Hippocampal sclerosis
Hippocampal sclerosis also commonly referred to as mesial temporal sclerosis, is the most common brain pathological lesion underyling intractable temporal lobe epilepsy 1. Hippocampal sclerosis is the commonest cause of drug-resistant epilepsy in adults, and is associated with alterations to structures and networks beyond the hippocampus 2. In a European series of 9523 patients with epilepsy undergoing surgery, hippocampal sclerosis was identified in 36.4%, long-term epilepsy-associated tumors in 23.6%, and focal cortical dysplasias in 19.8% 3. It is not known whether hippocampal sclerosis exists before the onset of epilepsy or whether it is caused by seizures 4. Its has been proposed that childhood seizures cause hippocampal sclerosis. Hippocampal sclerosis is seen in up to 65% of autopsy studies, although significantly less on imaging.
Hippocampal sclerosis is now recognized as one of the main causes of focal epilepsy and is present in approximately 10% of adults with new-onset focal epilepsy 5. Moreover, hippocampal sclerosis often causes refractory epilepsy and is the sole pathology in about a third of all surgical resections for epilepsy and is an associated pathology so-called dual pathology in approximately 5% 6. However, the hippocampus has a twofold interest in epilepsy, in addition to being a cause of epilepsy, the hippocampus is vulnerable to damage from seizure activity 2. In particular, prolonged seizures (status epilepticus) can result in hippocampal sclerosis 2. The hippocampus is also vulnerable to other insults including traumatic brain injury, and inflammation. Hippocampal sclerosis can occur in association with other brain lesions; the prevailing view is that it is probably a secondary consequence. In such instances, successful surgical treatment usually involves the resection of both the lesion and the involved hippocampus 2. This dichotomous role of hippocampal damage as the cause and result of seizures stems possibly from its physiological role in memory formation and neuronal plasticity 7.
Hippocampal sclerosis is histopathologically seen as segmental pyramidal cell loss in CA1, CA3, and CA4 regions, whereas CA2 pyramidal and dentate gyrus granule cells are most seizure resistant (see Figure 2) 8. Neuronal cell loss is associated with reactive astrogliosis causing tissue stiffening, which has been traditionally termed as “Ammon’s horn sclerosis” 9. Some of the proposed pathomechanisms include disruption of neuronal circuitries, causing aberrant mossy fiber sprouting and molecular rearrangement/plasticity of ion channel and neurotransmitter receptor expression 10. Abnormalities have also been noted in the dentate gyrus in the form of granule cell dispersion 11. Additionally, variable cell loss is also detectable in adjacent cortical regions, including subiculum, entorhinal cortex, and amygdala.
Experimental data have pointed to numerous neuroprotective strategies to prevent hippocampal sclerosis. Initial neuroprotective strategies aimed at glutamate receptors may be effective, but later, metabolic pathways, apoptosis, reactive oxygen species, and inflammation are involved, perhaps necessitating the use of interventions aimed at multiple targets 2. Some of the therapies that are used to treat status epilepticus may be neuroprotective. However, prevention of neuronal death does not necessarily prevent the later development of epilepsy or cognitive deficits. Perhaps, the most important intervention is the early, aggressive treatment of seizure activity, and the prevention of prolonged seizures.
