Frontotemporal dementia
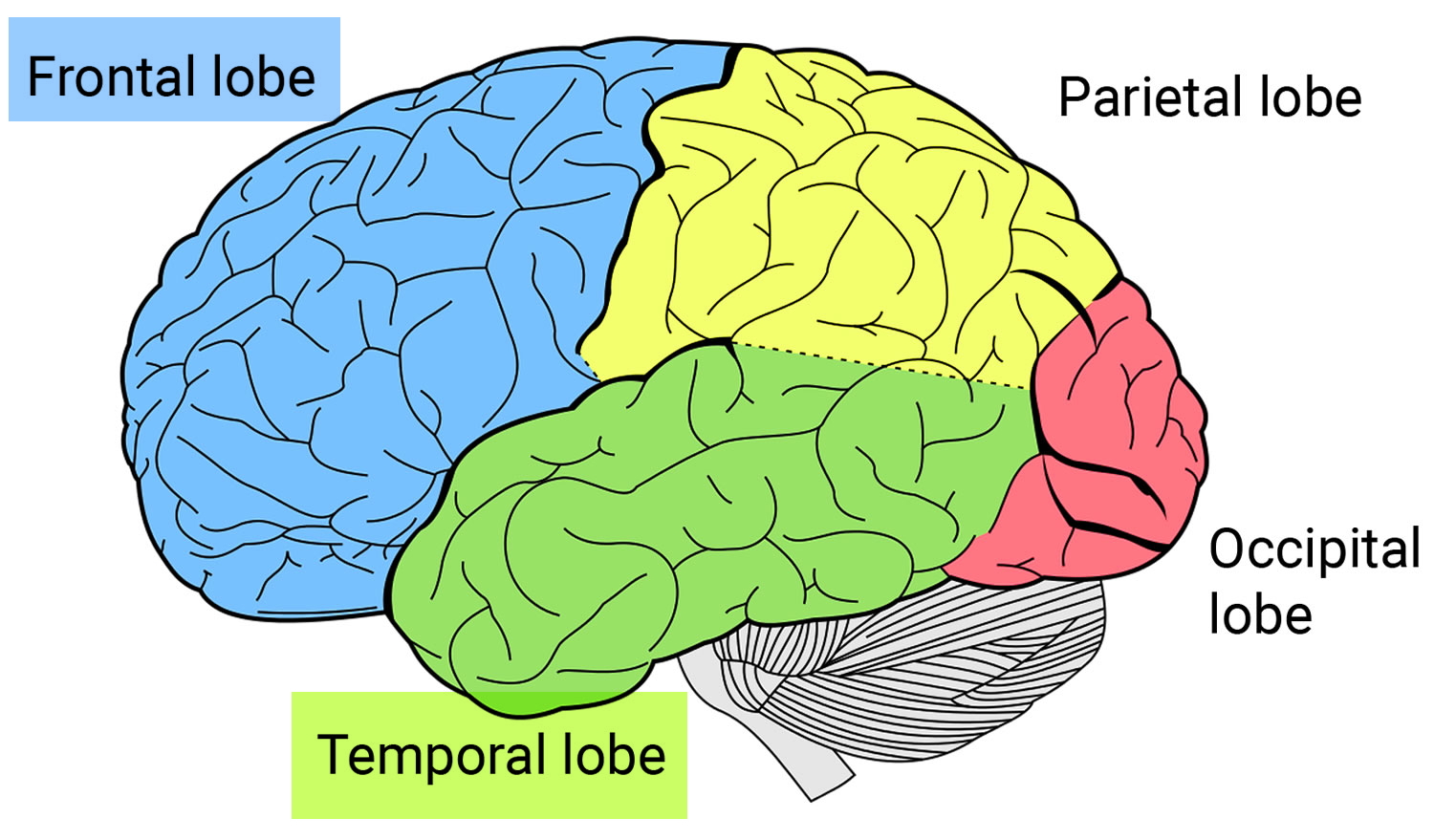
Frontotemporal dementia also known as frontotemporal lobe dementia, frontotemporal lobar degeneration (FTLD), FTD, frontotemporal dementia with parkinsonism or Pick’s disease, is an umbrella term for a group of brain disorders that primarily affect the frontal and temporal lobes of the brain 1. The frontal and temporal lobes of the brain are generally associated with personality, behavior and language. The frontal lobe is the largest lobe in the brain and is located right behind the forehead. The frontal lobe is critical for thinking, planning, decision making and other higher mental processes. The frontal lobe helps people to manage and control emotional responses. The temporal lobes are located below and to both sides of the frontal lobe. The temporal lobes are believed to be involved in semantic memory, or your knowledge of objects, people, words and faces. The temporal lobes also play a role in language and emotion. In frontotemporal dementia, the frontal and temporal lobes of the brain shrink (atrophy). Signs and symptoms vary, depending on which part of the brain is affected. Some people with frontotemporal dementia have dramatic changes in their personalities and become socially inappropriate, impulsive or emotionally indifferent, while others lose the ability to use language properly. Presently, the term frontotemporal dementia encompasses clinical disorders that include changes in behavior, language, executive function and often motor symptoms 2. Frontotemporal dementia usually causes changes in behavior or language problems at first. These come on gradually and get worse slowly over time. Eventually, most people will experience problems in both of these areas. Some people also develop physical problems and difficulties with their mental abilities. Frontotemporal dementia can be misdiagnosed as a psychiatric problem or as Alzheimer’s disease.
Frontotemporal dementia is the third most common form of dementia across all age groups, after Alzheimer’s disease and Lewy body dementia and is the second most common cause of early-onset dementia (age <65) and usually involves patients with age ranges from 45 to 65 3, 4. Frontotemporal dementia can occur in people as young as 20. But it usually begins between ages 40 and 60. The average age at which it begins is 54. People with frontotemporal dementia have abnormal substances called tangles, Pick bodies, and Pick cells and 3R tau proteins inside nerve cells in the damaged areas of the brain.
The first description of a patient with frontotemporal dementia was made by Arnold Pick in 1892 5; the patient had aphasia (aphasia affects a person’s ability to express and understand written and spoken language), lobar atrophy, and presenile dementia. In 1911, Alois Alzheimer recognized the characteristic association with Pick bodies and named the clinicopathological entity Pick’s disease 6, which led to the use of Pick’s disease as a synonym for frontotemporal dementia. In 1982, Mesulam described a language subtype of the disorder, later defined as primary progressive aphasia 7. It was later replaced by the term frontotemporal dementia based on distinct clinical and histopathological criteria, assigned by a research group from the United Kingdom and Sweden in 1994 8.
Frontotemporal dementia is classified into two distinct clinical types; behavior variant frontotemporal dementia (bvFTD) and language type frontotemporal dementia 4. The language type of frontotemporal dementia is further classified into non-fluent variant primary progressive aphasia (nfvPPA), and semantic variant primary progressive aphasia (svPPA) depending on the areas of neuronal loss in frontal and temporal lobes 9.
Frontotemporal dementia symptoms include marked changes in social behavior and personality, and/or problems with language. People with behavior changes may have disinhibition (with socially inappropriate behavior), apathy and loss of empathy, hyperorality (eating excessive amounts of food or attempting to consume inedible things), agitation, compulsive behavior, and various other changes 10. Examples of problems with language include difficulty speaking or understanding speech. Some people with frontotemporal dementia also develop a motor syndrome such as parkinsonism or motor neuron disease (which may be associated with various additional symptoms).
The exact cause of frontotemporal dementia is unknown, but it’s mainly a sporadic disease. Genetics also plays a key role where approximately 30-40% of cases are passed down through families 11, 12. Among them, 13.4% of cases have an autosomal dominant inheritance. Mutations in over 20 genes have been identified in the possible development of frontotemporal dementia 4. Commonly involved genes are MAPT gene (microtubules associated protein tau gene located on chromosome 17q21.32), GRN gene (granulin gene on chromosome 17q21.32) and hexanucleotide repeat expansion of the chromosome 9 open-reading-frame 72 (C9orf72) gene on chromosomal location 9p21.2 (responsible for cytoskeleton organization) 13, 14. It is also postulated that C9orf72 gene mutation is a continuum between frontotemporal dementia and amyotrophic lateral sclerosis (ALS or Lou Gehrig’s disease) and is responsible for the occurrence of both entities. If you have a family history of frontotemporal dementia, you may want to consider talking to your doctor about being referred to a geneticist and possibly having a genetic test to see if you’re at risk. Head trauma and thyroid disease have been linked with the development of frontotemporal dementia with a 3.3-fold higher risk and 2.5-fold higher risk, respectively 4.
Frontotemporal dementia is rare with estimation of 1.61 to 4.1 cases per one hundred thousand people annually 15. One study showed the prevalence of frontotemporal dementia, progressive supranuclear palsy and corticobasal syndrome was 10.8 per 100,000 with peak prevalence at the age range of 65 to 69 years 15. There is an estimated twenty to thirty thousand people in the United States with frontotemporal dementia at one time 16.
Changes in the Brain
Frontotemporal disorders affect the frontal and temporal lobes of the brain (see Brain image below). They can begin in the frontal lobe, the temporal lobe, or both. Initially, frontotemporal disorders leave other brain regions untouched, including those that control short-term memory 17.
The frontal lobes, situated above the eyes and behind the forehead both on the right and left sides of the brain, direct executive functioning. This includes planning and sequencing (thinking through which steps come first, second, third, and so on), prioritizing (doing more important activities first and less important activities last), multitasking (shifting from one activity to another as needed), and monitoring and correcting errors.
When functioning well, the frontal lobes also help manage emotional responses. They enable people to avoid inappropriate social behaviors, such as shouting loudly in a library or at a funeral. They help people make decisions that make sense for a given situation. When the frontal lobes are damaged, people may focus on insignificant details and ignore important aspects of a situation or engage in purposeless activities. The frontal lobes are also involved in language, particularly linking words to form sentences, and in motor functions, such as moving the arms, legs, and mouth.
The temporal lobes, located below and to the side of each frontal lobe on the right and left sides of the brain, contain essential areas for memory but also play a major role in language and emotions. They help people understand words, speak, read, write, and connect words with their meanings. They allow people to recognize objects and to relate appropriate emotions to objects and events. When the temporal lobes are dysfunctional, people may have difficulty recognizing emotions and responding appropriately to them.
Which lobe—and part of the lobe—is affected first determines which symptoms appear first. For example, if the disease starts in the part of the frontal lobe responsible for decision-making, then the first symptom might be trouble managing finances. If it begins in the part of the temporal lobe that connects emotions to objects, then the first symptom might be an inability to recognize potentially dangerous objects—a person might reach for a snake or plunge a hand into boiling water, for example.
Frontotemporal degeneration vs Alzheimer’s disease
Frontotemporal degeneration
- Different symptoms. Frontotemporal dementia brings a gradual, progressive decline in behavior, language or movement, with memory usually relatively preserved.
- Frontotemporal degeneration typically strikes younger people. Although age of onset ranges from 21 to 80, the majority of frontotemporal dementia cases occur between 45 and 64. Frontotemporal degeneration occurs in a younger age group than dementia of the Alzheimer type, with peak incidence occurring in individuals aged 55-65 years 18. Therefore, frontotemporal dementia has a substantially greater impact on work, family, and the economic burden faced by families than Alzheimer’s disease.
- Frontotemporal degeneration is less common and still far less known. Frontotemporal dementia’s estimated U.S. prevalence is around 60,000 cases 19, and many in the medical community remain unfamiliar with it. Frontotemporal dementia is frequently misdiagnosed as Alzheimer’s, depression, Parkinson’s disease, or a psychiatric condition. On average, it currently takes 3.6 years to get an accurate diagnosis.
Alzheimer’s disease
Alzheimer’s disease is the most common cause of dementia — a continuous decline in thinking, behavioral and social skills that disrupts a person’s ability to function independently.
Memory loss is the key symptom of Alzheimer’s disease. The early signs of Alzheimer’s disease may be forgetting or difficulty remembering recent events or conversations. As Alzheimer’s disease progresses, a person with Alzheimer’s disease will develop severe memory impairment and lose the ability to carry out everyday tasks.
At first, a person with Alzheimer’s disease may be aware of having difficulty with remembering things and organizing thoughts. A family member or friend may be more likely to notice how the symptoms worsen.
10 Early Signs and Symptoms of Alzheimer’s disease:
- Memory loss that disrupts daily life: One of the most common signs of Alzheimer’s disease, especially in the early stage, is forgetting recently learned information. Others include forgetting important dates or events, asking for the same information over and over, and increasingly needing to rely on memory aids (e.g., reminder notes or electronic devices) or family members for things they used to handle on their own.
- Challenges in planning or solving problems: Some people may experience changes in their ability to develop and follow a plan or work with numbers. They may have trouble following a familiar recipe or keeping track of monthly bills. They may have difficulty concentrating and take much longer to do things than they did before.
- Difficulty completing familiar tasks at home, at work or at leisure: People with Alzheimer’s often find it hard to complete daily tasks. Sometimes, people may have trouble driving to a familiar location, managing a budget at work or remembering the rules of a favorite game.
- Confusion with time or place: People with Alzheimer’s can lose track of dates, seasons and the passage of time. They may have trouble understanding something if it is not happening immediately. Sometimes they may forget where they are or how they got there.
- Trouble understanding visual images and spatial relationships: For some people, having vision problems is a sign of Alzheimer’s. They may have difficulty reading, judging distance and determining color or contrast, which may cause problems with driving.
- New problems with words in speaking or writing: People with Alzheimer’s may have trouble following or joining a conversation. They may stop in the middle of a conversation and have no idea how to continue or they may repeat themselves. They may struggle with vocabulary, have problems finding the right word or call things by the wrong name (e.g., calling a “watch” a “hand-clock”).
- Misplacing things and losing the ability to retrace steps: A person with Alzheimer’s disease may put things in unusual places. They may lose things and be unable to go back over their steps to find them again. Sometimes, they may accuse others of stealing. This may occur more frequently over time.
- Decreased or poor judgment: People with Alzheimer’s may experience changes in judgment or decision-making. For example, they may use poor judgment when dealing with money, giving large amounts to telemarketers. They may pay less attention to grooming or keeping themselves clean.
- Withdrawal from work or social activities: A person with Alzheimer’s may start to remove themselves from hobbies, social activities, work projects or sports. They may have trouble keeping up with a favorite sports team or remembering how to complete a favorite hobby. They also may avoid being social because of the changes they have experienced.
- Changes in mood and personality: The mood and personalities of people with Alzheimer’s can change. They can become confused, suspicious, depressed, fearful or anxious. They may be easily upset at home, at work, with friends or in places where they are out of their comfort zone.
Memory
Everyone has occasional memory lapses. It’s normal to lose track of where you put your keys or forget the name of an acquaintance. But the memory loss associated with Alzheimer’s disease persists and worsens, affecting the ability to function at work or at home.
People with Alzheimer’s may:
- Repeat statements and questions over and over
- Forget conversations, appointments or events, and not remember them later
- Routinely misplace possessions, often putting them in illogical locations
- Get lost in familiar places
- Eventually forget the names of family members and everyday objects
- Have trouble finding the right words to identify objects, express thoughts or take part in conversations
Thinking and reasoning
Alzheimer’s disease causes difficulty concentrating and thinking, especially about abstract concepts such as numbers.
