can be classified into the following three stages 65:
- Stage 1 – Sensory polyneuropathy
- Stage 2 – Progressive walking disability
- Stage 3 – Wheelchair bound or bedridden
The prognosis depends on the presence and identity of a TTR variant and the organ(s) involved. Patients with early-onset of variant-sequence TTR may die within a few years of diagnosis. Older patients with slowly progressive disease can live for decades after the onset of symptoms and may never develop life-threatening disease 62.
Penetrance of the individual TTR mutations vary. The penetrance of the same mutation in different geographic areas can also vary, for example, the Portuguese population showing much higher penetrance of the Val30Met mutation during middle age (80% at 50 years) compared with the French population (18% at 50 years) 63.
In contrast to light chain amyloidosis (AL), symptomatic cardiac involvement in familial amyloid polyneuropathy does not necessarily portend a poor prognosis. Median survival in cardiac light chain amyloidosis is about 6 months, but is several years in older patients with cardiac familial amyloid polyneuropathy, even in those with a TTR variant. Familial amyloid polyneuropathy usually proves fatal within 7–12 years from the onset of symptoms, most often due to cardiac dysfunction, infection, or cachexia 64.
Within most of the regions in which it is endemic, clinical onset of familial amyloid polyneuropathy often occurs before age 40 years with progressive sensory-motor and autonomic neuropathy, leading to cachexia and eventually death. Length-dependent small-fiber sensory and motor polyneuropathy with life-threatening autonomic dysfunction is a distinguishing feature of familial amyloid polyneuropathy in these areas. In addition, cardiac, renal, and ocular involvement are also common 63.
In nonendemic areas, and in endemic regions of Sweden, the onset of disease-related symptoms tends to be later in life, from age 50 years onward and with a male predominance for the late-onset familial amyloid polyneuropathy. Neuropathy tends to affect all fibers and may closely resemble chronic inflammatory demyelinating polyneuropathy (CIDP). Typically, sensory and motor neuropathy symptoms of upper and lower extremities occur, associated with mild autonomic symptoms 63.
References- Benson MD. The hereditary amyloidoses. Best Pract Res Clin Rheumatol. 2003 Dec;17(6):909-27. doi: 10.1016/j.berh.2003.09.001
- Hereditary amyloidosis. https://medlineplus.gov/ency/article/000368.htm
- Rowczenio DM, Noor I, Gillmore JD, Lachmann HJ, Whelan C, Hawkins PN, Obici L, Westermark P, Grateau G, Wechalekar AD. Online Registry for Mutations in Hereditary Amyloidosis Including Nomenclature Recommendations. Hum Mutat. Sep 2014; 35(9):E2402-12. http://www.ncbi.nlm.nih.gov/pubmed/25044787
- Adams D, Koike H, Slama M, Coelho T. Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol. 2019 Jul;15(7):387-404. doi: 10.1038/s41582-019-0210-4
- Hereditary Amyloidosis. http://amyloidosis.org/facts/familial
- Ikeda S, Nakazato M, Ando Y, Sobue G: Familial transthyretin-type amyloid polyneuropathy in Japan: clinical and genetic heterogeneity. Neurology. 2002, 58: 1001-1007. 10.1212/WNL.58.7.1001
- Holmgren G, Costa PM, Andersson C, Asplund K, Steen L, Beckman L, Nylander PO, Teixeira A, Saraiva MJ, Costa PP: Geographical distribution of TTR met30 carriers in northern Sweden: discrepancy between carrier frequency and prevalence rate. J Med Genet. 1994, 31: 351-354. 10.1136/jmg.31.5.351
- Benson M: Amyloidosis. The Metabolic and Molecular Bases of Inherited Disease. Edited by: Scriver CR, Beaudet AL, Valle D, Sly WS, Childs B, Kinzler KW, Vogelstein B. New York: McGraw-Hill; 2000:5345-5378.