Figure 1. Hippocampal sclerosis
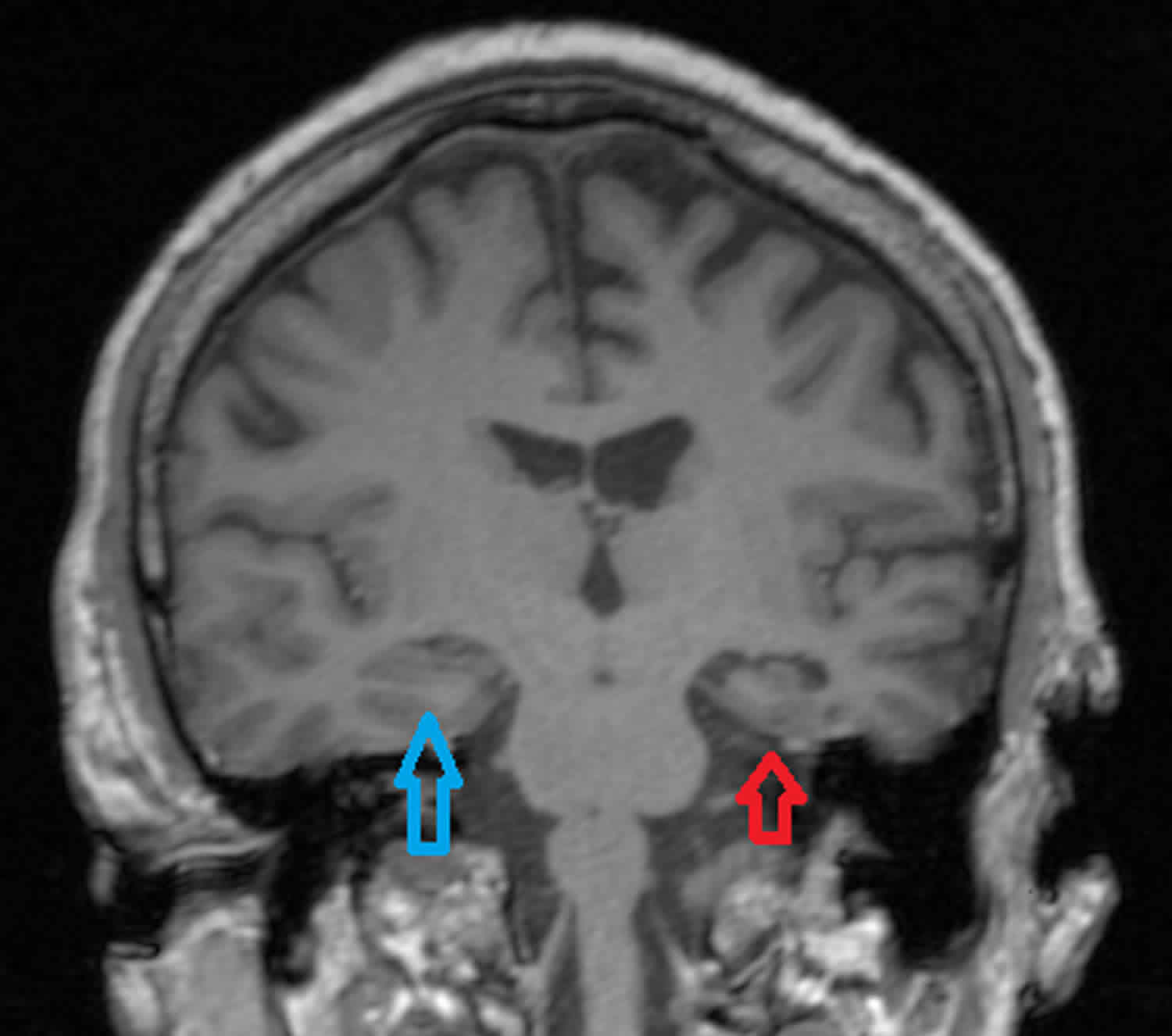
Footnote: Adult with epilepsy on medication. Moderate degree of volume loss of the left temporal lobe (temporal lobe atrophy) very much excessive for the patient’s age including extensive sclerosis of the hippocampus consistent with a structural cause for epilepsy. The left temporal horn is dilated measuring 8 mm vis-à-vis 2 mm for the normal contralateral side. Marked volume loss of the left hippocampus. The remainder of the brain is normal in appearance.