Multitasking is especially difficult, and it may be challenging to manage finances, balance checkbooks and pay bills on time. These difficulties may progress to an inability to recognize and deal with numbers.
Making judgments and decisions
The ability to make reasonable decisions and judgments in everyday situations will decline. For example, a person may make poor or uncharacteristic choices in social interactions or wear clothes that are inappropriate for the weather. It may be more difficult to respond effectively to everyday problems, such as food burning on the stove or unexpected driving situations.
Planning and performing familiar tasks
Once-routine activities that require sequential steps, such as planning and cooking a meal or playing a favorite game, become a struggle as the disease progresses. Eventually, people with advanced Alzheimer’s may forget how to perform basic tasks such as dressing and bathing.
Changes in personality and behavior
Brain changes that occur in Alzheimer’s disease can affect moods and behaviors. Problems may include the following:
- Depression
- Apathy
- Social withdrawal
- Mood swings
- Distrust in others
- Irritability and aggressiveness
- Changes in sleeping habits
- Wandering
- Loss of inhibitions
- Delusions, such as believing something has been stolen
Preserved skills
Many important skills are preserved for longer periods even while symptoms worsen. Preserved skills may include reading or listening to books, telling stories and reminiscing, singing, listening to music, dancing, drawing, or doing crafts.
These skills may be preserved longer because they are controlled by parts of the brain affected later in the course of the disease.
Current Alzheimer’s disease medications may temporarily improve symptoms or slow the rate of decline. These treatments can sometimes help people with Alzheimer’s disease maximize function and maintain independence for a time.
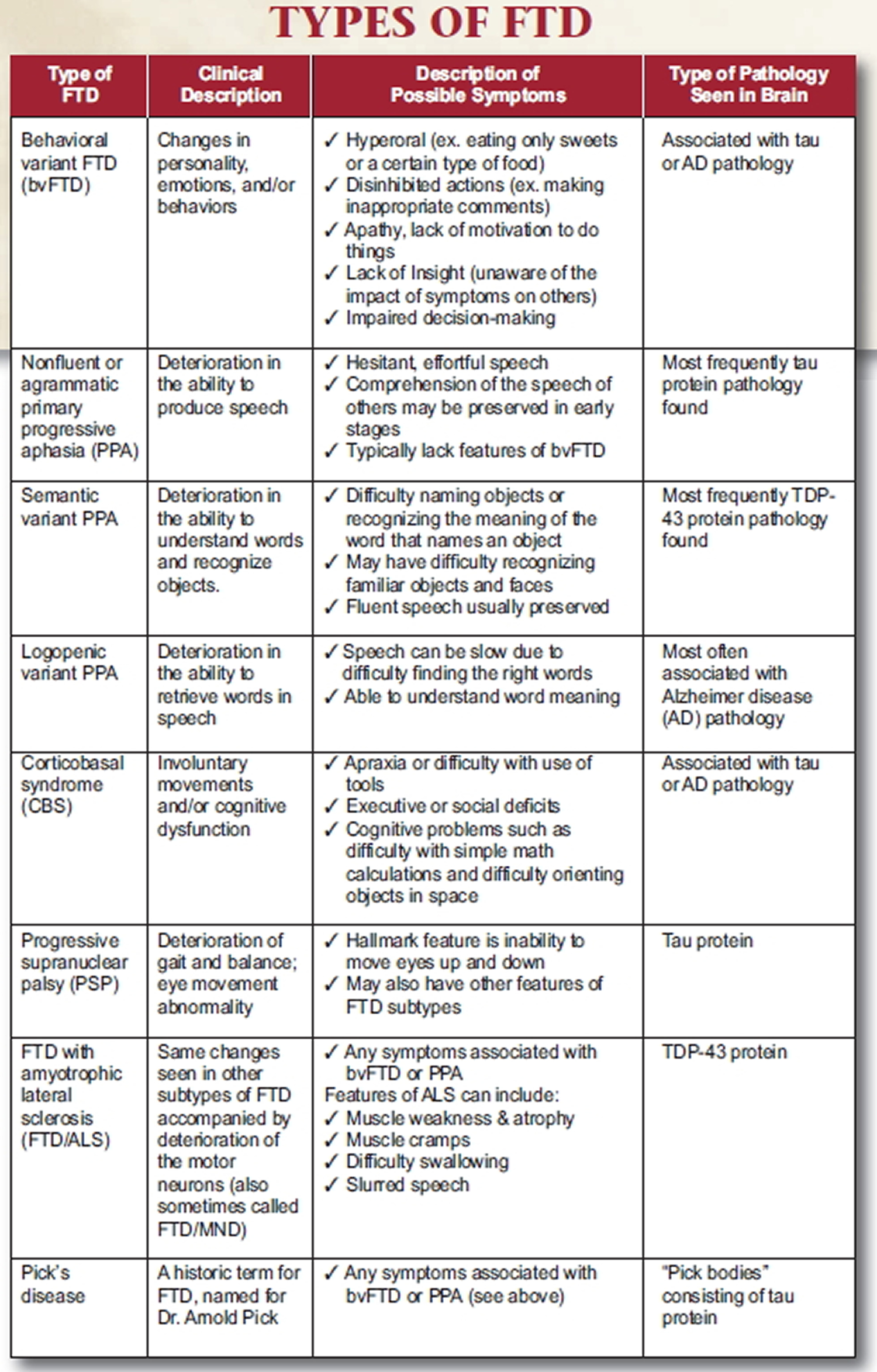
Types of frontotemporal dementia
Frontotemporal degeneration is the umbrella term for a range of disorders that impact the frontal and temporal lobes of the brain. Each disorder can be identified according to the symptoms that appear first and most prominently, whether in behavior (behavioral variant frontotemporal degeneration [bvFTD]), changes in the ability to speak and understand language (primary progressive aphasia [PPA]) or in movement (corticobasal syndrome, progressive supranuclear palsy). The clinical syndrome where FTD and amyotrophic lateral sclerosis (ALS) occur in the same person is referred to as ALS-Frontotemporal Spectrum Disorder (ALS-FTSD).
The most common types of FTD are:
- Behavioral variant frontotemporal dementia (bvFTD). Progressive behavior/personality decline—characterized by changes in personality, behavior, emotions, and judgment (called
behavioral variant frontotemporal dementia). - Primary progressive aphasia (PPA). Progressive language decline — Aphasia means difficulty communicating. This form has three subtypes:
- Progressive Nonfluent/Agrammatic variant PPA (nfvPPA) also called progressive nonfluent aphasia, which affects the ability to speak.
- Semantic variant PPA also called semantic dementia, which affects the ability to use and understand language.
- Logopenic variant PPA also known as PPA-L have difficulty finding words when they are speaking.
- Frontotemporal Dementia with Motor Neuron Disease (FTD-MND) also called Frontotemporal Degeneration with ALS (FTD-ALS).
- Frontotemporal degeneration and the motor neuron disease ALS can occur in the same individual. ALS also known as Lou Gehrig’s disease in the US, is sometimes used interchangeably with the term motor neuron disease (MND). Motor neuron disease is also used as a general term for a group of progressive neurological disorders in which there is a loss of nerve cells called motor neurons. These cells control voluntary muscle activity such as speaking, walking, and talking. Individuals may exhibit weakness, muscle loss (atrophy), tiny involuntary muscle twitches (fasciculation), difficulty speaking (dysarthria) and difficult swallowing (dysphagia). Involuntary control of muscle activity such as breathing and increased tightness and stiffness of muscles (spasticity) is also affected. When people have symptoms of frontotemporal degeneration and motor neuron disease, they may be described as having FTD associated with motor neuron disease (FTD-MND), or FTD associated with amyotrophic lateral sclerosis (FTD-ALS).
- Corticobasal Syndrome (CBS)
- Progressive Supranuclear Palsy (PSP)
In the early stages it can be hard to know which of these disorders a person has because symptoms and the order in which they appear can vary widely from one person to the next. Also, the same symptoms can appear later in different disorders. For example, language problems are most typical of primary progressive aphasia but can also appear later in the course of behavioral variant frontotemporal dementia (bvFTD)
Of the clinical syndromes, frontotemporal dementia with motor neuron disease (FTD-MND) is the most heritable and svPPA is the least heritable (Goldman et al., 2005).
Table 1. Types of Frontotemporal Dementia (FTD) or Frontotemporal Lobar Degeneration (FLTD)
Behavioral variant frontotemporal dementia (bvFTD)
Behavioral variant frontotemporal dementia (bvFTD) frequently referred to as frontotemporal dementia or Pick’s disease, is the most common form of FTD, is responsible for about half of all cases of frontotemporal degeneration (FTD) 21. Behavioral variant frontotemporal dementia (bvFTD) presents with early changes in behavior, personality, emotion and executive control 22. These changes can include new behavioral symptoms such as disinhibition, new compulsions, dietary changes or symptoms like apathy and lack of empathy. The hallmarks of behavioral variant frontotemporal dementia (bvFTD) are personality changes, apathy, and a progressive decline in socially appropriate behavior, judgment, self-control, and empathy. Unlike in Alzheimer’s disease, memory is usually relatively spared in bvFTD. People with bvFTD typically do not recognize the changes in their own behavior, or exhibit awareness or concern for the effect their behavior has on the people around them.
Many of these initial symptoms are easily mistaken for a psychiatric illness making bvFTD patients at high risk for misdiagnosis 23. In order to meet clinical criteria for a diagnosis of behavioral variant frontotemporal dementia (bvFTD), there needs to be a constellation of at least three symptoms fitting into the six categories which include: disinhibition, apathy, lack of empathy, compulsions, hyperorality and executive dysfunction 22.
The behavioral symptoms of bvFTD correlate with dysfunction in the paralimbic areas including medial frontal, orbital frontal, anterior cingulate and frontoinsular cortices 24. Right hemisphere atrophy is associated more with behavior changes 25. Von Economo neurons, present primarily in large brained, socially complex animals 26 are found in the pregenual anterior cingulate cortex, frontoinsular and orbital frontal cortex 27. Seeley has shown that Von Economo neurons are selectively vulnerable in bvFTD but not Alzheimer’s disease 27. These Von Economo neuron-containing areas within the salience network are selectively vulnerable and represent the epicenter of bvFTD pathology, before there is spread to other areas within the network 28. Neurodegeneration is thought to spread within the network with toxic protein aggregates moving from cell to cell in a prion-like manner 29.
It is imperative to take an excellent and thorough clinical history with the goal of identifying the brain region where the disease begins. New symptoms developing through time can localize the spread of neuropathologic changes with disease progression. There are heterogeneous clinical phenotypes with bvFTD based upon the regional spread in the individual patient. Executive control is lost once the neuropathology involves the dorsal lateral prefrontal cortex that interacts with bilateral parietal lobes 28.
Diagnostic criteria for behavioral variant frontotemporal dementia (bvFTD)
In 2011, an international consortium developed revised guidelines for the diagnosis of behavioral variant frontotemporal dementia (bvFTD) based on recent literature and collective experience 22. The following criteria delineates the new criteria for bvFTD (Behavioral Variant Frontotemporal Degeneration or Frontotemporal Dementia).
- I. Neurodegenerative disease
- The following symptom must be present to meet criteria for bvFTD
- A. Shows progressive deterioration of behaviour and/or cognition by observation or history (as provided by a knowledgeable informant).
- The following symptom must be present to meet criteria for bvFTD
- II. Possible bvFTD
- Three of the following behavioral/cognitive symptoms (A–F) must be present to meet criteria. Ascertainment requires that symptoms be persistent or recurrent, rather than single or rare events.
- A. Early* behavioral disinhibition [one of the following symptoms (A.1–A.3) must be present]:
- A.1. Socially inappropriate behaviour
- A.2. Loss of manners or decorum
- A.3. Impulsive, rash or careless actions
- B. Early apathy or inertia [one of the following symptoms (B.1–B.2) must be present]:
- B.1. Apathy
- B.2. Inertia
- C. Early loss of sympathy or empathy [one of the following symptoms (C.1–C.2) must be present]:
- C.1. Diminished response to other people’s needs and feelings
- C.2. Diminished social interest, interrelatedness or personal warmth
- D. Early perseverative, stereotyped or compulsive/ritualistic behavior [one of the following symptoms (D.1–D.3) must be present]:
- D.1. Simple repetitive movements
- D.2. Complex, compulsive or ritualistic behaviours
- D.3. Stereotypy of speech
- E. Hyperorality and dietary changes [one of the following symptoms (E.1–E.3) must be present]:
- E.1. Altered food preferences
- E.2. Binge eating, increased consumption of alcohol or cigarettes
- E.3. Oral exploration or consumption of inedible objects
- F. Neuropsychological profile: executive/generation deficits with relative sparing of memory and visuospatial functions [all of the following symptoms (F.1–F.3) must be present]:
- F.1. Deficits in executive tasks
- F.2. Relative sparing of episodic memory
- F.3. Relative sparing of visuospatial skills
- A. Early* behavioral disinhibition [one of the following symptoms (A.1–A.3) must be present]:
- Three of the following behavioral/cognitive symptoms (A–F) must be present to meet criteria. Ascertainment requires that symptoms be persistent or recurrent, rather than single or rare events.
- III. Probable bvFTD
- All of the following symptoms (A–C) must be present to meet criteria.
- A. Meets criteria for possible bvFTD
- B. Exhibits significant functional decline (by caregiver report or as evidenced by Clinical Dementia Rating Scale or Functional Activities Questionnaire scores)
- C. Imaging results consistent with bvFTD [one of the following (C.1–C.2) must be present]:
- C.1. Frontal and/or anterior temporal atrophy on MRI or CT
- C.2. Frontal and/or anterior temporal hypoperfusion or hypometabolism on PET or SPECT
- All of the following symptoms (A–C) must be present to meet criteria.
- IV. Behavioral variant FTD with definite FTLD Pathology
- Criterion A and either criterion B or C must be present to meet criteria.
- A. Meets criteria for possible or probable bvFTD
- B. Histopathological evidence of FTLD on biopsy or at post-mortem
- C. Presence of a known pathogenic mutation
- Criterion A and either criterion B or C must be present to meet criteria.
- V. Exclusionary criteria for bvFTD
- Criteria A and B must be answered negatively for any bvFTD diagnosis. Criterion C can be positive for possible bvFTD but must be negative for probable bvFTD.
- A. Pattern of deficits is better accounted for by other non-degenerative nervous system or medical disorders
- B. Behavioural disturbance is better accounted for by a psychiatric diagnosis
- C. Biomarkers strongly indicative of Alzheimer’s disease or other neurodegenerative process.
- Criteria A and B must be answered negatively for any bvFTD diagnosis. Criterion C can be positive for possible bvFTD but must be negative for probable bvFTD.
Footnote: *As a general guideline ‘early’ refers to symptom presentation within the first 3 years.