- Prevalence and incidence of rare diseases: Bibliographic data. https://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf
- Yamashita T, Hamidi Asl K, Yazaki M, Benson MD: A prospective evaluation of the transthyretin Ile122 allele frequency in an African-American population. Amyloid. 2005, 12: 127-130. 10.1080/13506120500107162
- Dharmarajan K, Maurer MS: Transthyretin Cardiac Amyloidoses in Older North Americans. J Am Geriatr Soc. 2012, 60: 765-774. 10.1111/j.1532-5415.2011.03868.x
- Zaros C, Genin E, Hellman U, Saporta MA, Languille L, Wadington-Cruz M, Suhr O, Misrahi M, Planté-Bordeneuve V. On the origin of the transthyretin Val30Met familial amyloid polyneuropathy. Ann Hum Genet. 2008 Jul;72(Pt 4):478-84. doi: 10.1111/j.1469-1809.2008.00439.x
- Conceição I, De Carvalho M. Clinical variability in type I familial amyloid polyneuropathy (Val30Met): comparison between late- and early-onset cases in Portugal. Muscle Nerve. 2007 Jan;35(1):116-8. doi: 10.1002/mus.20644
- Hellman U, Alarcon F, Lundgren HE, Suhr OB, Bonaiti-Pellie C, Plante-Bordeneuve V: Heterogeneity of penetrance in familial amyloid polyneuropathy, ATTR Val30Met, in the Swedish population. Amyloid. 2008, 15: 181-186. 10.1080/13506120802193720
- Planté-Bordeneuve V, Carayol J, Ferreira A, Adams D, Clerget-Darpoux F, Misrahi M, Said G, Bonaiti-Pellie C: Genetic study of transthyretin amyloid neuropathies: carrier risks among French and Portuguese families. J Med Genet. 2003, 40: e120-10.1136/jmg.40.11.e120
- Kato-Motozaki Y, Ono K, Shima K, Morinaga A, Machiya T, Nozaki I, Shibata-Hamaguchi A, Furukawa Y, Yanase D, Ishida C, et al: Epidemiology of familial amyloid polyneuropathy in Japan: Identification of a novel endemic focus. J Neurol Sci. 2008, 270: 133-140. 10.1016/j.jns.2008.02.019
- Benson MD, Buxbaum JN, Eisenberg DS, Merlini G, Saraiva MJM, Sekijima Y, Sipe JD, Westermark P. Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid. 2018 Dec;25(4):215-219. doi: 10.1080/13506129.2018.1549825 https://www.tandfonline.com/doi/full/10.1080/13506129.2018.1549825
- Cornwell GG 3rd, Murdoch WL, Kyle RA, Westermark P, Pitkänen P. Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am J Med. 1983 Oct;75(4):618-23. doi: 10.1016/0002-9343(83)90443-6
- Ueda M, Horibata Y, Shono M, Misumi Y, Oshima T, Su Y, Tasaki M, Shinriki S, Kawahara S, Jono H, Obayashi K, Ogawa H, Ando Y. Clinicopathological features of senile systemic amyloidosis: an ante- and post-mortem study. Mod Pathol. 2011 Dec;24(12):1533-44. doi: 10.1038/modpathol.2011.117
- Griffin JM, Maurer MS. Transthyretin cardiac amyloidosis: A treatable form of heart failure with a preserved ejection fraction. Trends Cardiovasc Med. 2019 Dec 17:S1050-1738(19)30166-5. doi: 10.1016/j.tcm.2019.12.003
- Sekijima Y, Yazaki M, Ueda M, Koike H, Yamada M, Ando Y. First nationwide survey on systemic wild-type ATTR amyloidosis in Japan. Amyloid. 2018 Mar;25(1):8-10. doi: 10.1080/13506129.2017.1409706
- Sekijima Y, Uchiyama S, Tojo K, Sano K, Shimizu Y, Imaeda T, Hoshii Y, Kato H, Ikeda S. High prevalence of wild-type transthyretin deposition in patients with idiopathic carpal tunnel syndrome: a common cause of carpal tunnel syndrome in the elderly. Hum Pathol. 2011 Nov;42(11):1785-91. doi: 10.1016/j.humpath.2011.03.004
- Yanagisawa A, Ueda M, Sueyoshi T, Okada T, Fujimoto T, Ogi Y, Kitagawa K, Tasaki M, Misumi Y, Oshima T, Jono H, Obayashi K, Hirakawa K, Uchida H, Westermark P, Ando Y, Mizuta H. Amyloid deposits derived from transthyretin in the ligamentum flavum as related to lumbar spinal canal stenosis. Mod Pathol. 2015 Feb;28(2):201-7. doi: 10.1038/modpathol.2014.102
