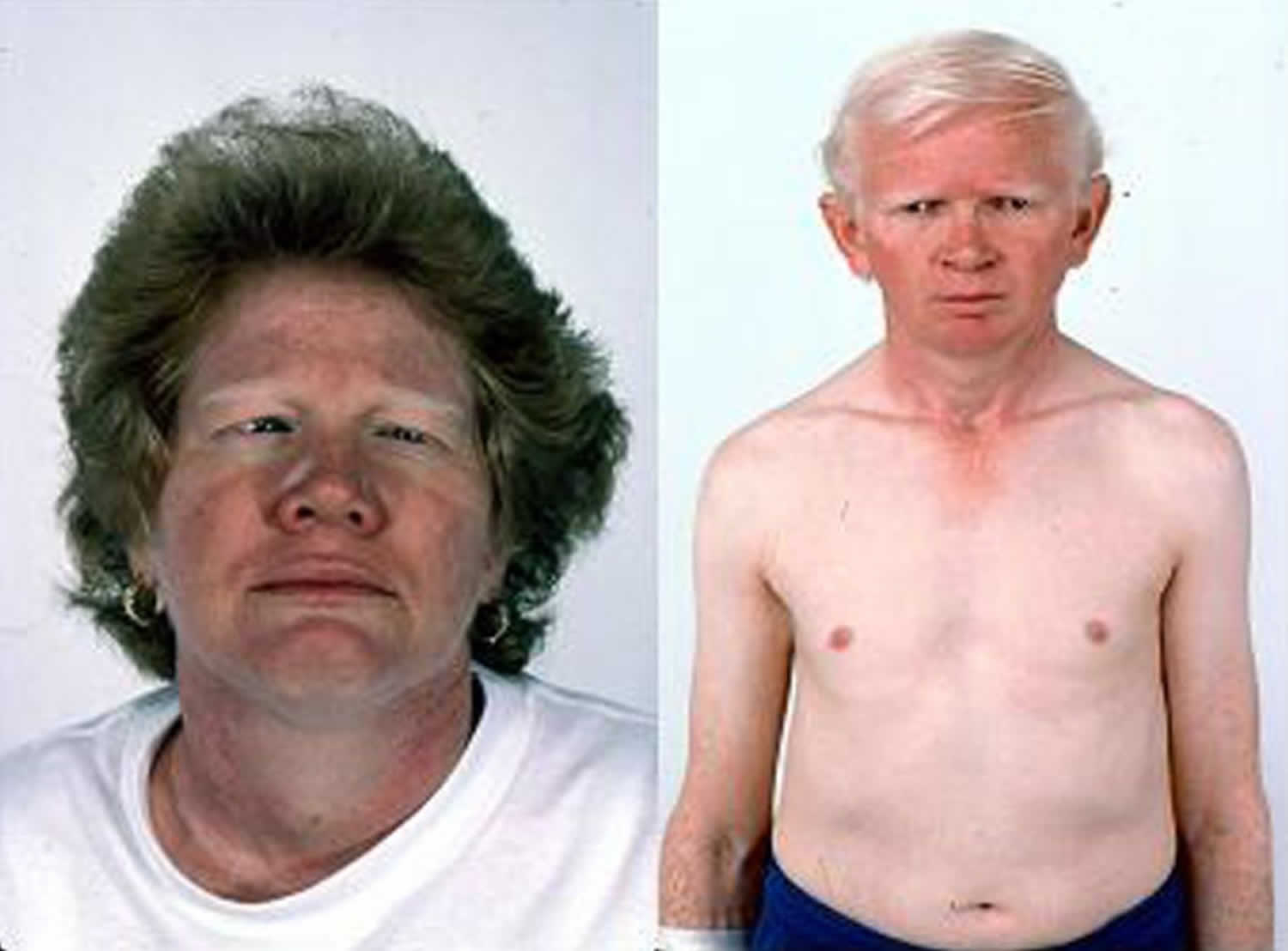
Hermansky Pudlak syndrome
Hermansky-Pudlak syndrome (HPS) also called delta storage pool disease or albinism with hemorrhagic diathesis and pigmented reticuloendothelial cells, is a rare inherited disorder characterized by a condition called oculocutaneous albinism (decreased skin pigmentation with visual impairment), which causes abnormally light coloring (pigmentation) of the skin, hair, and eyes and blood platelet dysfunction with prolonged bleeding 1. Affected individuals typically have fair skin and white or light-colored hair (albinism). Hermansky-Pudlak syndrome is the third most prevalent form of albinism. People with Hermansky-Pudlak syndrome have a higher than average risk of skin damage and skin cancers caused by long-term sun exposure. Oculocutaneous albinism reduces pigmentation of the colored part of the eye (iris) and the light-sensitive tissue at the back of the eye (retina). Reduced vision, rapid and involuntary eye movements (nystagmus), and increased sensitivity to light (photophobia) are also common in oculocutaneous albinism. In Hermansky-Pudlak syndrome, these vision problems usually remain stable after early childhood.