[Source 22 ]Behavioral variant frontotemporal dementia (bvFTD) symptoms
The following are possible symptoms of bvFTD:
- Disinhibition (a loss or lack of restraint based on social norms, leading to inappropriate behavior and impulsivity).
- Disinhibition includes socially inappropriate behavior such as invading interpersonal space, inappropriate touching or over-familiarity with strangers. There can be impulsive or careless actions like new onset gambling, stealing, poor decision-making without regards to the consequences (e.g. buying matching motor cycles for themselves and their 7 year old son). Disinhibition is linked to right orbital frontal cortex degeneration 30. New criminal behaviors are seen in 37%–54% of bvFTD patients 31. Loss of social decorum such as telling off color jokes, using crude language, and rudeness with lack of embarrassment is common in bvFTD. There can be dramatic change in self-awareness, with lack of insight into one’s disease 32. Behaviors may include:
- Making uncharacteristic rude or offensive comments
- Ignoring other people’s personal space
- Shoplifting, reckless spending
- Touching strangers or inappropriate sexual behavior
- Aggressive outbursts
- Disinhibition includes socially inappropriate behavior such as invading interpersonal space, inappropriate touching or over-familiarity with strangers. There can be impulsive or careless actions like new onset gambling, stealing, poor decision-making without regards to the consequences (e.g. buying matching motor cycles for themselves and their 7 year old son). Disinhibition is linked to right orbital frontal cortex degeneration 30. New criminal behaviors are seen in 37%–54% of bvFTD patients 31. Loss of social decorum such as telling off color jokes, using crude language, and rudeness with lack of embarrassment is common in bvFTD. There can be dramatic change in self-awareness, with lack of insight into one’s disease 32. Behaviors may include:
- Apathy (indifference or lack of interest in previously meaningful activities)
- Apathy can present in many ways 33. Affective apathy presents as indifference or not caring. Motor apathy manifests as decreased drive to move and less movement overall 34 whereas cognitive apathy is a loss of desire to engage in goal oriented activities 33. Symptoms of apathy may also include social withdrawal in work activities, family functions or hobbies. Patients often need prompting to stay engaged in conversation, do chores or even to move. Apathy is easily misinterpreted as depression. Atrophy in the medial prefrontal lobes and anterior cingulate have been correlated with apathy in bvFTD 35. Behaviors may include:
- Loss of interest in work, hobbies, and personal relationships
- Neglect of personal hygiene
- Loss of initiative
- Apathy can present in many ways 33. Affective apathy presents as indifference or not caring. Motor apathy manifests as decreased drive to move and less movement overall 34 whereas cognitive apathy is a loss of desire to engage in goal oriented activities 33. Symptoms of apathy may also include social withdrawal in work activities, family functions or hobbies. Patients often need prompting to stay engaged in conversation, do chores or even to move. Apathy is easily misinterpreted as depression. Atrophy in the medial prefrontal lobes and anterior cingulate have been correlated with apathy in bvFTD 35. Behaviors may include:
- Emotional blunting
- Loss of warmth, empathy, sympathy or concern for others. Behaviors may include:
- Indifference to important events (e.g., death of a family member or friend);
- Failure to recognize that loved ones are upset or unhappy
- Making cruel comments towards others
- Loss of warmth, empathy, sympathy or concern for others. Behaviors may include:
- Compulsive or ritualistic behaviors
- Single behaviors or routines that are performed over and over. These may include:
- Repeating words or phrases
- Hand rubbing, clapping
- Re-reading the same book over and over again
- Hoarding
- Walking to the same place at the same time every day
- Single behaviors or routines that are performed over and over. These may include:
- Changes in eating habits or diet
- Excessive, compulsive or inappropriate eating and drinking, or other pronounced changes in dietary preferences.
- Binge eating
- Carbohydrate craving
- Eating only specific foods
- Increased or first-time use of tobacco products
- Hyperorality
- Excessive water or alcohol consumption
- Attempting to consume inedible objects
- Excessive, compulsive or inappropriate eating and drinking, or other pronounced changes in dietary preferences.
- Deficits in executive function
- Poor decision-making, judgment, problem-solving, and organizational skills. Examples include:
- Difficulty planning the day’s activities
- Questionable financial decisions
- On-the-job mistakes that may be uncharacteristic
- Poor decision-making, judgment, problem-solving, and organizational skills. Examples include:
- Lack of insight
- As noted above, failure to recognize changes in behavior or exhibit awareness of effects of behavior on others. Behaviors may include:
- Blaming others for consequences of socially unacceptable behavior; e.g., job loss
- Anger at limitations on activities
- As noted above, failure to recognize changes in behavior or exhibit awareness of effects of behavior on others. Behaviors may include:
- Other symptoms
- Agitation, emotional instability. These may be conveyed through:
- Pacing
- Frequent and abrupt mood changes
- Agitation, emotional instability. These may be conveyed through:
Primary Progressive Aphasias
Primary Progressive Aphasia (PPA) is characterized predominantly by the gradual loss of the ability to speak, read, write, and understand what others are saying.
Primary progressive aphasia (PPA) is diagnosed when three criteria are met:
- There is a gradual impairment of language (not just speech). Speech problems alone are not sufficient for diagnosing primary progressive aphasia (PPA). When the dominant problem is speech rather than language, the diagnosis is progressive apraxia of speech rather than primary progressive aphasia (PPA).
- The language problem is initially the only impairment.
- The underlying cause is a neurodegenerative disease.
Experts further subdivide primary progressive aphasia (PPA) into three clinical subtypes based on the specific language skills that are most affected.
- Nonfluent/Agrammatic variant PPA (nfvPPA) also called progressive nonfluent aphasia. A person has more and more trouble producing speech. Eventually, the person may no longer be able to speak at all. He or she may eventually develop movement symptoms similar to those seen in corticobasal syndrome. In this case, individuals lose grammar (the small connecting words) but have preserved language comprehension for specific items/objects. This causes speech to become effortful, hesitant, and sentence length becomes progressively truncated. Writing and language comprehension may be affected in the same manner. This subtype of PPA is usually associated with tau pathology.
- Semantic variant PPA also called semantic dementia, a person slowly loses the ability to understand single words and sometimes to recognize the faces of familiar people and common objects. Here, the meaning of specific words is lost, and both comprehension of the word and the ability to retrieve the name of an object may be lost. Speech remains fluent and grammar is good, however, paraphasias (word substitution errors) are common. This subtype of PPA is usually associated with TDP-43 pathology.
- Logopenic variant PPA, a person has trouble finding the right words during conversation but can understand words and sentences. The person does not have problems with grammar. In these individuals, speech is slow, but grammar and comprehension are less affected. Impairment in the repetition of multisyllabic words and particularly phrases is a key feature. Sound substitution (phonemic) paraphasias are also seen in this group, as in a false word that rhymes with the intended word. Logopenic aphasia is usually associated with an underlying Alzheimer’s pathology.
Nonfluent/Agrammatic Primary Progressive Aphasia
Patients with nonfluent/agrammatic variant primary progressive aphasias (nfvPPA) also called progressive nonfluent aphasia find it increasingly difficult to speak yet can still recall the meanings of individual words. The inability to form sounds with their lips and tongue is caused by degeneration of the parts of the brain that control certain related muscles; the muscles themselves, however, are unaffected. The technical term for such problems is apraxia of speech. As a result, their speech becomes slow and effortful (effortful speech) and they may appear to be physically struggling to produce words. Over time, the speech becomes labored, slow, slurred, and choppy with disrupted prosody. The patient begins making inconsistent speech sound errors without awareness of this deficit. There can be inconsistent insertions, deletions, distortions, and substitutions in speech sounds. If a patient is asked to repeat a complexly structured word like “catastrophe” five times in a row, he or she will demonstrate a motor speech apraxia by repeating this word differently each time 36. Improper use of grammar is frequently present in nonfluent/agrammatic variant primary progressive aphasias (nfvPPA), but can be subtle or even absent in the early stages of the illness. People with nonfluent/agrammatic variant primary progressive aphasias (nfvPPA) make many mistakes while speaking, including omitting small grammatical words, using word endings and verb tenses incorrectly, and/or mixing up the order of words in sentences. Eventually, some may develop difficulty swallowing as well as more widespread motor symptoms similar to those seen in the movement-predominant forms of FTD such as corticobasal syndrome.
Patients with nonfluent/agrammatic variant primary progressive aphasias (nfvPPA) can often have an aphemia (a type of aphasia characterized by the inability to express ideas in spoken words), but can still communicate correctly with written language. Eventually agrammatism will involve both verbal and written language. Effortful speech often occurs before clear apraxia of speech or agrammatism 37. The neuroanatomical correlate for the symptoms of nonfluent/agrammatic variant primary progressive aphasias (nfvPPA) are Broadman’s Area 44, 45 (Broca’s area) in the left inferior frontal gyrus and the anterior insula 38. As time progresses, patients will have decreased verbal output and eventually become non-verbal. Mutism in nonfluent/agrammatic variant primary progressive aphasias (nfvPPA) is correlated to a larger lesion expanding beyond the typical inferior frontal and insular regions involved early in nfvPPA 39.
The defining symptoms of nonfluent/agrammatic variant primary progressive aphasias (nfvPPA):
- Apraxia. Difficulty producing movements of lips and tongue needed for speech. This results in distorted or incorrect speech sounds with slow, labored speech, and groping movements of the face and mouth in an effort to produce the correct sound.. Effortful speech is often the first symptom. Multisyllabic words are the most difficult to produce.
- Agrammatism. Omitting words in sentences, especially short connecting words (e.g., “to,” “from,” “the.” The order of words in sentences is often incorrect. Errors are made in the use of word endings, verb tenses and pronouns. Speech becomes restricted to short, simple phrases that are difficult for listener to understand because of omissions and errors. As examples: the affected individual may use “seed” instead of “saw” or “throwed” instead of “threw.” They may say: “Today…go lunch…ah…sister” for “today I am going to lunch with my sister.”
- Impaired comprehension of complex sentences. Single-word comprehension is unaffected, but the ability to understand long or grammatically difficult sentences is reduced. Persons with primary progressive aphasia (PPA) may find it increasingly difficult to understand what they experience as “too much” verbal information, i.e. watching television or understanding conversation in a group setting.
- Mutism. The affected person does not speak at all.
- Difficulty swallowing. Develops later in the progression of the disease.
- Motor symptoms. Parkinson’s disease-like movement deficits can occur. The person affected may experience slow, stiff movement, lose balance or fall easily, have difficulty using an arm or leg, and experience restricted up-and-down eye movement.
Diagnostic criteria for nonfluent/agrammatic variant PPA (nfvPPA)
- I. Clinical diagnosis of nonfluent/agrammatic variant PPA:
- At least one of the following core features must be present:
- Agrammatism in language production
- Effortful, halting speech with inconsistent speech sound errors and distortions (apraxia of speech)
- At least 2 of 3 of the following other features must be present:
- Impaired comprehension of syntactically complex sentences
- Spared single-word comprehension
- Spared object knowledge
- At least one of the following core features must be present:
- II. Imaging-supported nonfluent/agrammatic variant diagnosis
- Both of the following criteria must be present:
- Clinical diagnosis of nonfluent/agrammatic variant PPA
- Imaging must show one or more of the following results:
- Predominant left posterior frontoinsular atrophy on MRI or
- Predominant left posterior frontoinsular hypoperfusion or hypometabolism on SPECT or PET
- Both of the following criteria must be present:
- III. Nonfluent/agrammatic variant PPA with definite pathology
- Clinical diagnosis (criterion A below) and either criterion B or C must be present:
- Clinical diagnosis of nonfluent/agrammatic variant PPA
- Histopathologic evidence of a specific neurodegenerative pathology (e.g., frontotemporal lobar degeneration-tau, frontotemporal lobar degeneration-TDP [frontotemporal lobar degeneration with TDP-43], Alzheimer’s disease, other)
- Presence of a known pathogenic mutation
- Clinical diagnosis (criterion A below) and either criterion B or C must be present:
Semantic Variant Primary Progressive Aphasia
Semantic Variant Primary Progressive Aphasia (svPPA) also known as PPA-S, is characterized by the progressive loss of the meanings of words 40. If there are additional major problems in identifying objects or faces, the condition is also called semantic dementia. Other language skills, including the ability to produce speech and to repeat phrases and sentences spoken by others, are unaffected. However, although the affected person may continue to speak fluently, their speech becomes vague and difficult to understand because many words are omitted or substituted. As the disorder progresses, people with semantic variant primary progressive aphasia (svPPA) may also exhibit changes in behavior similar to those seen in bvFTD, such as disinhibition and rigid food preferences.
If the left temporal lobe is involved, as in left svPPA, symptoms are predominantly language-based with a slow loss of semantic knowledge. When the right temporal lobe is primarily involved (right svPPA), behavioral symptoms predominate 9. As time progresses both temporal lobes become involved and symptoms begin to overlap 41. By five to seven years, patients get more frontal lobe structures involved and develop symptoms of bvFTD with disinhibition, change in food preference and weight gain 41. Patients with semantic variant primary progressive aphasia (svPPA), tend to have slow progression and can live a decade or more after symptom onset 42. Of the core clinical phenotypes in frontotemporal dementia, svPPA is the least likely to have a genetic cause underlying the etiology 43.
Right temporal svPPA patients present with prominent behavioral changes that include emotional distance, irritability, social isolation, bizarre alterations in dress, compulsions, disruption of sleep, appetite and libido 41. Patients with right temporal svPPA can often lack social pragmatics. They can be viscous, with telling long-winded stories, interrupting loved ones and not picking up on normal social cues that they may be acting inappropriately. It is thought that this inability to pick up social cues has to do with an inability to read the emotions on faces. Worsening degrees of right amygdala atrophy has been correlated with impairment in processing facial emotions 44.
Patients with either left or right temporal svPPA can have new compulsions. Left svPPA patients have been shown to have compulsions directed towards visual or non-verbal stimuli where objects are devoid of verbal information like coins or pictures. Right svPPA have compulsions around games with words and symbols 41.
An interesting phenomenon is the development of new artistic abilities that has been observed in svPPA. Left svPPA patients have developed new visual abilities in painting, drawing, music, and gardening 45. Right svPPA patients can have emergent abilities in writing despite their progressive neurodegenerative condition 46. One patient with bilateral, right worse than left, svPPA was studied in depth at UCSF. As the right temporal lobe became more involved, he showed decline in emotional perception displayed through his new skill of painting 47. The topics of the painting would show couples and landscapes, but as his disease progressed the facial expressions became less genuine. Painted couples would stand next to each other, not holding hands, with bizarre smiles showing teeth 47. One could theorize that this case illustrates the difficulty the patient had understanding emotions in others as the right temporal lobe involvement progressed.
Semantic variant primary progressive aphasia (svPPA) clinical features
- Anomia. An inability to recall the names of objects; difficulty “finding the right word.” The person affected may not be able to name a picture of a truck, or may substitute another word in the same category such as “car” for “truck.”
- Reduced single-word comprehension. The person affected is unable to recall what words mean, especially words that are less familiar or less frequently used. For example, she or he may ask “What is a truck?” When asked to bring an orange, the patient may come back with an apple because the meaning of the word ‘orange’ is lost. This does not mean that the object is not recognized, as shown by the fact that the patient will not try to eat an orange without peeling it.