- Marcoux J, Mangione PP, Porcari R, Degiacomi MT, Verona G, Taylor GW, Giorgetti S, Raimondi S, Sanglier-Cianférani S, Benesch JL, Cecconi C, Naqvi MM, Gillmore JD, Hawkins PN, Stoppini M, Robinson CV, Pepys MB, Bellotti V. A novel mechano-enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Mol Med. 2015 Oct;7(10):1337-49. doi: 10.15252/emmm.201505357
- Pinto MV, Milone M, Mauermann ML, Dyck PJB, Alhammad R, McPhail ED, Grogan M, Liewluck T. Transthyretin amyloidosis: Putting myopathy on the map. Muscle Nerve. 2020 Jan;61(1):95-100. doi: 10.1002/mus.26723
- Koike H, Katsuno M. Expanding the spectrum of transthyretin amyloidosis. Muscle Nerve. 2020 Jan;61(1):3-4. doi: 10.1002/mus.26741
- Stangou AJ, Heaton ND, Rela M, Pepys MB, Hawkins PN, Williams R. Domino hepatic transplantation using the liver from a patient with familial amyloid polyneuropathy. Transplantation. 1998 Jun 15;65(11):1496-8. doi: 10.1097/00007890-199806150-00016
- Holmgren G, Steen L, Ekstedt J, Groth CG, Ericzon BG, Eriksson S, Andersen O, Karlberg I, Nordén G, Nakazato M, et al. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30). Clin Genet. 1991 Sep;40(3):242-6. doi: 10.1111/j.1399-0004.1991.tb03085.x
- Misumi Y, Ueda M, Masuda T, Tsuda Y, Nomura T, Okada M, Inoue Y, Tasaki M, Obayashi K, Yamashita T, Ando Y. Characteristics of acquired transthyretin amyloidosis: A case series and review of the literature. Neurology. 2019 Oct 22;93(17):e1587-e1596. doi: 10.1212/WNL.0000000000008360
- Koike H, Katsuno M. Transthyretin Amyloidosis: Update on the Clinical Spectrum, Pathogenesis, and Disease-Modifying Therapies. Neurol Ther. 2020;9(2):317-333. doi:10.1007/s40120-020-00210-7 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7500251
- Koike H, Kiuchi T, Iijima M, Ueda M, Ando Y, Morozumi S, Tomita M, Kawagashira Y, Watanabe H, Katsuno M, Shimoyama Y, Okazaki Y, Kamei H, Sobue G. Systemic but asymptomatic transthyretin amyloidosis 8 years after domino liver transplantation. J Neurol Neurosurg Psychiatry. 2011 Nov;82(11):1287-90. doi: 10.1136/jnnp.2010.218958
- Barreiros AP, Geber C, Birklein F, Galle PR, Otto G. Clinical symptomatic de novo systemic transthyretin amyloidosis 9 years after domino liver transplantation. Liver Transpl. 2010 Jan;16(1):109. doi: 10.1002/lt.21928
- Koike H, Misu K, Sugiura M, Iijima M, Mori K, Yamamoto M, Hattori N, Mukai E, Ando Y, Ikeda S, Sobue G. Pathology of early- vs late-onset TTR Met30 familial amyloid polyneuropathy. Neurology. 2004 Jul 13;63(1):129-38. doi: 10.1212/01.wnl.0000132966.36437.12
- Koike H, Sobue G. What is the prototype of familial amyloid polyneuropathy? J Neurol Neurosurg Psychiatry. 2014 Jul;85(7):713. doi: 10.1136/jnnp-2013-306241
- Ando Y, Nakamura M, Araki S. Transthyretin-related familial amyloidotic polyneuropathy. Arch Neurol. 2005 Jul. 62(7):1057-62.
- Vita G, Mazzeo A, Di Leo R, Ferlini A. Recurrent syncope as persistently isolated feature of transthyretin amyloidotic polyneuropathy. Neuromuscul Disord. 2005 Mar;15(3):259-61. doi: 10.1016/j.nmd.2004.10.015
- Sekijima Y. Hereditary Transthyretin Amyloidosis. 2001 Nov 5 [Updated 2021 Jun 17]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2022. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1194
- Ando Y, Suhr OB. Autonomic dysfunction in familial amyloidotic polyneuropathy (FAP). Amyloid. 1998 Dec. 5(4):288-300.