Figure 2. Hippocampal sclerosis
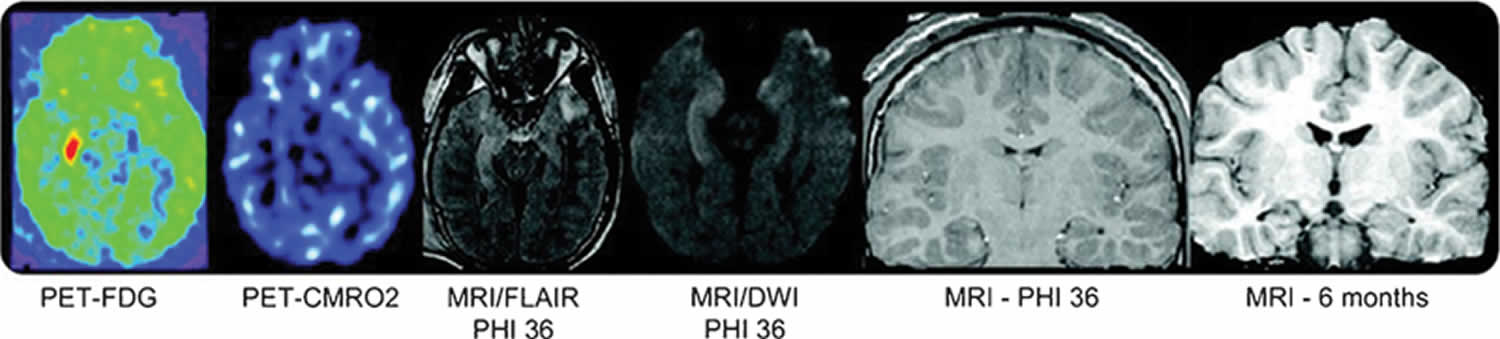
Footnote: Hippocampal atrophy ipsilateral to the seizure focus. The patient has increased glucose metabolism in the right hippocampus without a similar increase in CMRO2 (oxidative metabolism positron emission tomography [PET]). The hyperintensity on the fluid-attenuated inversion recovery (FLAIR) sequence was due to acute seizure activity and not traumatic hemorrhage. Magnetic resonance imaging scan of brain at 6 months shows right hippocampal atrophy and also right temporal lobe atrophy.
Abbreviations: CMRO2 = oxidative metabolism positron emission tomography (PET); FDG = fluorodeoxyglucose PET; PIH = postinjury hour.
[Source 12 ]Figure 3. Hippocampal sclerosis histopathology
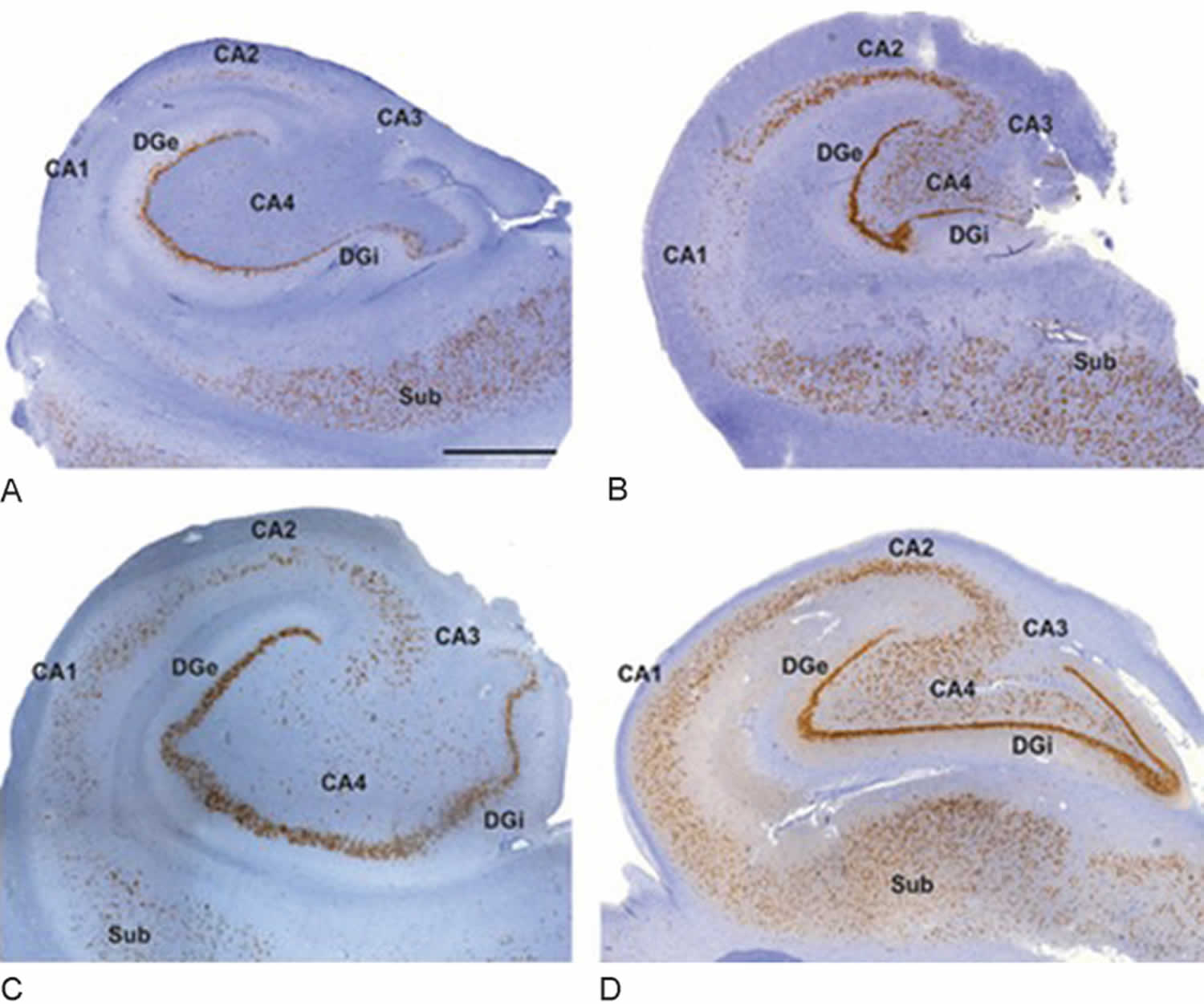
Footnote: Histopathologic subtypes of hippocampal sclerosis. (A) International League Against Epilepsy (ILAE) hippocampal sclerosis type 1 shows pronounced preferential pyramidal cell loss in both CA4 and CA1 sectors. Damage to sectors CA3 and CA2 is more variable but frequently visible. (B) ILAE hippocampal sclerosis type 2 (CA1 predominant neuronal cell loss and gliosis): This is a rarer and atypical hippocampal sclerosis pattern characterized by neuronal loss primarily involving CA1 compared with other subfields where damage