- Impaired object knowledge. Being unable to remember what a familiar object is or how it is used. For example, the person affected may be unable to identify common kitchen utensils and how they are used in cooking. This is very unusual at initial stages of semantic PPA but may appear later.
- Surface dyslexia/dysgraphia. Difficulty reading and writing words that do not follow pronunciation or spelling rules; such words are spelled or spoken “as if” they followed the rules. For example, the person affected may write “no” instead of “know,” or read “broad” as “brode.”
The initial features of classic left temporal semantic variant primary progressive aphasia (svPPA) are anomia and single word comprehension deficits 37. The earliest symptom is often poor comprehension of single low frequency words such as “giraffe” with initial preservation of higher frequency words like “dog”. As symptoms progress, patients also lose semantic knowledge about objects 37, so if a patient does not know the word “giraffe,” they will not improve with phonemic cues or semantic hints like “animal with a long neck.” When svPPA patients are asked to read irregularly spelled words (yacht, gnat), they will sound them out, attempting phonetic “regularization”, having lost the knowledge or their unconventional sound (i.e. y-act, ga-nat), a phenomenon known as surface dyslexia 37. In svPPA, repetition is spared, speech apraxia is not present, and syntax and grammar remain impressively intact. If the mesial temporal lobes are involved, memory can be affected but executive function and visuospatial skills typically are preserved 48. Clinical research criteria for language predominant (left) svPPA must include the core features of impaired confrontational naming and impaired single word comprehension. There also must be three of the following four criteria: impaired object knowledge, surface dyslexia, spared repetition, and spared speech production 37.
Diagnostic criteria for semantic variant PPA (svPPA)
- I. Clinical diagnosis of semantic variant PPA
- Both of the following core features must be present:
- Impaired confrontation naming
- Impaired single-word comprehension
- At least three of the following other diagnostic features must be present:
- Impaired object knowledge, particularly for low-frequency or low-familiarity items
- Surface dyslexia or dysgraphia
- Spared repetition
- Spared speech production (grammar and motor speech)
- Both of the following core features must be present:
- II. Imaging-supported semantic variant PPA diagnosis
- Both of the following criteria must be present:
- Clinical diagnosis of semantic variant PPA
- Imaging must show one or more of the following results:
- Predominant anterior temporal lobe atrophy
- Predominant anterior temporal hypoperfusion or hypometabolism on SPECT or PET
- Both of the following criteria must be present:
- III. Semantic variant PPA with definite pathology
- Clinical diagnosis (criterion A below) and either criterion B or C must be present:
- A. Clinical diagnosis of semantic variant PPA
- B. Histopathologic evidence of a specific neurodegenerative pathology (e.g., frontotemporal lobar degeneration-tau, frontotemporal lobar degeneration-TDP [frontotemporal lobar degeneration with TDP-43], Alzheimer’s disease, other)
- C. Presence of a known pathogenic mutation
- Clinical diagnosis (criterion A below) and either criterion B or C must be present:
Logopenic Variant PPA
Logopenic Variant PPA (lvPPA) also known as PPA-L have difficulty finding words when they are speaking. As a result, they may speak slowly and hesitate frequently as they search for the right word. Unlike people with semantic variant PPA, however, they are still able to recall the meanings of words. Unlike people with agrammatic PPA, speech can be perfectly fluent during small talk but then becomes hesitant and halting when the person needs to be specific or use a more unfamiliar word. Speech is usually not effortful or distorted. The logopenic variant PPA (lvPPA) is also characterized by a narrow attention span for words that compromise the ability to repeat phrases and sentences. As the disease progresses, affected individuals may develop problems comprehending complex sentences.
Logopenic variant PPA (lvPPA) clinical features:
- Impaired single-word retrieval. Difficulty finding the right word while speaking.
- Pauses and hesitations due to time needed for word retrieval
- Extended description (circumlocution) may be substituted for a forgotten word
- Impaired repetition. More difficulty with longer phrases and sentences.
- Phonological speech errors. Mistakes in speech sounds, including omissions and substitutions. For example, the person affected may substitute sounds made with the tip of the tongue such as “t” or “d” for sounds made near the throat such as “k” or “g”: “tup” instead of “cup” or “dap” instead of “gap.” They may omit final consonants: “slee” instead of “sleep.”
- Phonological paraphasias. Substitution of a non-word with some of the same sounds for a legitimate word. For example, the person affected may say “lelephone” for “telephone.”
- Poor comprehension of complex sentences. With single-word comprehension spared.
- Difficulty swallowing. May develop later in the progression of the disease.
Diagnostic criteria for logopenic variant PPA (lvPPA)
Doctors will consider a diagnosis of logopenic variant PPA (lvPPA) based on the following symptoms:
- Impaired single-word retrieval in spontaneous speech
- Impaired repetition of phrases and sentences
AND at least three of the following:
- Phonological speech errors
- Single-word comprehension and object knowledge unaffected
- Physical ability to form words (motor speech) unaffected
- Simple but correct grammar
Corticobasal syndrome
Corticobasal syndrome belongs to the category of FTD disorders that primarily affect movement. To meet the most recent clinical criteria for probable corticobasal syndrome, a patient must have an asymmetric presentation with two of the following motor symptoms: limb rigidity or akinesia, limb dystonia, or limb myoclonus, as well as two of the following higher cortical symptoms: orobuccal or limb apraxia, cortical sensory deficit, or alien limb phenomena 50. A person affected by corticobasal syndrome may present with cognitive, motor, or language symptoms as the first sign 51. Development of a second and/or third category of symptoms makes it easier for the physician to recognize the illness as corticobasal syndrome. Individuals with corticobasal syndrome are easier to diagnose if they are showing limb apraxia, such as no longer being able to use the remote control for the television set, or not being able to retrieve mail from the mailbox. Initial symptoms of corticobasal syndrome often begin around age 60 (although there is variability in the age of onset) and then become bilateral as the disease progresses. A person with corticobasal syndrome may first present with a language disorder and develop motor symptoms over time.
Movement deficits in corticobasal syndrome often begin on one side of the body, but eventually both sides are affected. In addition to motor symptoms, people with corticobasal syndrome may exhibit changes in behavior and language skills common to bvFTD and PPA, particularly as the disease progresses.
Some symptoms of both corticobasal syndrome and progressive supranuclear palsy (PSP), another FTD disorder associated with a decline in motor function, resemble those often seen in people with Parkinson’s disease. These features are sometimes referred to as “atypical Parkinsonism.”
Like all FTD disorders, corticobasal syndrome is associated with degeneration of the brain’s frontal and temporal lobes. In addition, several regions deeper in the brain that play important roles in initiating, controlling, and coordinating movement are also affected.
The term corticobasal degeneration (CBD) is applied to cases which have a particular type of tauopathy at autopsy. Some cases of corticobasal syndrome prove to have Alzheimer’s pathology instead.
Signs and symptoms of corticobasal syndrome include:
- Limb Apraxia
- Inability to compel a hand, arm, or leg to carry out a desired motion although the muscle strength needed to complete the action is maintained
- Difficulty completing familiar purposeful activities, such as opening a door, operating the television remote, or using kitchen tools
- Tripping or falling
- Akinesia/bradykinesia. Absence (akinesia) or abnormally slow (bradykinesia) movement.
- Rigidity. Stiffness, resistance to movement.
- Dystonia. Uncontrollable muscle contraction that causes an arm or leg to twist involuntarily or to assume an abnormal posture.
- Cognitive symptoms. These may include:
- Alien limb phenomenon – sensation that an arm or leg is not part of the body, accompanied by inability to control movement of the limb
- Acalculia – inability to carry out simple mathematical calculations, such as adding or subtracting
- Visuospatial deficits – difficulty orienting in space.
Progressive Supranuclear Palsy
Progressive supranuclear palsy (PSP) previously known as Steele-Richardson-Olszewski syndrome, belongs to the category of frontotemporal dementia that primarily affect movement. Some symptoms of both progressive supranuclear palsy (PSP) and corticobasal syndrome, another FTD disorder associated with a decline in motor function, resemble those often seen in people with Parkinson’s disease. These features are sometimes referred to as “atypical Parkinsonism.”
The earliest motor symptoms are stiffness in the axial muscles, the neck and trunk, along with poor balance and more frequent falls. The earliest visual signs are a decrease in upward vertical movement of the eyes (vertical saccades) and a progressive inability to move the eyes, including opening or closing the eyes. progressive supranuclear palsy can also affect coordination, and movement of the mouth, tongue, and throat. In addition to motor symptoms, people with progressive supranuclear palsy may exhibit changes in behavior and language skills common to bvFTD and PPA, particularly as the disease progresses.
Patients can present with predominant executive dysfunction, personality changes, reduced mental speed and attention deficits giving a more frontal behavior syndrome that is not emphasized in the current progressive supranuclear palsy criteria 52. Patients presenting with frontal behavior symptoms and without falls or supranuclear gaze palsy in their first year of presentation are likely to be among the 20% of progressive supranuclear palsy patients that are clinically misdiagnosed 53. Clinical presentations of bvFTD or nfvPPA can progress to progressive supranuclear palsy or alternatively, an initial presentation of progressive supranuclear palsy can progress into bvFTD or nonfluent/agrammatic variant primary progressive aphasias (nfvPPA) 54. Williams 53 has proposed a system to further differentiate progressive supranuclear palsy. He classifies Richardson’s syndrome as the classic PSP-S described above. PSP-Parkinsonism (PSP-P), is the most common non Steele-Richardson PSP subtype, which is similar to Richardson’s syndrome but has tremor and mild responsiveness to Levodopa. PSP-pure akinesia with gait freezing (PSP-PAGF) is a PSP-S subtype which can have a slower disease course in spite of severe atrophy in the globus pallidus, substantia nigra and subthalamic nucleus. There are also PSP-Corticobasal Syndrome (PSP-CBS) as well as PSP-progressive nonfluent aphasia (PSP-PNFA) 53.
The 1996 consensus clinical research criteria for possible progressive supranuclear palsy defines the following core features 55:
- onset after age 40,
- gradual progression,
- either vertical supranuclear gaze palsy or slow vertical saccades, and
- postural instability with falls in the first year.
Patients must also have no exclusion criteria. Probable progressive supranuclear palsy is inclusive of both a vertical supranuclear gaze palsy and postural instability with falls 55. Severe impairment of postural reflexes and bradykinesia make progressive supranuclear palsy falls extremely dangerous since patients are often unable to protect themselves from objects during the fall. The falls in progressive supranuclear palsy can be multifactorial from either the impaired eye movements, postural instability, impulsivity or all three symptoms.
Progressive supranuclear palsy (PSP) clinical features
- Supranuclear gaze palsies. The person affected may experience an inability to move or aim the eyes vertically (particularly downward) or horizontally (left and right). They may experience rapid involuntary eye movements, or difficulty blinking or excessive blinking. Supranuclear gaze palsies may be experienced as blurring. The person affected may experience difficulty reading, make poor eye contact during conversations, have difficulty going down stairs, or experience impaired vision while driving.
- Postural instability. Difficulty maintaining balance, which can lead to frequent, unexplained falls.
- Gait instability. An unsteady, awkward gait.
- Akinesia/bradykinesia. Absence of movement (akinesia) or abnormally slow (bradykinesia) movement.
- Rigidity. Stiffness, resistance to movement.
- Dysphagia. Difficulty swallowing, including gagging or choking. This can lead to aspiration pneumonia.
- Dysarthria. Slurred or slowed speech due to difficulty moving the muscles controlling the lips, tongue and jaw.
- Behavioral and emotional symptoms that may occur in progressive supranuclear palsy:
- A progressive deterioration in the diagnosed person’s ability to control or adjust their behavior appropriately in different social contexts is the hallmark of the behavior changes, and results in the embarrassing, inappropriate social situations that can be one of the most disturbing facets of FTD and related disorders.
- In addition to the depression, apathy and inability to control emotions noted above, progressive supranuclear palsy patients may manifest emotional blunting or indifference toward others and a lack of insight into changes in their own behavior.
- Cognitive symptoms. Progressive supranuclear palsy patients may suffer increasing impairment in “executive functions,” such as distractibility, mental rigidity and inflexibility, impairments in planning and problem solving, and poor financial judgment. progressive supranuclear palsy patients may also have memory problems. They also develop progressive language disturbance.
Frontotemporal dementia and Motor Neuron Disease
FTD-ALS (frontotemporal dementia with amyotrophic lateral sclerosis) is a combination of bvFTD and ALS (commonly called Lou Gehrig’s disease). Symptoms include the behavioral and/or language changes seen in bvFTD as well as the progressive muscle weakness seen in ALS. Symptoms of either disease may appear first, with other symptoms developing over time. 15% of FTD patients and up to 30% of patients with motor neuron disease experience overlap between the two syndromes 56. The coexistence of two disorders may be under recognized, as patients tend to present to either a neuromuscular disease clinic or a dementia clinic 57. To meet criteria for motor neuron disease, an frontotemporal dementia patient needs upper and lower motor neuron signs on physical examination or electromyogram (EMG) 58. The El Escorial criteria are one of the most widely accepted criteria for the diagnosis of ALS but the Awaji criteria are also used which deemphasizes the use of EMG to make an earlier diagnosis 58. If an frontotemporal dementia patient presents with fasciculations, muscle weakness, trouble swallowing, spastic tone, hyperreflexia, or pathological laughing or crying, this should raise concern for motor neuron disease and prompt a neuromuscular work up. If patients with ALS presents with symptoms concerning for bvfrontotemporal dementia, this should prompt a cognitive work up. There is a shorter survival in patients with both frontotemporal dementia and motor neuron disease since patients with frontotemporal dementia symptoms are less compliant with standard ALS treatments 59. Mutations in certain genes have been found in some patients with FTD-ALS.
As many as 20% of frontotemporal dementia patients develop signs of motor neuron disease (MND). Likewise approximately half of amyotrophic lateral sclerosis (ALS) patients develop some fronto-executive problems 60. A smaller number of these patients develop full-blown FTD-ALS. It is now recognized that the C9orf72 gene is the most common gene causing hereditary FTD, ALS and ALS with FTD (see Genetics section).
With or without the gene expansion, the addition of motor neuron disease to frontotemporal dementia is a compromising factor which greatly reduces median survival to less than 3 years 61. Behavioral symptoms complicate the management of dysphagia as well as respiratory dysfunction as respiration therapy and percutaneous endoscopic gastrostomy (PEG) feeding tubes are not well tolerated by the patient. PEG shows little survival benefit 62.
Although significant phenotypic heterogeneity exists among C9orf72 carriers, the majority present with either bvFTD or FTD-ALS. PPA variants including nonfluent/agrammatic and semantic are rare. Prominent psychosis with delusions and hallucinations are relatively common 63). Research into the clinical features associated with the C9orf72 mutation is active and on-going.
Frontotemporal dementia symptoms
Signs and symptoms of frontotemporal dementia can be different from one individual to the next. Signs and symptoms get progressively worse over time, usually over years. Clusters of symptom types tend to occur together, and people may have more than one cluster of symptom types.