People with Hermansky-Pudlak syndrome also have problems with blood clotting (coagulation) that lead to easy bruising and prolonged bleeding.
Some individuals with Hermansky-Pudlak syndrome develop breathing problems due to a lung disease called pulmonary fibrosis, which causes scar tissue to form in the lungs. The symptoms of pulmonary fibrosis usually appear during an individual’s early thirties and rapidly worsen. Individuals with Hermansky-Pudlak syndrome who develop pulmonary fibrosis often do not live for more than a decade after they begin to experience breathing problems.
Other, less common features of Hermansky-Pudlak syndrome include inflammation of the large intestine (granulomatous colitis) and kidney failure.
There are nine different types of Hermansky-Pudlak syndrome, which can be distinguished by their signs and symptoms and underlying genetic cause. Types 1 and 4 are the most severe forms of the disorder. Types 1, 2, and 4 are the only types associated with pulmonary fibrosis. Individuals with type 3, 5, or 6 have the mildest symptoms. Little is known about the signs, symptoms, and severity of types 7, 8, and 9.
Hermansky-Pudlak syndrome is a rare disorder in most populations and is estimated to affect 1 in 500,000 to 1,000,000 individuals worldwide 2. Hermansky Pudlak syndrome affects males and females in equal numbers. In Puerto Rico, Hermansky Pudlak syndrome type 1 and Hermansky Pudlak syndrome type 3 are the most common subtypes. This is because certain populations in Puerto Rico were established by a small number of individuals who carried the silent gene (founder effect) 3. Hermansky-Pudlak syndrome type 1 is more common in the northwestern Puerto Rico, where about 1 in 1,800 people are affected. Hermansky-Pudlak syndrome type 3 is common in people from central Puerto Rico.
One in 21 Puerto Ricans in the northwest of the island are carriers of the gene mutation (>160,000 people) 4. Thus, it is estimated that seven or eight children with Hermansky Pudlak syndrome will be born in Puerto Rico island each year 3. In Puerto Rico, Hermansky Pudlak syndrome type 1 arises mainly as a result of a duplication/frameshift (16 base pairs) on exon 15 of the HPS1 gene, accounting for 45% of all Hermansky Pudlak syndrome cases globally and most cases on the island 3.
Groups of affected individuals have been identified in many other regions, including India, Japan, the United Kingdom, and Western Europe. Outside of Puerto Rico, Hermansky Pudlak syndrome type 1 is caused by various mutations within the HPS1 gene loci 5. Another 25% of Hermansky Pudlak syndrome cases on the island occur from a deletion (3,904 base pairs) in HPS3 gene. HPS3 is believed to have developed in the 1880s–1890s in the central mountainous region of Puerto Rico 3. Consistent with the genetic drift expected over hundreds of years, more recent studies have suggested that carrier frequencies for HPS1 and HPS3 are similar across Puerto Rico 6.