- Plante-Bordeneuve V, Lalu T, Misrahi M, et al. Genotypic-phenotypic variations in a series of 65 patients with familial amyloid polyneuropathy. Neurology. 1998 Sep. 51(3):708-14.
- Hagiwara K, Ochi H, Suzuki S, et al. Highly selective leptomeningeal amyloidosis with transthyretin variant Ala25Thr. Neurology. April 2009. 72:1358-60.
- Ericzon BG, Wilczek HE, Larsson M, Wijayatunga P, Stangou A, Pena JR, Furtado E, Barroso E, Daniel J, Samuel D, Adam R, Karam V, Poterucha J, Lewis D, Ferraz-Neto BH, Cruz MW, Munar-Ques M, Fabregat J, Ikeda S, Ando Y, Heaton N, Otto G, Suhr O. Liver Transplantation for Hereditary Transthyretin Amyloidosis: After 20 Years Still the Best Therapeutic Alternative? Transplantation. 2015 Sep;99(9):1847-54. doi: 10.1097/TP.0000000000000574
- Koike H, Hashimoto R, Tomita M, Kawagashira Y, Iijima M, Nakamura T, Watanabe H, Kamei H, Kiuchi T, Sobue G. Impact of aging on the progression of neuropathy after liver transplantation in transthyretin Val30Met amyloidosis. Muscle Nerve. 2012 Dec;46(6):964-70. doi: 10.1002/mus.23480
- Koike H, Katsuno M. Ultrastructure in Transthyretin Amyloidosis: From Pathophysiology to Therapeutic Insights. Biomedicines. 2019 Feb 5;7(1):11. doi: 10.3390/biomedicines7010011
- Koike H, Ikeda S, Takahashi M, Kawagashira Y, Iijima M, Misumi Y, Ando Y, Ikeda SI, Katsuno M, Sobue G. Schwann cell and endothelial cell damage in transthyretin familial amyloid polyneuropathy. Neurology. 2016 Nov 22;87(21):2220-2229. doi: 10.1212/WNL.0000000000003362
- Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang CC, Ueda M, Kristen AV, Tournev I, Schmidt HH, Coelho T, Berk JL, Lin KP, Vita G, Attarian S, Planté-Bordeneuve V, Mezei MM, Campistol JM, Buades J, Brannagan TH 3rd, Kim BJ, Oh J, Parman Y, Sekijima Y, Hawkins PN, Solomon SD, Polydefkis M, Dyck PJ, Gandhi PJ, Goyal S, Chen J, Strahs AL, Nochur SV, Sweetser MT, Garg PP, Vaishnaw AK, Gollob JA, Suhr OB. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018 Jul 5;379(1):11-21. doi: 10.1056/NEJMoa1716153
- Benson MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, Planté-Bordeneuve V, Barroso FA, Merlini G, Obici L, Scheinberg M, Brannagan TH 3rd, Litchy WJ, Whelan C, Drachman BM, Adams D, Heitner SB, Conceição I, Schmidt HH, Vita G, Campistol JM, Gamez J, Gorevic PD, Gane E, Shah AM, Solomon SD, Monia BP, Hughes SG, Kwoh TJ, McEvoy BW, Jung SW, Baker BF, Ackermann EJ, Gertz MA, Coelho T. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018 Jul 5;379(1):22-31. doi: 10.1056/NEJMoa1716793
- Benson MD, Kluve-Beckerman B, Zeldenrust SR, Siesky AM, Bodenmiller DM, Showalter AD, Sloop KW. Targeted suppression of an amyloidogenic transthyretin with antisense oligonucleotides. Muscle Nerve. 2006 May;33(5):609-18. doi: 10.1002/mus.20503
- Geary RS, Norris D, Yu R, Bennett CF. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv Drug Deliv Rev. 2015 Jun 29;87:46-51. doi: 10.1016/j.addr.2015.01.008
- Miller SR, Sekijima Y, Kelly JW. Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab Invest. 2004 May;84(5):545-52. doi: 10.1038/labinvest.3700059
- Berk JL, Suhr OB, Obici L, Sekijima Y, Zeldenrust SR, Yamashita T, Heneghan MA, Gorevic PD, Litchy WJ, Wiesman JF, Nordh E, Corato M, Lozza A, Cortese A, Robinson-Papp J, Colton T, Rybin DV, Bisbee AB, Ando Y, Ikeda S, Seldin DC, Merlini G, Skinner M, Kelly JW, Dyck PJ; Diflunisal Trial Consortium. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013 Dec 25;310(24):2658-67. doi: 10.1001/jama.2013.283815
- Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, Kristen AV, Grogan M, Witteles R, Damy T, Drachman BM, Shah SJ, Hanna M, Judge DP, Barsdorf AI, Huber P, Patterson TA, Riley S, Schumacher J, Stewart M, Sultan MB, Rapezzi C; ATTR-ACT Study Investigators. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med. 2018 Sep 13;379(11):1007-1016. doi: 10.1056/NEJMoa1805689
- Obici L, Adams D. Acquired and inherited amyloidosis: Knowledge driving patients’ care. J Peripher Nerv Syst. 2020 Jun;25(2):85-101. doi: 10.1111/jns.12381
- Coelho T, Maia LF, Martins da Silva A, Waddington Cruz M, Planté-Bordeneuve V, Lozeron P, Suhr OB, Campistol JM, Conceição IM, Schmidt HH, Trigo P, Kelly JW, Labaudinière R, Chan J, Packman J, Wilson A, Grogan DR. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012 Aug 21;79(8):785-92. doi: 10.1212/WNL.0b013e3182661eb1
- Barroso FA, Judge DP, Ebede B, Li H, Stewart M, Amass L, Sultan MB. Long-term safety and efficacy of tafamidis for the treatment of hereditary transthyretin amyloid polyneuropathy: results up to 6 years. Amyloid. 2017 Sep;24(3):194-204. doi: 10.1080/13506129.2017.1357545
- Sekijima Y. Hereditary Transthyretin Amyloidosis. 2001 Nov 5 [Updated 2018 Dec 20]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1194
- Plante-Bordeneuve V. Transthyretin familial amyloid polyneuropathy: an update. J Neurol. 2018 Apr;265(4):976-983. doi: 10.1007/s00415-017-8708-4
- Ando Y, Coelho T, Berk JL, Cruz MW, Ericzon BG, Ikeda S, Lewis WD, Obici L, Planté-Bordeneuve V, Rapezzi C, Said G, Salvi F. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013 Feb 20;8:31. doi: 10.1186/1750-1172-8-31
- Pinto MV, Barreira AA, Bulle AS, Freitas MRG, França MC Jr, Gondim FAA, Marrone CD, Marques W Jr, Nascimento OJM, Rotta FT, Pupe C, Waddington-Cruz M. Brazilian consensus for diagnosis, management and treatment of transthyretin familial amyloid polyneuropathy. Arq Neuropsiquiatr. 2018 Sep;76(9):609-621. doi: 10.1590/0004-282X20180094
- Ando, Y., Coelho, T., Berk, J.L. et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis 8, 31 (2013). https://doi.org/10.1186/1750-1172-8-31
- Transthyretin-Related Amyloidosis. https://emedicine.medscape.com/article/335301-overview
- Obici, L., Kuks, J. B., Buades, J., Adams, D., Suhr, O. B., Coelho, T., Kyriakides, T., & European Network for TTR-FAP (ATTReuNET) (2016). Recommendations for presymptomatic genetic testing and management of individuals at risk for hereditary transthyretin amyloidosis. Current opinion in neurology, 29 Suppl 1(Suppl 1), S27–S35. https://doi.org/10.1097/WCO.0000000000000290
- Adams D, Samuel D, Goulon-Goeau C, et al. The course and prognostic factors of familial amyloid polyneuropathy after liver transplantation. Brain. 2000 Jul. 123 (Pt 7):1495-504.
- Parman Y, Adams D, Obici L, et al. Sixty years of transthyretin familial amyloid polyneuropathy (TTR-FAP) in Europe: where are we now? A European network approach to defining the epidemiology and management patterns for TTR-FAP. Curr Opin Neurol. 2016. 29 (suppl 1):S000–S000.
- Adams D, Suhr OB, Hund E, Obici L, et al. First European consensus for diagnosis, management, and treatment of transthyretin familial amyloid polyneuropathy. Curr Opin Neurol. 2016. 29 (suppl1):S000-S000.
- Transthyretin-Related Amyloidosis. https://emedicine.medscape.com/article/335301-overview#a3





{kind=link}