Signs of frontotemporal dementia can include:
- Personality and behavior changes – acting inappropriately or impulsively, appearing selfish or unsympathetic, neglecting personal hygiene, overeating, or loss of motivation
- Language problems – speaking slowly, struggling to make the right sounds when saying a word, getting words in the wrong order, or using words incorrectly
- Problems with mental abilities – getting distracted easily, struggling with planning and organisation
- Memory problems – these only tend to occur later on, unlike more common forms of dementia, such as Alzheimer’s disease
There may also be physical problems, such as slow or stiff movements, loss of bladder or bowel control (usually not until later on), muscle weakness or difficulty swallowing.
These problems can make daily activities increasingly difficult, and the person may eventually be unable to look after themselves.
Behavior and personality changes
Many people with frontotemporal dementia develop a number of unusual behaviors they’re not aware of. The most common signs of frontotemporal dementia involve extreme changes in behavior and personality. These can include:
- being insensitive or rude
- increasingly inappropriate social behavior
- acting impulsively or rashly
- loss of inhibitions
- lack of judgment
- seeming subdued
- losing interest (apathy) in people and things, which can be mistaken for depression
- losing drive and motivation
- inability to empathise with others, seeming cold and selfish
- repetitive compulsive behaviors, such as humming, smacking lips, hand-rubbing, clapping and foot-tapping, or routines such as walking exactly the same route repetitively
- a change in food preferences, such as suddenly liking sweet foods, overeating and poor table manners
- eating inedible objects
- compulsively wanting to put things in the mouth
- compulsive eating, alcohol drinking and/or smoking
- neglecting personal hygiene
As the condition progresses, people with frontotemporal dementia may become socially isolated and withdrawn.
Speech and language problems
Some subtypes of frontotemporal dementia lead to language problems or impairment or loss of speech. Primary progressive aphasia, semantic dementia and progressive agrammatic (nonfluent) aphasia are all considered to be frontotemporal dementia.
Problems caused by these conditions include:
- Increasing difficulty in using and understanding written and spoken language, such as having trouble finding the right word to use in speech or naming objects
- Trouble naming things, possibly replacing a specific word with a more general word such as “it” for pen
- Using words incorrectly – for example, calling a sheep a dog
- No longer knowing word meanings
- Forgetting the meaning of common words
- Loss of vocabulary
- Having hesitant speech that may sound telegraphic
- Making mistakes in sentence construction
- Difficulty making the right sounds to say words
- Repeating a limited number of phrases
- Slow, hesitant speech
- Getting words in the wrong order
- Automatically repeating things other people have said
Some people gradually lose the ability to speak, and can eventually become completely mute.
Problems with mental abilities
Problems with thinking do not tend to occur in the early stages of frontotemporal dementia, but these often develop as the condition progresses. These can include:
- difficulty working things out and needing to be told what to do
- poor planning, judgement and organization
- becoming easily distracted
- thinking in a rigid and inflexible way
- losing the ability to understand abstract ideas
- difficulty recognizing familiar people or objects
- memory difficulties, although this is not common early on
Dysexecutive syndrome
Another frontotemporal dementia subtype is the dysexecutive syndrome. Here, an affected individual suffers from problems with complex reasoning and problem solving. Planning, sequencing and goal- directed behavior are involved. Problems with controlling attention and disinhibition underlie the syndrome; individuals can become stimulus-bound. These individuals cannot multitask, and they need guidance to remain on simple tasks including basic activities of daily living. In contrast, memory testing remains close to normal and these individuals may do well on simple mental status tests.
Motor disorders
Rarer subtypes of frontotemporal dementia are characterized by problems with movement similar to those associated with Parkinson’s disease or amyotrophic lateral sclerosis (ALS).
Motor-related problems may include:
- Tremor
- Rigidity
- Muscle spasms or twitches
- Slow, stiff movements, similar to Parkinson’s disease
- Poor coordination
- Difficulty swallowing
- Loss of bladder control
- Loss of bowel control
- Muscle weakness
- Inappropriate laughing or crying
- Falls or walking problems
Some people have frontotemporal dementia overlapping with other neurological (nerve and brain) problems, including:
- motor neurone disease – causes increasing weakness, usually with muscle wasting
- corticobasal degeneration – causes problems controlling limbs, loss of balance and co-ordination, slowness and reduced mobility
- progressive supranuclear palsy – causes problems with balance, movement, eye movements and swallowing.
Frontotemporal dementia complications
According to survey-based studies, sleep, or nighttime behavior disturbance is demonstrated by 33%-76% of patients with frontotemporal dementia. these are more likely to be reported in patients with primary progressive aphasia than behavior variant frontotemporal dementia (bvFTD). Similar is the case in the caregivers of primary progressive aphasia patients. All sleep disorders require sleep hygiene and routine maintenance. Also, there is a risk of an overlapping syndrome with frontotemporal dementia, such as frontotemporal dementia-motor neuron disease, frontotemporal dementia-progressive supranuclear palsy, and frontotemporal dementia-amyotrophic lateral sclerosis. All require both pharmacologic and non-pharmacologic approaches as well as treatment of underlying disease.
- Insomnia: reported in 48% of cases
- Sleep-disordered breathing: reported in 68% of cases 64
- Excessive daytime sleepiness: reported in 64% of cases
- Restless leg syndrome: reported in 8% of cases
- Risk of falls 65
- Hypersexuality
- Eating disorders, (such as hyperphagia/binge eating, lack of appetite, a particular food preference) 66
Frontotemporal dementia causes
In frontotemporal dementia, the frontal and temporal lobes of the brain shrink. In addition, there is an abnormal buildup of altered brain proteins in the frontal and temporal lobes of the brain. What causes these changes is usually unknown. In about 60% of people, they are often the first person is their family to be affected by frontotemporal degeneration. These people are classified as having a sporadic or nonfamilial form of the frontotemporal dementia. Researchers do not know why these people have developed frontotemporal degeneration. Their family members do not appear to be at an increased risk of developing the disorder. However, there is a strong genetic component to frontotemporal degeneration. In about 40% percent of cases there is a history of neurodegeneration or psychiatric illness in the family 12. In some of these people, the cause of the disorder is unknown. Researchers do not know whether there is an altered gene in these families, but the disorder occurs with more frequency than it otherwise would by chance. One theory is that, in these families, frontotemporal degeneration occurs because of the interaction of multiple factors (multifactorial cause). This might mean that there is a gene or genes that cause people to be more likely (predisposed) to develop a disorder, but the disorder will not develop unless these gene(s) are triggered or “activated” under certain circumstances such as particular environmental factors. However, despite research, no evidence has been found linking any potential environmental factor to the development of frontotemporal degeneration. There is no known risk factor for the disorder other than a family history of frontotemporal degeneration or a similar disorder. Recently, researchers have confirmed shared genetics and molecular pathways between frontotemporal dementia and amyotrophic lateral sclerosis (ALS). More research needs to be done to understand the connection between these conditions, however.
The two most common proteins involved are the tau protein and the transactive response DNA binding protein-43 (TDP-43). These proteins are normally found in the brain. Their specific functions are complex and not fully understood. However, these proteins are critical to the proper health and function of nerve cells (neurons). In individuals with frontotemporal dementia these proteins are misfolded (misshapen); as a result, they clump together. As these protein clumps, or aggregates, build up in the brain, they interfere with or disrupt the normal functioning of nerve cells, which ultimately die. Nerve cell death within the frontal and temporal lobes causes these areas of the brain shrink (atrophy).
These clumps of misfolded proteins may be referred to as “bodies” or “inclusions”. So the terms, tau bodies or tau inclusions, refer to nerve cells that contain these clumps of misfolded proteins. These inclusions may have distinctive features even when the same protein is involved. For examples, Pick’s disease refers to distinctive, round, silver-staining inclusions called Pick’s bodies. Most inclusions associated with frontotemporal dementia contain either variations of tau protein or TDP-43.
In about 10-25% of people diagnosed with frontotemporal dementia, an altered gene has been identified. Mutations in over 20 genes have been identified in the possible development of frontotemporal dementia 4. Genes provide instructions for creating proteins that play a critical role in many functions of the body. When an alteration of a gene occurs, the protein product may be faulty, inefficient, or absent. Depending upon the functions of the particular protein, this can affect many organ systems of the body, including the brain.
The major genes that have been linked to frontotemporal degeneration include the MAPT gene (microtubules associated protein tau gene located on chromosome 17q21.32), GRN gene (granulin gene on chromosome 17q21.32) and hexanucleotide repeat expansion of the chromosome 9 open-reading-frame 72 (C9orf72) gene on chromosomal location 9p21.2 (responsible for cytoskeleton organization) 13, 14. The MAPT gene provides instructions for making a protein called tau. This protein is found throughout the nervous system, including in nerve cells (neurons) in the brain. It is involved in assembling and stabilizing microtubules, which are rigid, hollow fibers that make up the cell’s structural framework (the cytoskeleton). Microtubules help cells maintain their shape, assist in the process of cell division, and are essential for the transport of materials within cells. The GRN gene produces the brain protein known as progranulin, which is important for optimal functioning of the protein TDP-43. Progranulin appears to be critical for the survival of nerve cells (neurons). The C9ORF72 gene also produces a protein that is found in nerve cells although its role in not well understood. It is also postulated that C9orf72 gene mutation is a continuum between frontotemporal dementia and amyotrophic lateral sclerosis and is responsible for the occurrence of both entities. These three genes account for the majority of people with inherited frontotemporal degeneration. In rare instances, additional genes have been identified as causing frontotemporal degeneration including the VCP, TARDBP, FUS, CHMP2B and TBK1 genes.
The altered genes that cause frontotemporal degeneration are usually inherited in an autosomal dominant manner. Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother. Dominant genetic disorders occur when only a single copy of an abnormal gene is necessary for the appearance of the disease. The altered gene can be inherited from either parent, or can be the result of a new mutation (gene change) in the affected individual. The risk of passing the abnormal gene from affected parent to offspring is 50% for each pregnancy regardless of the sex of the resulting child. If you have a family history of frontotemporal dementia, you may want to consider talking to your doctor about being referred to a geneticist and possibly having a genetic test to see if you’re at risk.
Head trauma and thyroid disease have been linked with the development of frontotemporal dementia with a 3.3-fold higher risk and 2.5-fold higher risk, respectively 4.
Is Frontotemporal dementia hereditary?
In the majority of cases, frontotemporal dementia is not being inherited within the family 67. However, people with both inherited and sporadic (not inherited) frontotemporal lobe dementia exhibit the same clinical symptoms, which makes evaluation of the family history the most sensitive tool for determining the likelihood of a genetic cause.
While scientists have found gene mutations that are linked to frontotemporal lobe dementia, most cases of frontotemporal lobe dementia are sporadic, meaning that there is no known family history of frontotemporal lobe dementia. Current genetic research shows that if a diagnosed individual has no family history of frontotemporal lobe dementia or related neurological conditions, there is less than a 10% chance that they carry a mutation in a currently known frontotemporal lobe dementia gene.
However, approximately 40% of individuals with frontotemporal lobe dementia do have a family history that includes at least one other relative who also has or had a neurodegenerative disease. In approximately 15-40% of all frontotemporal lobe dementia cases, a genetic cause (e.g., a gene mutation) can be identified as the likely cause of the disease and in most cases it is an inherited mutation.
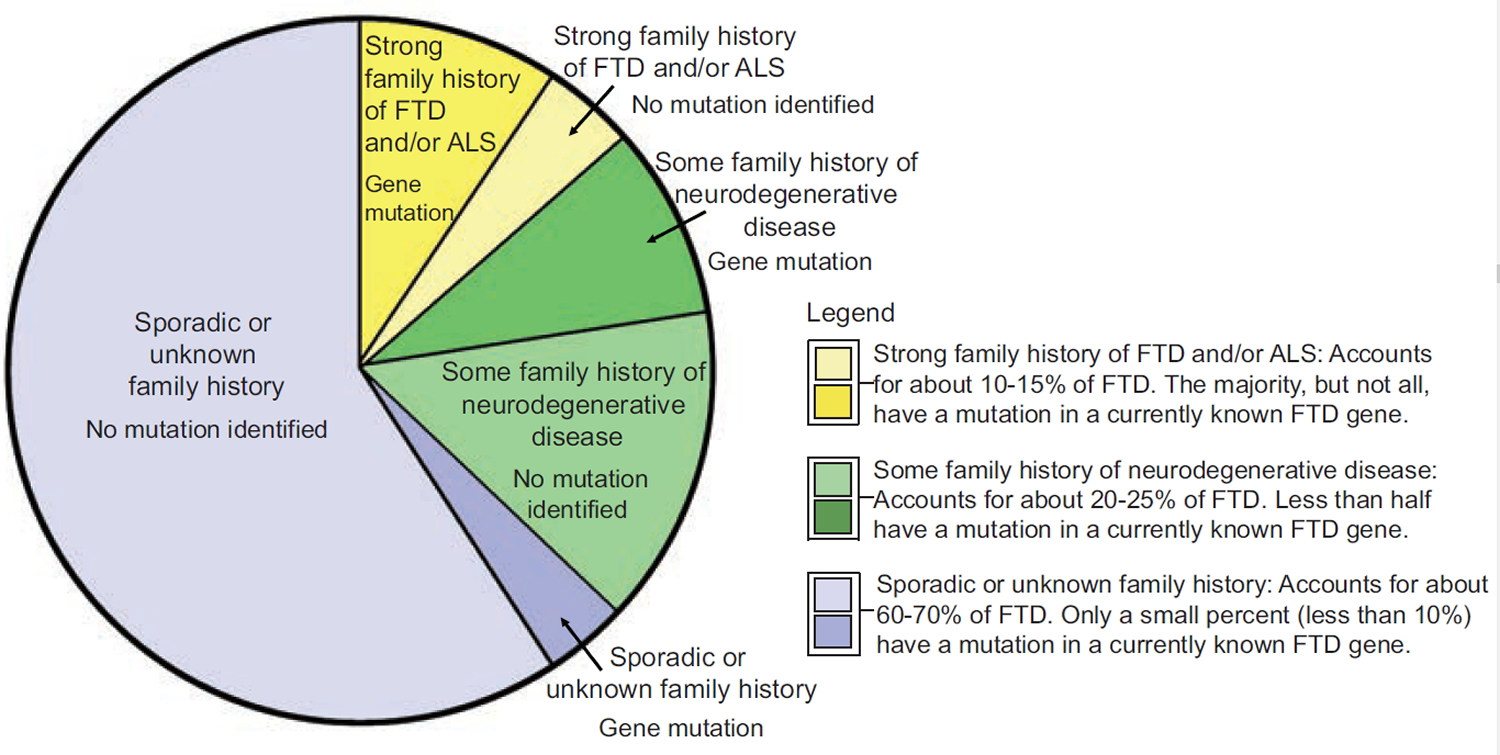
As seen in the pie chart image, families that include a history of multiple relatives with FTD (frontotemporal dementia) and/or ALS are the most likely to have an underlying genetic cause, while cases with sporadic or unknown family histories are the least likely. There are also families that fall in the middle, meaning that there is at least one relative who has or had a related neurodegenerative disease like Alzheimer’s disease or Parkinson disease, but not a clear-cut family history of FTD or ALS in close relatives. In short, the greater the family history of FTD or an associated disease like ALS, the more likely genetics plays a role. It is important to note that not all individuals with a family history of neurodegenerative disease will have an identifiable gene mutation. This could be because the responsible gene has not yet been identified, or because the disease is not actually due to a gene mutation.