As Hermansky-Pudlak syndrome is the result of a defective gene, no curative treatment is possible.
Important management considerations include:
- Sun protection measures from an early age to prevent sun damage and skin cancers
- Referral for specialist assessments including dermatology, ophthalmology, respiratory and hematology
- Platelet transfusions before surgical procedures
- Genetic counseling for patients and their families.
Witkop and co-workers 7 reported that 76% of patients with Hermansky Pudlak syndrome die of causes directly related to the Hermansky Pudlak syndrome. The leading cause of death was pulmonary fibrosis in 50% of patients. Up to 13% of patients died from hemorrhagic episodes. Another 13% of patients with the syndrome died from complications of granulomatous enteropathic disease.
Figure 1. Hermansky Pudlak syndrome
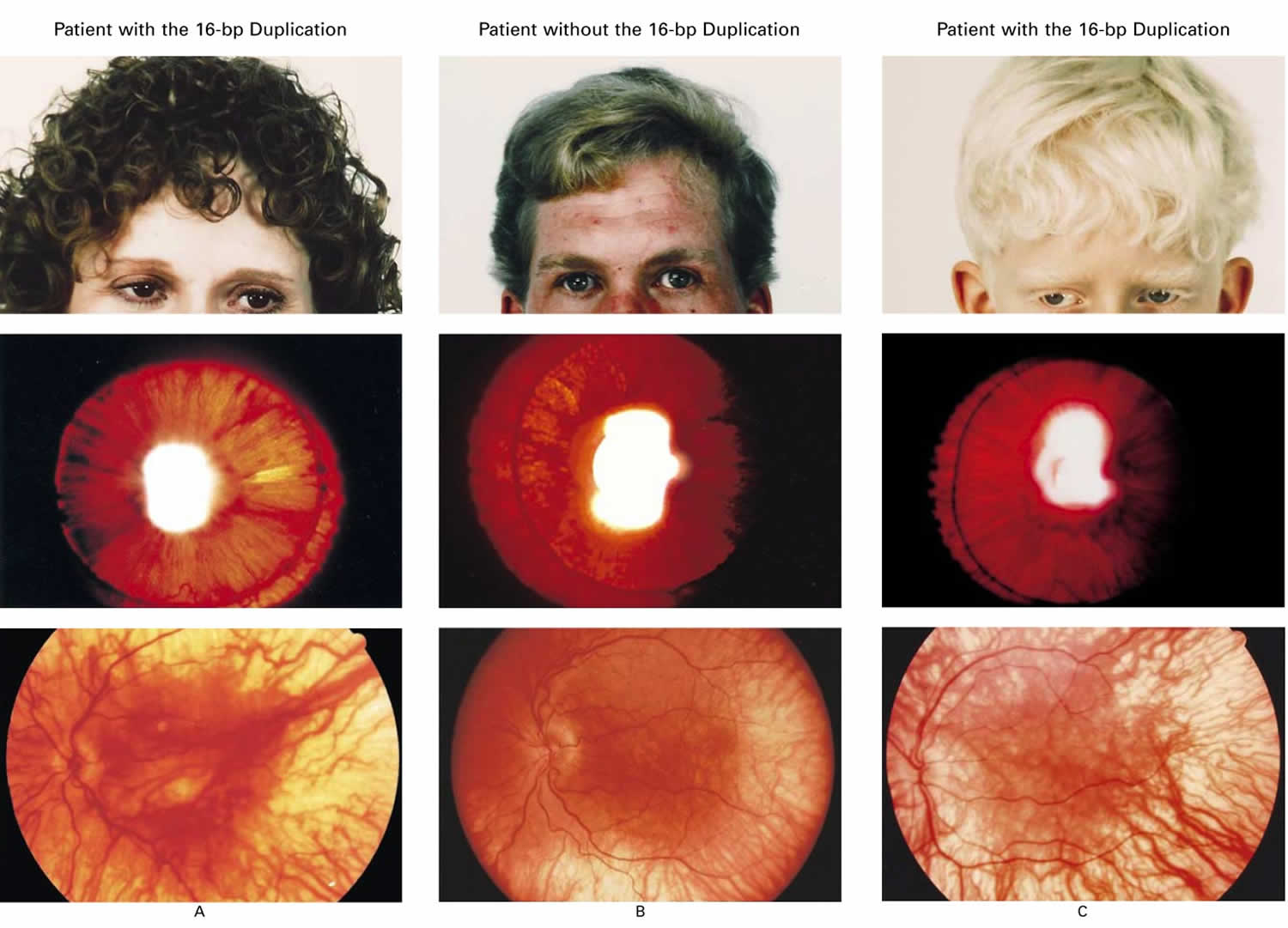
Footnote: Variability in pigmentation in three patients with Hermansky–Pudlak Syndrome. The top panels show skin and hair pigmentation, the middle panels iris transillumination, and the bottom panels choroid and retina pigmentation. Two of the patients are homozygous for the 16-bp duplication (Panels A and C), and one patient does not have the duplication (Panel B). The degree of pigmentation varied widely in both types of patients and did not distinguish members of one group from those of the other. In normal persons, pigment prevents transillumination of the iris and light appears only through the pupil. In patients with Hermansky–Pudlak syndrome, light transilluminating the iris appears orange and the areas of residual pigmentation are black.
[Source 8 ]Lysosomes are organelles that contain substances capable of breaking down various structures such as proteins, lipids, carbohydrates and nucleic acids.
Lysosome-related organelles (LROs) are structures found within specific types of cells (eg, melanocytes) that share some similarities with lysosomes. They have various functions, including involvement in processes of pigmentation, blood clotting and immunity.
Hermansky Pudlak syndrome causes