is often not detectable by visual inspection (C) ILAE hippocampal sclerosis type 3 (CA4 predominant neuronal cell loss and gliosis): This is characterized by restricted cell loss mostly in CA4. (D) No hippocampal sclerosis, gliosis only. All stainings represent NeuN immunohistochemistry with hematoxylin counterstaining using 4-μm-thin paraffin embedded sections. DGe/DGI, external/internal limbs of dentate gyrus; Sub, subiculum. Scale bar in A = 1,000 μm (applies also to B–D).
[Source 13 ]Hippocampal sclerosis classification
Several classification systems have been proposed for hippocampal sclerosis. The most widely used is the new International League Against Epilepsy (ILAE) classification system (see Table 1), which divides hippocampal sclerosis into three types based on a semi-quantitative survey of segmental cell loss within hippocampal subfields. International League Against Epilepsy (ILAE) hippocampal sclerosis as type 1, previously termed classical hippocampal sclerosis, occurring in 60 to 80% of resections has both CA1 and CA4 loss; ILAE Type 2 has predominant CA1 sclerosis, occurring in 5 to 10% and ILAE Type 3 also termed end-folium sclerosis mainly involving CA4 and dentate gyrus, occurring in 4 to 7% and gliosis without hippocampal sclerosis, which may be seen in up to 20% of temporal lobe resections 14.