There are many descriptive words used in the medical setting when discussing family history. These words are often used interchangeably, making it potentially confusing to understand each term’s meaning and significance. For example, the term “hereditary” indicates that a trait or disease can be directly transmitted between parent and offspring. “Familial” is a very broad term used to indicate that more than one person in a family has a trait or disease. “Familial” denotes that there is a possibility of a genetic cause due to apparent clustering of affected individuals in a family. This may occur by chance or due to a genetic mutation (hereditary cause). When a genetic counselor looks at a person’s family history and draws a pedigree, they look for patterns of disease in the family and depending on the number and type of affected individuals, a “pattern of inheritance” is assigned.
At this time, all known genetic forms of frontotemporal dementia are inherited in an autosomal dominant fashion (Figure 1). “Autosomal” refers to the first 22 pairs of chromosomes, which are identical in both males and females. “Dominant” inheritance means that only one copy of a gene has to have a mutation to cause disease. Typically, the copy of the gene with the mutation is inherited from a parent, and most individuals with an autosomal dominant condition will have a family history of the disease. A child of a person with a dominant mutation has a 50% chance of inheriting the copy of the gene with the mutation and a 50% chance of inheriting the copy of the gene without the mutation.
It is believed that there are likely other factors, both genetic and environmental, that influence an individual’s risk of developing FTD. There may be genes or specific mutations that do not directly cause FTD, but rather increase susceptibility to disease. This type of genetic finding is referred to as a risk factor. It is also likely that a combination of both genetic and environmental factors influence the risk of disease; this is often referred to as multifactorial inheritance. Research is underway to determine other risk factors that play a role in the development of FTD.
Figure 1. Autosomal dominant inheritance pattern
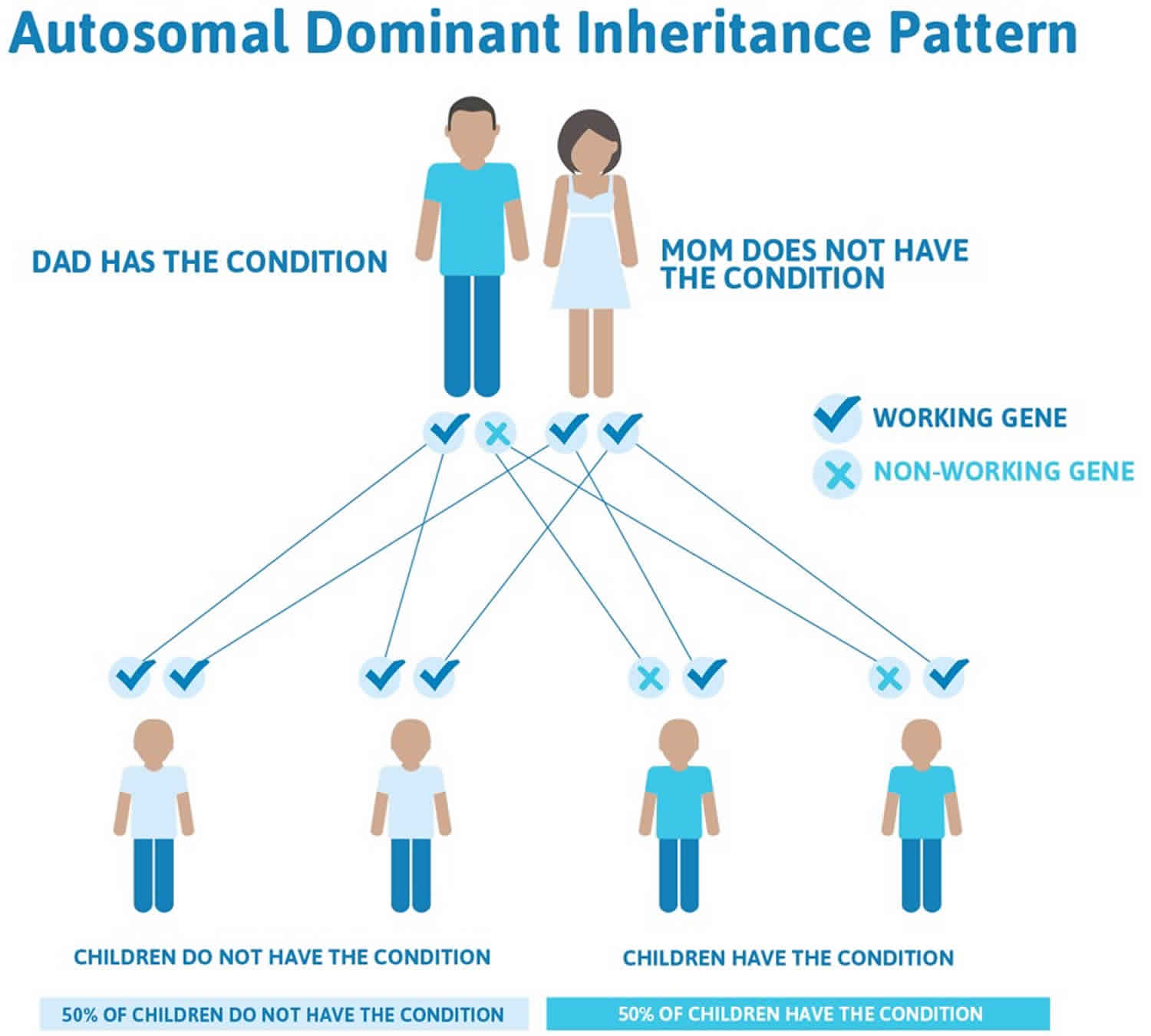
Frontotemporal dementia genetics
In about 10-25% of people diagnosed with frontotemporal lobar degeneration an altered gene has been identified. Genes are the segments of DNA that contain “recipes” for making the proteins that carry out the functions needed to keep cells alive. Variants are changes in the DNA sequence of a gene. Like typos in a cookbook recipe, variants lead to changes in the instructions for making the protein encoded by the gene.
Scientists have discovered several different genes that, when mutated, can lead to frontotemporal disorders:
- C9ORF72 gene — Most common gene associated with familial frontotemporal degeneration and familial ALS. It also occurs in some cases of sporadic ALS. This mutation can cause a frontotemporal disorder, ALS, or both conditions in a person. Individuals with the C9ORF72 mutation have abnormal accumulations of the TDP-43 protein in affected neurons. The genetic change in the C9orf72 gene is typically a repeat of a DNA segment (GGGGCC). This DNA segment is typically repeated from 2 to about 24 times for each of us. For people with familial frontotemporal degeneration or ALS, however, this GGGGCC segment is repeated more than 60 times. The abnormally expanded GGGGCC segment disrupts the C9orf72 gene, leading to a cascade of events which result in abnormal accumulation of TDP-43 protein. Repeat sizes between 24 and 60 are considered “intermediate,” but it is not clear at this time whether, or how frequently, “intermediate” expansions cause frontotemporal dementia or ALS.
- Tau gene also called the MAPT gene — The MAPT (Microtubule-associated protein tau) gene provides instructions for making a protein called tau. Tau typically plays a role in the assembly and stabilization of nerve cells in the brain cells. A mutation in MAPT gene causes abnormalities in the tau proteins, which forms tangles inside neurons and ultimately leads to the destruction of brain cells. Inheriting a mutation in MAPT gene means a person will almost surely develop frontotemporal degeneration, usually the bvFTD form, but the exact age of onset and symptoms cannot be predicted.
- GRN gene — The GRN gene provides instructions for making a protein called progranulin (GRN). A mutation in the GRN gene can lead to lower production of the protein progranulin, which in turn causes TDP-43, a cellular protein, to go awry in brain cells. Many frontotemporal degeneration can result, though bvFTD is the most common. The GRN gene can cause different symptoms in different family members and cause the disease to begin at different ages. People with GRN genetic variants have abnormal accumulation of a protein called TDP-43 in the brain, although the relationship between GRN variants, decreased levels of progranulin, and abnormal accumulation of TDP-43 protein is not fully understood today.
- VCP, CHMP2B, TARDBP, FUS, SQSTM1, CHCHD10, TBK1, OPTN, CCNF, and TIA1 genes — Mutations in these genes lead to very rare familial types of frontotemporal disorders. TARDBP and FUS gene mutations are more often associated with hereditary ALS.
Scientists are still working to find other genes that contribute to frontotemporal dementia. As with any human disease, there may be environmental or genetic risk factors that could increase susceptibility to a disease. At this time, there are no known risk factors that could indicate a susceptibility to FTD, but ongoing research is working to better understand risk factors for FTD.
All known genetic forms of frontotemporal dementia are inherited in an autosomal dominant fashion (see Figure 1), which means that the children of an affected person have a 50% chance of inheriting the genetic variant associated with frontotemporal dementia. If an individual has a mutation in one of the known FTD associated genes, there is a 50% chance of passing on the mutation and a 50% chance of NOT passing on the mutation to his/her child. This same 50/50 risk applies to every child of that individual. If the child does inherit the mutation, he or she will most likely develop symptoms of the disease at some point in adulthood, but the age of onset, severity, and type of symptoms cannot be predicted at this time.
Genetic testing can be done either with blood or saliva being collected and used to isolate a sample of DNA. This DNA sample can then be analyzed for mutations in the specific genes of interest. For individuals with an unknown genetic cause, a complete analysis for any known disease-causing genetic changes would need to be performed. However, for individuals that already know the specific gene mutation that is present in their family, the genetic test can be targeted to just check for that known family mutation.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Table 2. Frontotemporal dementia genetics and pathological correlation in frontotemporal degeneration
| Prevalence among familial FTD cases | Geographic prevalence of mutation carriers | Atrophy patterns | Common clinical presentations | FTLD proteinopathy | Mechanisms of neurodegeneration | |
|---|---|---|---|---|---|---|
| C9orf72 (9p21.2) | 13–50% | Scandinavia, west Europe, USA, Australia, rare in Asia | Symmetrical, orbitofrontal, medial and dorsolateral frontal, followed by temporal lobes, parietal and occipital lobes, cerebellum, posterior thalamus | BV-FTD FTD-MND ALS Parkinsonism Late-onset psychosis | TDP type B (less commonly type A) dipeptide repeat proteins in neocortex, thalamus, cerebellum, and hippocampus | Endosomal trafficking dysregulation; RNA-foci formation and impaired transcription processing; dipeptide repeat protein toxicity |
| MAPT (17q21.1) | 5–20% | Northwest Europe, USA | Symmetric frontal, anterior cingulate cortex, insular, anterior, and medial temporal lobe | BV-FTD Parkinsonism | Tau (often unclassifiable, occasionally resembling Pick’s disease), corticobasal degeneration or progressive supranuclear palsy | Altered microtubule stabilisation and/or increased propensity of tau self-aggregation |
| GRN (17q21.32) | 5–20% | England, central and southern Europe, USA | Asymmetrical, anterior temporal, temporo-parietal, frontal (left > PPA; right > BV-FTD), anterior cingulate cortex, insular | BV-FTD NFV-PPA Parkinsonism CBS | TDP type A | Impaired neurotrophic function, promotion of inflammation, impaired lysosomal-mediated protein degradation |
| TARDBP (1p36.22) | Rare | Italy, France, North America, Japan, China, Australia | Symmetrical frontal (type A, DLPFC; type B, VMPFC), OFC, temporal atrophy | ALS FTD-MND | TDP type A or B | Impaired RNA-binding, transcription, translation, alternative splicing, RNA transport and stabilisation |
| FUS (16p11.2) | Rare | Worldwide | Frontal and temporal atrophy with striking striatal atrophy | ALS FTD-MND | FUS | Impaired RNA-binding, transcription, translation, alternative splicing, RNA transport and stabilisation |
| VCP (9p13.3) | Rare | West Europe, USA, Brazil, Korea, Australia | Frontal, temporal, and parietal lobes, especially prefrontal cortex and superior temporal gyrus; hippocampus, caudate nucleus, amygdala | BV-FTD FTD-MND Inclusion body myopathy Paget’s disease of the bone | TDP type D | Impaired ubiquitin-proteasome mediated protein degradation and autophagy |
| CHMP2B (3p11.2) | Rare | Denmark | Generalised cortical atrophy, mostly severe in frontal and temporal cortices | BV-FTD FTD-MND | Ubiquitin proteasome system proteins | Impaired endosomal-lysosomal and autophagic protein degradation pathway |
Footnote: Pathology of frontotemporal lobar degeneration in patients with mutations in named genes, with chromosomal locations indicated in parentheses.
Abbreviations: FTD=frontotemporal dementia; FTLD=frontotemporal lobar degeneration; BV=behavioral variant; SV=semantic variant; NFV=non-fluent variant; PPA=primary progressive aphasia; MND=motor neuron disease; ALS=amyotrophic lateral sclerosis; TDP=TAR DNA-binding protein 43; FUS=fused-in-sarcoma; CBS=corticobasal syndrome; DLPFC=dorsolateral prefrontal cortex; VMPFC=ventromedial prefrontal cortex; OFC=orbitofrontal cortex.
[Source 2 ]Who can get genetic testing for frontotemporal dementia?
Any individual with frontotemporal dementia who is interested in genetic testing may get tested, although a genetic cause is more likely in those individuals who also have a family history of frontotemporal dementia. Anyone considering genetic testing or who has a family history of frontotemporal dementia should seek genetic counseling prior to genetic testing. The test is most beneficial when a family member with frontotemporal dementia is the first person tested. Once a mutation has been identified, other members of the family may choose to be tested for the same mutation even if they don’t have symptoms of the disease (presymptomatic genetic testing).
Identifying a gene that causes FTD in an affected individual will not provide an answer for curing the disease, but may help prevent the patient from being treated for the wrong diagnosis. Additionally, if an unaffected person finds out that they inherited a FTD gene mutation, there is no treatment available at this time to prevent the onset of disease.
Testing a person who is currently not showing symptoms but who is at risk for inheriting a known family mutation is called presymptomatic (or predictive) testing. For pre-symptomatic testing, a mutation must have been first identified in the family so that the clinician knows the appropriate test to order and can properly interpret the results. Individuals in the same family may have differing opinions on whether or not to pursue pre symptomatic testing. The pursuit of pre-symptomatic testing is a very personal decision that may be confusing and emotionally difficult. Therefore, genetic counseling and psychological evaluation are usually required to help the individual throughout the testing process.
The interpretation of genetic test results can be confusing. It is recommended that anyone who has genetic testing for frontotemporal dementia discuss the results with a genetic counselor or another medical genetics professional.