At least ten genes (HPS1, AP3B1, HPS3, HPS4, HPS5, HPS6, DTNBP1, BLOC1S3, PLDN, and AP3D1) are associated with Hermansky-Pudlak syndrome 1. These genes provide instructions for making proteins that are used to make four distinct protein complexes. These protein complexes play a role in the formation and movement (trafficking) of a group of cell structures called lysosome-related organelles (LROs). Lysosome-related organelles are very similar to compartments within the cell called lysosomes, which digest and recycle materials. However, lysosome-related organelles perform specialized functions and are found only in certain cell types. Lysosome-related organelles have been identified in pigment-producing cells (melanocytes), blood-clotting cells (platelets), and lung cells.
Mutations in one of 10 genes (HPS1, AP3B1, HPS3, HPS4, HPS5, HPS6, DTNBP1, BLOC1S3, PLDN, and AP3D1) associated with Hermansky-Pudlak syndrome prevent the formation of lysosome-related organelles or impair the functioning of these cell structures. In general, mutations in genes that involve the same protein complex cause similar signs and symptoms. People with Hermansky-Pudlak syndrome have oculocutaneous albinism because the lysosome-related organelles within melanocytes cannot produce and distribute the substance that gives skin, hair, and eyes their color (melanin). Bleeding problems are caused by the absence of lysosome-related organelles within platelets, which affects the ability of platelets to stick together and form a blood clot. Mutations in some of the genes that cause Hermansky-Pudlak syndrome affect the normal functioning of lysosome-related organelles in lung cells, leading to pulmonary fibrosis.
Mutations in the HPS1 gene cause approximately 75 percent of the Hermansky-Pudlak syndrome cases from Puerto Rico. About 45 percent of affected individuals from other populations have mutations in the HPS1 gene. Mutations in the HPS3 gene are found in about 25 percent of affected people from Puerto Rico and in approximately 20 percent of affected individuals from other areas. The other genes associated with Hermansky-Pudlak syndrome each account for a small percentage of cases of this condition.
In some people with Hermansky-Pudlak syndrome, the genetic cause of the disorder is unknown.
Hermansky Pudlak syndrome inheritance pattern
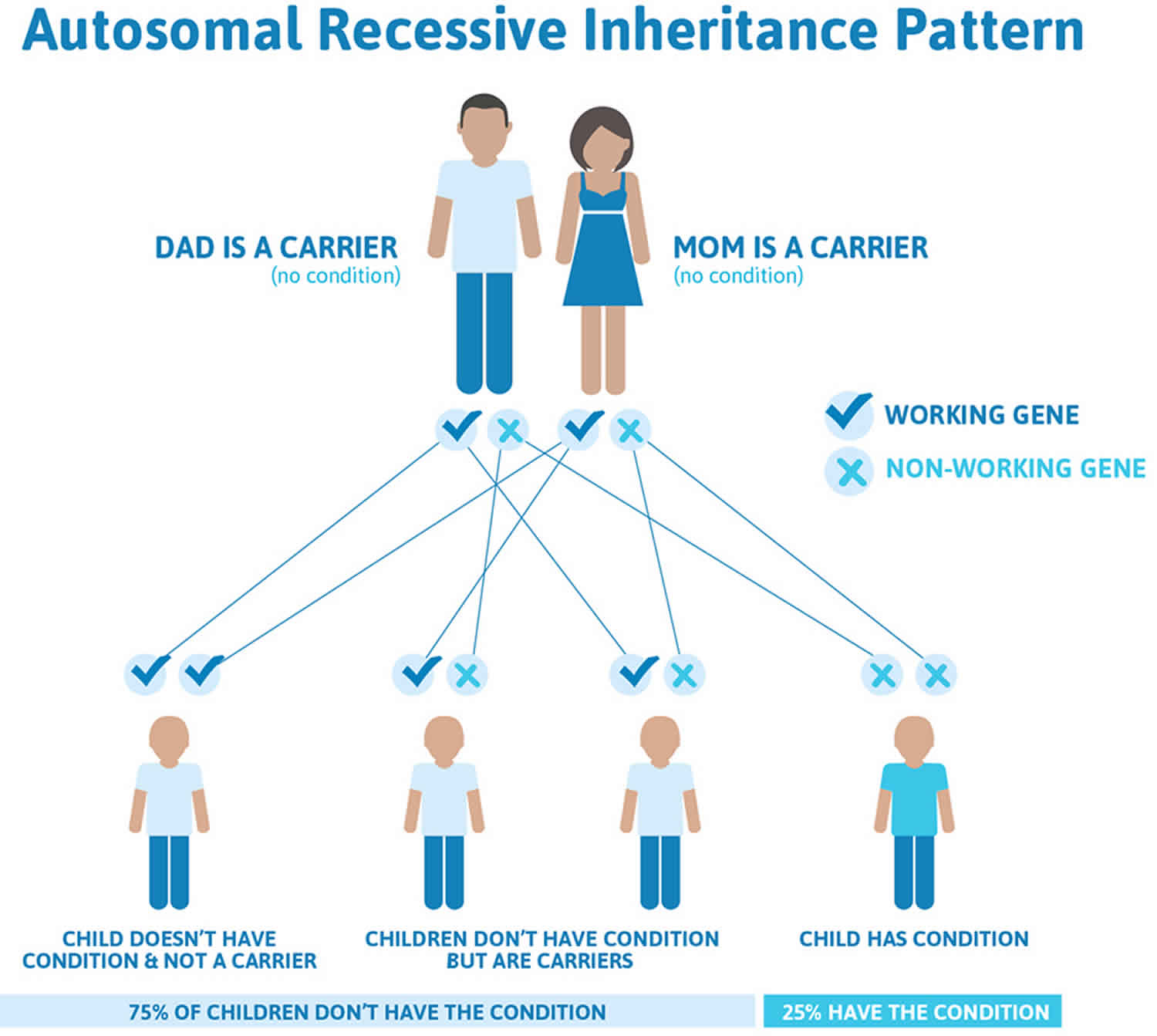
Hermansky Pudlak syndrome is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 2 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 2. Hermansky Pudlak syndrome autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Hermansky Pudlak syndrome symptoms
Hermanksy-Pudlak syndrome is characterized by:
- Oculocutaneous albinism. Cutaneous findings in Hermansky-Pudlak syndrome include:
- Varying degrees of skin and hair color pigmentary dilution, compared to first-degree relatives
- Freckles and lentigines in sun-exposed areas
- Actinic keratosis and skin cancers, especially squamous cell carcinoma.