Frontotemporal dementia diagnosis
There’s no single test for frontotemporal dementia. A diagnosis of frontotemporal degeneration is based upon identification of characteristic symptoms, a detailed patient and family history, a thorough clinical evaluation and a variety of specialized tests. The disorder can be especially challenging to diagnose early because symptoms of frontotemporal dementia often overlap with those of other conditions. In addition, early in the course of the disorder, people with the behavioral variant of frontotemporal degeneration tend to score very well on neuropsychological testing.
Blood tests
Blood tests to help rule out other conditions, such as liver or kidney disease, your doctor may order blood tests.
Sleep study
Some symptoms of obstructive sleep apnea (memory and thinking problems and behavioral changes) can be similar to those of frontotemporal dementia. If you also have symptoms of sleep apnea (loud snoring and pauses in breathing while sleeping), your doctor may have you undergo a sleep study to rule out obstructive sleep apnea as a cause of your symptoms.
Neuropsychological testing
A neuropsychological evaluation involves an interview and certain tests, often pencil and paper type tests. This evaluation will allow a physician to assess behavior, language, memory, visual-spatial and other cognitive functions. This type of testing is especially helpful in determining the type of dementia at an early stage. The pattern of testing abnormality may help distinguish frontotemporal dementia from other causes of dementia.
Brain scans
By looking at images of the brain, doctors may be able to pinpoint any visible conditions — such as clots, bleeding or tumors — that may be causing signs and symptoms.
- Magnetic resonance imaging (MRI). An MRI machine uses radio waves and a strong magnetic field to produce detailed images of the brain.
- Fluorodeoxyglucose positron emission tracer (FDG-PET) scan. This test uses a low-level radioactive tracer attached to glucose (blood sugar) that’s injected into the blood. This allows physicians to see the chemical activity (metabolism) in the brain. The tracer can help show areas of the brain where nutrients are poorly metabolized. Areas of low metabolism can show where degeneration has occurred in the brain, which can help doctors diagnose the type of dementia. Individuals with frontotemporal degeneration exhibit reduced chemical activity (hypometabolism) in the frontal and temporal areas of the brain, a finding that can distinguish the disorder from Alzheimer disease. More recently, an amyloid PET tracer has been developed that can bind to misfolded amyloid proteins accumulated in the brain. This can support or rule out a diagnosis of Alzheimer’s disease which can then be ruled out during the diagnostic process for frontotemporal dementia. A spinal tap (lumbar puncture) can be performed to measure amyloid and tau proteins. This test can also support or decrease the likelihood of an Alzheimer’s diagnosis.
- Single-photon emission computed tomography (SPECT) scan. In SPECT, physicians use a radioactive substance and a special camera to create 3-D images of internal areas of the body, including the brain. SPECT can reveal characteristic changes such as reduced blood flow in certain areas of the brain.
Lumbar puncture
Lumbar puncture (spinal tap) to test the spinal fluid (fluid that surrounds and supports the brain and spine); this may be useful to rule out Alzheimer’s disease as the cause of symptoms.
FTD genetic testing
Frontotemporal dementia genetic testing can confirm a diagnosis of frontotemporal degeneration in certain people. Molecular genetic testing can detect mutations in the MAPT, GRN or C9ORF72 genes known to cause frontotemporal dementia, but testing is currently available only as a diagnostic service at specialized laboratories or as part of a clinical research study. Genetic testing should only be performed after genetic counseling.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Frontotemporal dementia treatments
There’s currently no cure for frontotemporal dementia or any treatment that will slow it down. Drugs used to treat or slow Alzheimer’s disease don’t seem to be helpful for people with frontotemporal dementia, and some may worsen the symptoms of frontotemporal dementia. But certain medications and speech therapy can help manage symptoms of frontotemporal dementia, possibly for several years.
Frontotemporal dementia treatments include:
- medicines – to control some of the behavioural problems
- therapies – such as physiotherapy, occupational therapy, and speech and language therapy for problems with movement, everyday tasks and communication
- dementia activities – such as memory cafes, which are drop-in sessions for people with memory problems and their carers to get support and advice
- support groups – who can offer tips on managing symptoms from dementia experts and people living with frontotemporal dementia, and their families
Medications
- Antidepressants. Some types of antidepressants, such as trazodone, may reduce the behavioral problems associated with frontotemporal dementia. Selective serotonin reuptake inhibitors (SSRIs) — such as citalopram (Celexa), paroxetine (Paxil) or sertraline (Zoloft) — also have been effective in some people.
- Antipsychotics. Antipsychotic medications, such as olanzapine (Zyprexa) or quetiapine (Seroquel), are sometimes used to treat the behavioral problems of frontotemporal dementia. However, these medications must be used with caution in people with dementia due to the risk of serious side effects, including an increased risk of death.
Therapy
People experiencing language difficulties may benefit from speech therapy to learn alternate strategies for communication.
Managing Behavior
The behaviors of a person with bvFTD can upset and frustrate family members and other caregivers. It is natural to grieve for the “lost person,” but it is also important to learn how to best live with the person he or she has become. Understanding changes in personality and behavior and knowing how to respond can reduce caregivers’ frustration and help them cope with the challenges of caring for a person with a frontotemporal disorder.
Managing behavioral symptoms can involve several approaches. To ensure the safety of a person and his or her family, caregivers may have to take on new responsibilities or arrange care that was not needed before. For example, they may have to drive the person to appointments and errands, care for young children, or arrange for help at home.
It is helpful, though often difficult, to accept rather than challenge people with behavioral symptoms. Arguing or reasoning with them will not help because they cannot control their behaviors or even see that they are unusual or upsetting to others. Instead, be as sensitive as possible and understand that it’s the illness “talking.”
Frustrated caregivers can take a “timeout”—take deep breaths, count to 10, or leave the room for a few minutes.
To deal with apathy, limit choices and offer specific choices. Open-ended questions (“What would you like to do today?”) are more difficult to answer than specific ones (“Do you want to go to the movies or the shopping center today?”).
Maintaining the person’s schedule and modifying the environment can also help. A regular schedule is less confusing and can help people sleep better. If compulsive eating is an issue, caregivers may have to supervise eating, limit food choices, lock food cabinets and the refrigerator, and distract the person with other activities. To deal with other compulsive behaviors, caregivers may have to change schedules or offer new activities.
Medications are available to treat certain behavioral symptoms. Antidepressants called selective serotonin reuptake inhibitors are commonly prescribed to treat social disinhibition and impulsive behavior. Patients with aggression or delusions sometimes take low doses of antipsychotic medications. The use of Alzheimer’s disease medications to improve behavioral and cognitive symptoms in people with bvFTD and related disorders is being studied, though results so far have been mixed, with some medications making symptoms worse. If a particular medication is not working, a doctor may try another. Always consult a doctor before changing, adding, or stopping a drug.
Treating Language Problems
Treatment of primary progressive aphasia (PPA) has two goals—maintaining language skills and using new tools and other ways to communicate. Treatment tailored to a person’s specific language problem and stage of PPA generally works best. Since language ability declines over time, different strategies may be needed as the illness progresses.
To communicate without talking, a person with PPA may use a communication notebook (an album of photos labeled with names of people and objects), gestures, and drawings. Some people find it helpful to use or point to lists of words or phrases stored in a computer or personal digital assistant.
Caregivers can also learn new ways of talking to someone with PPA. For example, they can speak slowly and clearly, use simple sentences, wait for responses, and ask for clarification if they don’t understand something.
A speech-language pathologist who knows about PPA can test a person’s language skills and determine the best tools and strategies to use. Note that many speech-language pathologists are trained to treat aphasia caused by stroke, which requires different strategies from those used with PPA. (See the Resources section starting on page 27 to find speech-language pathologists and other experts who know about frontotemporal disorders.)
Managing Movement Problems
No treatment can slow down or stop frontotemporal-related movement disorders, though medications and physical and occupational therapy may provide modest relief.
For people with corticobasal syndrome (CBS), movement difficulties are sometimes treated with medications for Parkinson’s disease. But these medicines offer only minimal or temporary improvement. Physical and occupational therapy may help people with CBS move more easily. Speech therapy may help them manage language symptoms.
For people with progressive supranuclear palsy (PSP), sometimes Parkinson’s disease drugs provide temporary relief for slowness, stiffness, and balance problems. Exercises can keep the joints limber, and weighted walking aids— such as a walker with sandbags over the lower front rung—can help maintain balance. Speech, vision, and swallowing difficulties usually do not respond to any drug treatment. Antidepressants have shown modest success. For people with abnormal eye movements.
People with FTD-ALS typically decline quickly over the course of 2 to 3 years. During this time, physical therapy can help treat muscle symptoms, and a walker or wheelchair may be useful. Speech therapy may help a person speak more clearly at first. Later on, other ways of communicating, such as a speech synthesizer, can be used. The ALS symptoms of the disorder ultimately make it impossible to stand, walk, eat, and breathe on one’s own.
For any movement disorder caused by FTLD, a team of experts can help patients and their families address difficult medical and caregiving issues. Physicians, nurses, social workers, and physical, occupational, and speech therapists who are familiar with frontotemporal disorders can ensure that people with movement disorders get appropriate medical treatment and that their caregivers can help them live as well as possible.
Home remedies
You’ll need to have caregivers, as your condition progresses, to assist with daily life activities, maintain your safety, provide transportation and help with finances. Your doctor will discuss lifestyle changes with you, such as when you may need to stop driving a car or let someone you trust take over your finances. Regular cardiovascular exercise may help improve your mood and thinking skills.
It may be helpful to make some adjustments in your home to make daily living tasks easier and reduce your chance of injuries, such as removing rugs or raising toilets.
In some cases, caregivers can reduce behavioral problems by changing the way they interact with people with dementia. Ask your loved one’s doctor about any available resources that provide training in caring for someone with dementia. Possible changes in interaction include:
- Avoiding events or activities that trigger the undesirable behavior
- Removing negative environmental cues, such as the car keys
- Maintaining a calm environment
- Providing structured routines
- Simplifying daily tasks
- Distracting and redirecting attention from problem behaviors
Care Plans
Before treatment starts, your current and future health and social care needs will be assessed, and a care plan drawn up.
This is a way of ensuring you receive the right treatment for your needs. It involves identifying areas where you may need some assistance.
These may be:
- what support you or your carer need for you to remain as independent as possible – including whether you might need care at home or in a nursing home
- whether there are any changes that need to be made to your home to make it easier to live in
- whether you need any financial assistance
Support and other therapies
In addition to medication, there are a number of therapies and practical measures that can help make everyday living easier for someone with dementia.
These include:
- occupational therapy – to identify problem areas in everyday life, such as getting dressed, and help work out practical solutions
- speech and language therapy – to help improve any communication or swallowing problems
- physiotherapy – to help with movement difficulties
- relaxation techniques – such as massage, and music or dance therapy
- social interaction, leisure activities and other dementia activities – such as memory cafés, which are drop-in sessions for people with memory problems and their carers to get support and advice
- strategies for challenging behaviour – such as distraction techniques, a structured daily routine, and activities like doing puzzles or listening to music
incontinence products if needed
It may also be helpful to get in touch with a support group, such as Rare Dementia Support, the Alzheimer’s Society or Dementia Organization.
End of life and legal issues
If you’ve been diagnosed with dementia, you might want to make arrangements for your care that take into account the decline in your mental abilities.
This may include ensuring your wishes are upheld if you’re not able to make decisions for yourself.
You may want to consider:
- drawing up an advance decision – this makes your treatment preferences known in case you’re unable to do this in the future
- having a plan for where you want to receive treatment as your condition becomes more advanced
- giving a relative lasting power of attorney – this is the power to make decisions about you if you’re unable to.
Frontotemporal dementia prognosis
How quickly frontotemporal dementia gets worse varies from person to person and is very difficult to predict.
People with the condition can become socially isolated as the illness progresses. They may not want to spend time in the company of others, or may behave in rude or insulting ways. The disease progresses steadily and often rapidly, ranging from less than 2 years in some individuals to more than 10 years in others.
Eventually some individuals with frontotemporal dementia will need 24-hour care and monitoring at home or in an institutionalized care setting. Home-based help will usually be needed at some stage, and some people will eventually need care in a nursing home.
The average survival time after symptoms start is around 8 to 10 years. But this is highly variable and some people live much longer than this.
If you or a loved one has been diagnosed with dementia, remember you’re not alone. The National Institutes of Health, National Institute on Aging and social services, as well as voluntary organisations and specialist support groups, can provide advice and support for you and your family.
The mortality risk associated with frontotemporal dementia is six-fold as compared to Alzheimer’s disease, which has a four-fold risk 14.
References- Mulkey M. Understanding Frontotemporal Disease Progression and Management Strategies. Nurs Clin North Am. 2019 Sep;54(3):437-448. doi: 10.1016/j.cnur.2019.04.011
- Bang, J., Spina, S., & Miller, B. L. (2015). Frontotemporal dementia. Lancet (London, England), 386(10004), 1672–1682. https://doi.org/10.1016/S0140-6736(15)00461-4
- Vieira, R. T., Caixeta, L., Machado, S., Silva, A. C., Nardi, A. E., Arias-Carrión, O., & Carta, M. G. (2013). Epidemiology of early-onset dementia: a review of the literature. Clinical practice and epidemiology in mental health : CP & EMH, 9, 88–95. https://doi.org/10.2174/1745017901309010088
- Khan I, De Jesus O. Frontotemporal Lobe Dementia. [Updated 2021 Aug 30]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK559286
- Pick A. Über die Beziehungen der senilen Hirnatrophie zur Aphasie. Prager Med Wochenschr. 1892;17:165–67.
- Alzheimer A. Über eigenartige Krankheitsfälle der späteren Alters. Z Gesamte Neurol Psychiatr. 1911;4:356–85.
- Mesulam MM. Primary progressive aphasia. Ann Neurol. 2001 Apr;49(4):425-32.
- Clinical and neuropathological criteria for frontotemporal dementia. The Lund and Manchester Groups. (1994). Journal of neurology, neurosurgery, and psychiatry, 57(4), 416–418. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1072868/pdf/jnnpsyc00034-0016.pdf
- Olney, N. T., Spina, S., & Miller, B. L. (2017). Frontotemporal Dementia. Neurologic clinics, 35(2), 339–374. https://doi.org/10.1016/j.ncl.2017.01.008
- Frontotemporal dementia. https://rarediseases.org/gard-rare-disease/frontotemporal-dementia
- Rohrer, J D et al. “The heritability and genetics of frontotemporal lobar degeneration.” Neurology vol. 73,18 (2009): 1451-6. https://doi.org/10.1212/WNL.0b013e3181bf997a
- Shpilyukova YA, Fedotova EY, Illarioshkin SN. [Genetic Diversity in Frontotemporal Dementia]. Mol Biol (Mosk). 2020 Jan-Feb;54(1):17-28. Russian. doi: 10.31857/S0026898420010139
- Borroni, B., Benussi, A., Premi, E., Alberici, A., Marcello, E., Gardoni, F., Di Luca, M., & Padovani, A. (2018). Biological, Neuroimaging, and Neurophysiological Markers in Frontotemporal Dementia: Three Faces of the Same Coin. Journal of Alzheimer’s disease : JAD, 62(3), 1113–1123. https://doi.org/10.3233/JAD-170584
- Bretag-Norris R, Gallur L, Flynn P. Heterogeneity in the psychiatric presentation of behavioural variant frontotemporal dementia (bvFTD). Australas Psychiatry. 2019 Oct;27(5):491-495. doi: 10.1177/1039856219860031
- Coyle-Gilchrist, I. T., Dick, K. M., Patterson, K., Vázquez Rodríquez, P., Wehmann, E., Wilcox, A., Lansdall, C. J., Dawson, K. E., Wiggins, J., Mead, S., Brayne, C., & Rowe, J. B. (2016). Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology, 86(18), 1736–1743. https://doi.org/10.1212/WNL.0000000000002638
- Knopman, D. S., & Roberts, R. O. (2011). Estimating the number of persons with frontotemporal lobar degeneration in the US population. Journal of molecular neuroscience : MN, 45(3), 330–335. https://doi.org/10.1007/s12031-011-9538-y
- National Institute of Neurological Disorders and Stroke. Frontotemporal Disorders: Hope Through Research. https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Hope-Through-Research/Frontotemporal-Disorders
- Hodges JR, Davies R, Xuereb J, Kril J, Halliday G. Survival in frontotemporal dementia. Neurology. 2003 Aug 12. 61(3):349-54.