- Visual impairment.Visual disorders in Hermanksy-Pudlak syndrome include:
- Photophobia (light sensitivity)
- Reduced visual acuity/ refractive errors
- Color vision deficiency
- Nystagmus (involuntary eye movements)
- Strabismus (horizontal and vertical eye deviations)
- Bleeding tendency due to platelet dysfunction.
Systemic conditions associated with Hermansky-Pudlak syndrome include:
- Lung fibrosis (scarring of the lungs), which may lead to reduced life expectancy
- Granulomatous inflammation of the large bowel (similar to Crohn’s disease)
- Kidney disease
- Heart disease.
The symptoms of Hermansky Pudlak syndrome are present at birth. Many babies have involuntary eye movements. The first symptoms of Hermansky Pudlak syndrome often include easy bruising, bleeding gums, nose bleeds, and excessive bleeding after surgery or accidents. The classic symptoms of Hermansky-Pudlak syndrome include the lack of color (pigmentation) in the skin, hair, and eyes (oculocutaneous albinism), and dysfunction of blood platelets leading to prolonged bleeding (storage pool-deficient platelets).
The skin, hair, and eyes of a person with Hermansky Pudlak syndrome may vary in color from very pale to almost normal coloring. Eyesight is almost always impaired, commonly with visual acuities of 20/200 or worse (i.e., legally blind). The blood storage abnormality may cause excessive bleeding, especially in women during menstruation. Bleeding may become life-threatening, and aspirin makes the bleeding worse. Approximately one-sixth of Hermansky Pudlak syndrome patients develop inflammation of the colon, with bleeding. Gastrointestinal problems including stomach pain, diarrhea, and weight loss may occur in the teens. Patients with type 1, type 2, or type 4 Hermansky Pudlak syndrome develop a lung disease called pulmonary fibrosis (scarring of the lungs) that can lead to death in their thirties, forties, or fifties.
The deposits of fatty-like substance ceroid lipofuscin may occur within the cells of many organs such as the lungs, colon, heart, and kidneys. This by itself is not known to cause symptoms.
Hermansky Pudlak syndrome diagnosis
Hermansky Pudlak syndrome is diagnosed by the clinical features of hypopigmentation of the skin and hair, characteristic eye findings, and characteristic appearance of the blood platelets under an electron microscope. Electron microscopy of platelets demonstrates the virtual absence of dense bodies, which are required for normal platelet aggregation. Molecular genetic testing for mutations in the HPS1, AP3B1, HPS3, HPS4, HPS5, HPS6, DTNBP1, BLOC1S3, PLDN, and AP3D1 genes is available to confirm the diagnosis.
Hermansky Pudlak syndrome treatment
As Hermansky Pudlak syndrome is the result of a defective gene, no curative treatment is possible. Treatment of Hermansky Pudlak syndrome patients with excessive bleeding may consist of transfusions of normal blood platelets. Women with excessive menstrual bleeding (menorrhagia) can be treated with oral contraceptives. The drug desmopressin acetate (DDAVP) can also be administered to patients with acute bleeding and has proved effective for some patients with this symptom. Individuals with Hermansky Pudlak syndrome should avoid blood anticoagulants, such as aspirin.