- Frontotemporal Dementia. https://www.psp.org/iwanttolearn/prime-of-life-brain-disease/ftd/
- The Association for Frontotemporal Degeneration. Genetics of FTD. https://www.theaftd.org/wp-content/uploads/2009/03/Final-FTD-Genetics-Brochure-with-Cover-8.2.2012.pdf
- Behavioral Variant FTD. https://www.theaftd.org/what-is-ftd/behavioral-variant-ftd-bvftd
- Rascovsky, K., Hodges, J. R., Knopman, D., Mendez, M. F., Kramer, J. H., Neuhaus, J., van Swieten, J. C., Seelaar, H., Dopper, E. G., Onyike, C. U., Hillis, A. E., Josephs, K. A., Boeve, B. F., Kertesz, A., Seeley, W. W., Rankin, K. P., Johnson, J. K., Gorno-Tempini, M. L., Rosen, H., Prioleau-Latham, C. E., … Miller, B. L. (2011). Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain : a journal of neurology, 134(Pt 9), 2456–2477. https://doi.org/10.1093/brain/awr179
- Woolley, J. D., Khan, B. K., Murthy, N. K., Miller, B. L., & Rankin, K. P. (2011). The diagnostic challenge of psychiatric symptoms in neurodegenerative disease: rates of and risk factors for prior psychiatric diagnosis in patients with early neurodegenerative disease. The Journal of clinical psychiatry, 72(2), 126–133. https://doi.org/10.4088/JCP.10m06382oli
- Seeley, W. W., Crawford, R., Rascovsky, K., Kramer, J. H., Weiner, M., Miller, B. L., & Gorno-Tempini, M. L. (2008). Frontal paralimbic network atrophy in very mild behavioral variant frontotemporal dementia. Archives of neurology, 65(2), 249–255. https://doi.org/10.1001/archneurol.2007.38
- Rosen, H. J., Allison, S. C., Schauer, G. F., Gorno-Tempini, M. L., Weiner, M. W., & Miller, B. L. (2005). Neuroanatomical correlates of behavioural disorders in dementia. Brain : a journal of neurology, 128(Pt 11), 2612–2625. https://doi.org/10.1093/brain/awh628
- Seeley WW, Carlin DA, Allman JM, Macedo MN, Bush C, Miller BL, Dearmond SJ. Early frontotemporal dementia targets neurons unique to apes and humans. Ann Neurol. 2006 Dec;60(6):660-7. doi: 10.1002/ana.21055
- Seeley W. W. (2008). Selective functional, regional, and neuronal vulnerability in frontotemporal dementia. Current opinion in neurology, 21(6), 701–707. https://doi.org/10.1097/WCO.0b013e3283168e2d
- Seeley, W. W., Menon, V., Schatzberg, A. F., Keller, J., Glover, G. H., Kenna, H., Reiss, A. L., & Greicius, M. D. (2007). Dissociable intrinsic connectivity networks for salience processing and executive control. The Journal of neuroscience : the official journal of the Society for Neuroscience, 27(9), 2349–2356. https://doi.org/10.1523/JNEUROSCI.5587-06.2007
- Walker, L. C., & Jucker, M. (2015). Neurodegenerative diseases: expanding the prion concept. Annual review of neuroscience, 38, 87–103. https://doi.org/10.1146/annurev-neuro-071714-033828
- Tekin S, Cummings JL. Frontal-subcortical neuronal circuits and clinical neuropsychiatry: an update. J Psychosom Res. 2002 Aug;53(2):647-54. doi: 10.1016/s0022-3999(02)00428-2
- Liljegren, M., Naasan, G., Temlett, J., Perry, D. C., Rankin, K. P., Merrilees, J., Grinberg, L. T., Seeley, W. W., Englund, E., & Miller, B. L. (2015). Criminal behavior in frontotemporal dementia and Alzheimer disease. JAMA neurology, 72(3), 295–300. https://doi.org/10.1001/jamaneurol.2014.3781
- Rankin, K. P., Baldwin, E., Pace-Savitsky, C., Kramer, J. H., & Miller, B. L. (2005). Self awareness and personality change in dementia. Journal of neurology, neurosurgery, and psychiatry, 76(5), 632–639. https://doi.org/10.1136/jnnp.2004.042879
- Chow, T. W., Binns, M. A., Cummings, J. L., Lam, I., Black, S. E., Miller, B. L., Freedman, M., Stuss, D. T., & van Reekum, R. (2009). Apathy symptom profile and behavioral associations in frontotemporal dementia vs dementia of Alzheimer type. Archives of neurology, 66(7), 888–893. https://doi.org/10.1001/archneurol.2009.92
- Merrilees, J., Hubbard, E., Mastick, J., Miller, B. L., & Dowling, G. A. (2009). Rest-activity and behavioral disruption in a patient with frontotemporal dementia. Neurocase, 15(6), 515–526. https://doi.org/10.1080/13554790903061371
- Holroyd CB, Yeung N. Motivation of extended behaviors by anterior cingulate cortex. Trends Cogn Sci. 2012;16(2):122–128. doi: 10.1016/j.tics.2011.12.008
- Ogar J, Willock S, Baldo J, Wilkins D, Ludy C, Dronkers N. Clinical and anatomical correlates of apraxia of speech. Brain Lang. 2006;97(3):343–350. doi: 10.1016/j.bandl.2006.01.008
- Gorno-Tempini, M. L., Hillis, A. E., Weintraub, S., Kertesz, A., Mendez, M., Cappa, S. F., Ogar, J. M., Rohrer, J. D., Black, S., Boeve, B. F., Manes, F., Dronkers, N. F., Vandenberghe, R., Rascovsky, K., Patterson, K., Miller, B. L., Knopman, D. S., Hodges, J. R., Mesulam, M. M., & Grossman, M. (2011). Classification of primary progressive aphasia and its variants. Neurology, 76(11), 1006–1014. https://doi.org/10.1212/WNL.0b013e31821103e6
- Gorno-Tempini, M. L., Dronkers, N. F., Rankin, K. P., Ogar, J. M., Phengrasamy, L., Rosen, H. J., Johnson, J. K., Weiner, M. W., & Miller, B. L. (2004). Cognition and anatomy in three variants of primary progressive aphasia. Annals of neurology, 55(3), 335–346. https://doi.org/10.1002/ana.10825
- Gorno-Tempini ML, Ogar JM, Brambati SM, Wang P, Jeong JH, Rankin KP, Miller BL. Anatomical correlates of early mutism in progressive nonfluent aphasia. Neurology. 2006;67(10):1849–1851. doi: 10.1212/01.wnl.0000237038.55627.5b
- Semantic Variant PPA. https://www.theaftd.org/what-is-ftd/primary-progressive-aphasia/semantic-variant-ppa-svppa
- Seeley, W. W., Bauer, A. M., Miller, B. L., Gorno-Tempini, M. L., Kramer, J. H., Weiner, M., & Rosen, H. J. (2005). The natural history of temporal variant frontotemporal dementia. Neurology, 64(8), 1384–1390. https://doi.org/10.1212/01.WNL.0000158425.46019.5C
- Hodges JR, Mitchell J, Dawson K, Spillantini MG, Xuereb JH, McMonagle P, Patterson K. Semantic dementia: demography, familial factors and survival in a consecutive series of 100 cases. Brain. 2010;133(Pt 1):300–306. doi: 10.1093/brain/awp248
- Flanagan, E. P., Baker, M. C., Perkerson, R. B., Duffy, J. R., Strand, E. A., Whitwell, J. L., Machulda, M. M., Rademakers, R., & Josephs, K. A. (2015). Dominant frontotemporal dementia mutations in 140 cases of primary progressive aphasia and speech apraxia. Dementia and geriatric cognitive disorders, 39(5-6), 281–286. https://doi.org/10.1159/000375299
- Rosen HJ, Perry RJ, Murphy J, Kramer JH, Mychack P, Schuff N, Weiner M, Levenson RW, Miller BL. Emotion comprehension in the temporal variant of frontotemporal dementia. Brain. 2002 Oct;125(Pt 10):2286-95. doi: 10.1093/brain/awf225
- Miller BL, Boone K, Cummings JL, Read SL, Mishkin F. Functional correlates of musical and visual ability in frontotemporal dementia. Br J Psychiatry. 2000 May;176:458-63. doi: 10.1192/bjp.176.5.458
- Miller BL. Frontotemporal Dementia. Oxford University Press; 2014.
- Liu, A., Werner, K., Roy, S., Trojanowski, J. Q., Morgan-Kane, U., Miller, B. L., & Rankin, K. P. (2009). A case study of an emerging visual artist with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurocase, 15(3), 235–247. https://doi.org/10.1080/13554790802633213
- Hodges JR, Patterson K, Ward R, Garrard P, Bak T, Perry R, Gregory C. The differentiation of semantic dementia and frontal lobe dementia (temporal and frontal variants of frontotemporal dementia) from early Alzheimer’s disease: a comparative neuropsychological study. Neuropsychology. 1999 Jan;13(1):31-40. doi: 10.1037//0894-4105.13.1.31
- Logopenic Variant PPA. https://www.theaftd.org/what-is-ftd/primary-progressive-aphasia/logopenic-variant-ppa
- Armstrong, M. J., Litvan, I., Lang, A. E., Bak, T. H., Bhatia, K. P., Borroni, B., Boxer, A. L., Dickson, D. W., Grossman, M., Hallett, M., Josephs, K. A., Kertesz, A., Lee, S. E., Miller, B. L., Reich, S. G., Riley, D. E., Tolosa, E., Tröster, A. I., Vidailhet, M., & Weiner, W. J. (2013). Criteria for the diagnosis of corticobasal degeneration. Neurology, 80(5), 496–503. https://doi.org/10.1212/WNL.0b013e31827f0fd1
- Corticobasal Syndrome. https://www.theaftd.org/what-is-ftd/corticobasal-syndrome
- Donker Kaat L, Boon AJ, Kamphorst W, Ravid R, Duivenvoorden HJ, van Swieten JC. Frontal presentation in progressive supranuclear palsy. Neurology. 2007;69(8):723–729. doi: 10.1212/01.wnl.0000267643.24870.26
- Williams DR, Lees AJ. Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol. 2009 Mar;8(3):270-9. doi: 10.1016/S1474-4422(09)70042-0
- Kertesz A, McMonagle P. Behavior and cognition in corticobasal degeneration and progressive supranuclear palsy. J Neurol Sci. 2010;289(1–2):138–143. doi: 10.1016/j.jns.2009.08.036
- Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, Goetz CG, Golbe LI, Grafman J, Growdon JH, Hallett M, Jankovic J, Quinn NP, Tolosa E, Zee DS. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996 Jul;47(1):1-9. doi: 10.1212/wnl.47.1.1
- Lomen-Hoerth C. Clinical phenomenology and neuroimaging correlates in ALS-FTD. J Mol Neurosci. 2011 Nov;45(3):656-62. doi: 10.1007/s12031-011-9636-x
- Lomen-Hoerth C. Clinical phenomenology and neuroimaging correlates in ALS-FTD. J Mol Neurosci. 2011;45(3):656–662. doi: 10.1007/s12031-011-9636-x
- Geevasinga N, Loy CT, Menon P, de Carvalho M, Swash M, Schrooten M, Vucic S. Awaji criteria improves the diagnostic sensitivity in amyotrophic lateral sclerosis: A systematic review using individual patient data. Clin Neurophysiol. 2016;127(7):2684–2691. doi: 10.1016/j.clinph.2016.04.005
- Olney RK, Murphy J, Forshew D, Garwood E, Miller BL, Langmore S, Kohn MA, Lomen-Hoerth C. The effects of executive and behavioral dysfunction on the course of ALS. Neurology. 2005 Dec 13;65(11):1774-7. doi: 10.1212/01.wnl.0000188759.87240.8b
- Lomen-Hoerth C, Murphy J, Langmore S, Kramer JH, Olney RK, Miller B. Are amyotrophic lateral sclerosis patients cognitively normal? Neurology. 2003: 60 (7), 1094–1097.
- Olney RK, Murphy J, Forshew D, Garwood E, Miller BL, Langmore S, et al. The effects of executive and behavioral dysfunction on the course of ALS. Neurology. 2005; 65:1774–7.
- Alagiakrishnan K, Bhanji RA, Kurian M, Evaluation and management of oropharyngeal dysphagia in different types of dementia: a systematic review. Arch Gerontol Geriatr. 2013 Jan-Feb; 56(1):1-9.
- Boeve BF, Boylan KB, Graff-Radford NR, DeJesus-Hernandez M, Knopman DS, Pedraza O, Vemuri P, Jones D, Lowe V, Murray ME, Dickson DW, Josephs KA, Rush BK, Machulda MM, Fields JA, Ferman TJ, Baker M, Rutherford NJ, Adamson J, Wszolek ZK, Adeli A, Savica R, Boot B, Kuntz KM, Gavrilova R, Reeves A, Whitwell J, Kantarci K, Jack CR Jr, Parisi JE, Lucas JA, Petersen RC. Rademakers R. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain. 2012 Mar;135 (Pt 3):765-83.
- McCarter SJ, St Louis EK, Boeve BF. Sleep Disturbances in Frontotemporal Dementia. Curr Neurol Neurosci Rep. 2016 Sep;16(9):85. doi: 10.1007/s11910-016-0680-3
- Burrell JR, Hodges JR. Falls in frontotemporal dementia and related syndromes. Handb Clin Neurol. 2018;159:195-203. doi: 10.1016/B978-0-444-63916-5.00012-4
- Aiello M, Silani V, Rumiati RI. You stole my food! Eating alterations in frontotemporal dementia. Neurocase. 2016 Aug;22(4):400-9. doi: 10.1080/13554794.2016.1197952
- The Association for Frontotemporal Degeneration. Genetics of FTD. https://www.theaftd.org/understandingftd/genetics




{kind=link}