Patients with Hermansky Pudlak syndrome types 1, 2, or 4 who develop pulmonary fibrosis may eventually need a lung transplant. For these individuals, platelet transfusions should be used sparingly, since multiple such transfusions can induce antibodies that could cause graft rejection.
Genetic counseling is recommended for affected individuals and their families. Other treatment is symptomatic and supportive.
Hermansky Pudlak syndrome prognosis
Clinical studies show that over 70% of patients with Hermansky Pudlak syndrome die of causes directly related to the syndrome. Systemic complications of Hermansky Pudlak syndrome lead to an average patient survival age of 30-50 years. Death usually results from complications, such as pulmonary fibrosis in more than 50% of patients with Hermansky Pudlak syndrome; hemorrhagic episodes in over 15% of patients with the syndrome; and granulomatous colitis in 15% of patients with Hermansky Pudlak syndrome.
Clinical studies have reported that most patients with the syndrome are legally blind. Best-corrected visual acuity in patients with the syndrome ranges from 20/60 to 20/400.
Patients with Hermansky Pudlak syndrome have skin diseases. Clinical studies report that 80% of patients with Hermansky Pudlak syndrome have freckles or lentigines. Solar keratoses, squamous cell, and basal cell carcinoma have been reported.
Visual prognosis: Most patients with Hermansky Pudlak syndrome are legally blind. Low vision aids may benefit these patients. Presenile cataracts may further reduce vision in patients with the syndrome.
Pulmonary fibrosis is the most common cause of morbidity and mortality in patients with the syndrome. Pulmonary complications shorten the life span in patients with Hermansky Pudlak syndrome.
Multiple blood transfusions increase the risk of blood-borne infections in patients with Hermansky Pudlak syndrome.
References- Hermansky Pudlak Syndrome. https://rarediseases.org/rare-diseases/hermansky-pudlak-syndrome
- Hermansky-Pudlak syndrome. https://medlineplus.gov/genetics/condition/hermansky-pudlak-syndrome
- Santiago Borrero PJ, Rodriguez-Perez Y, Renta JY, Izquierdo NJ, Del Fierro L, Munoz D, Molina NL, Ramírez S, Pagán-Mercado G, Ortíz I, et al. Genetic testing for oculocutaneous albinism type 1 and 2 and Hermansky–Pudlak syndrome type 1 and 3 mutations in Puerto Rico. J Invest Dermatol. 2006;126:85–90.
- Witkop CJ, Nuñez Babcock M, Rao GH, Gaudier F, Summers CG, Shanahan F, Harmon KR, Townsend D, Sedano HO, King RA, et al. Albinism and Hermansky-Pudlak syndrome in Puerto Rico. Bol Asoc Med P R. 1990 Aug;82(8):333-9.
- Carmona-Rivera C, Golas G, Hess RA, Cardillo ND, Martin EH, O’Brien K, Tsilou E, Gochuico BR, White JG, Huizing M, et al. Clinical, molecular, and cellular features of non–Puerto Rican Hermansky–Pudlak syndrome patients of Hispanic descent. J Invest Dermatol. 2011;131:2394–2400.
- Hurford MT, Sebastiano C. Hermansky–Pudlak syndrome: report of a case and review of the literature. Int J Clin Exp Pathol. 2008;1:550–554.
- Witkop CJ, Nunez Babcock M, Rao GH, et al. Albinism and Hermansky-Pudlak syndrome in Puerto Rico. Bol Asoc Med P R. 1990 Aug. 82(8):333-9.
- Genetic Defects and Clinical Characteristics of Patients with a Form of Oculocutaneous Albinism (Hermansky–Pudlak Syndrome). N Engl J Med 1998; 338:1258-1265 DOI: 10.1056/NEJM199804303381803





{kind=link}