The International League Against Epilepsy (ILAE) classification is important because it may relate to surgical outcome–with the no hippocampal sclerosis but gliosis and the type 2 hippocampal sclerosis having the poorest outcomes (∼40% completely seizure free with 2-year follow-up) compared with approximately 70% seizure free with type 1 hippocampal sclerosis 15.
The other important question is whether the pattern of damage relates to the nature of the precipitating event, age at the time of the precipitating event, and duration of epilepsy and seizure frequency. Interestingly, seizure frequency and duration of epilepsy had minimal impact on the type of hippocampal sclerosis. However, events before the age of 3 years tended to be associated with type 1. Type 3 hippocampal sclerosis and no hippocampal sclerosis were less strongly associated with the occurrence of identifiable preceding events, which tended to happen later, during adolescence 14.
From a clinical perspective, it would be ideal to identify the pattern of cell loss before resection. Although magnetic resonance imaging (MRI) studies can reliably determine the severity of hippocampal sclerosis (confirmed by histopathology) 16, automated programs with conventional clinical MRI (1.5 T or 3 T) cannot yet reliably differentiate changes in specific hippocampal subfields 17. The severity of hippocampal sclerosis does not, however, directly translate to surgical success, but may impact the clinical pattern of the seizures, with more severe hippocampal sclerosis being associated with symptoms and signs that are usually ascribed to extratemporal lobe epilepsies 18. The challenge of identifying specific patterns of cell loss may be surmounted by increasingly sophisticated acquisition protocols at higher field strengths.
Neuronal loss and gliosis may extend beyond the hippocampus and affect the amygdala, parahippocampal gyrus, and entorhinal cortex 19. Entorhinal cortex neuronal loss has also been described in the absence of hippocampal sclerosis 20. This, and the observation that the entorhinal cortex can alone maintain seizure-like activity 21, suggests that there may an important role for this region in the generation of mesial temporal seizures.
Hippocampal sclerosis is also associated with brain abnormalities further afield, with neuronal loss reported in the thalamus 22. Indeed, thalamic atrophy seems a common finding 23, indicating that there are more widespread structural changes associated with hippocampal sclerosis, supporting the concept of medial temporal lobe epilepsy as a network phenomenon involving disparate brain areas 24. It is unclear whether the more widespread structural and functional abnormalities associated with hippocampal sclerosis are part of the syndrome or the result of secondary involvement of these regions due to repeated seizures generated from medial temporal structures. However, more widespread structural abnormalities detected by MRI do seem to predict poorer surgical outcomes 25.
Table 1. International League Against Epilepsy classification of hippocampal sclerosis
| Type 1 | CA1: > 80% cell loss CA2: 30–50% cell loss CA3, 30–90% cell loss CA4 40–90% cell loss Dentate gyrus 50–60% granule cell loss |
| Type 2 | Predominant neuronal loss in CA1, affecting almost 80% of pyramidal cells. All other sectors show mild cell loss barely visible by qualitative microscopic inspection. |
| Type 3 | Predominant cell loss in CA4 (∼50% cell loss) and the dentate gyrus (35% cell loss), whereas CA3 (< 30%), CA2 (< 25%), and CA1 (< 20%) are only moderately affected |
| Type 4 | No hippocampal sclerosis and gliosis |
Hippocampal sclerosis causes
Hippocampal sclerosis can have more than one cause, and often the cause is a complex interplay between genetic background and environmental insults. Controversy exists as to the causative mechanism: is hippocampal sclerosis a result of temporal lobe epilepsy or vice versa? In children with newly diagnosed epilepsy, only ~ 1% have evidence of hippocampal sclerosis on imaging 26. Furthermore, in adults 3-10% of cases of hippocampal sclerosis demonstrate bilateral changes even though symptoms may be unilateral 1.
In the same year that Sommer was proposing hippocampal sclerosis to be the cause of epilepsy, Pfleger 27 published evidence that the hippocampus is particularly vulnerable to damage by seizures. Pfleger 27 described hemorrhagic lesions in the mesial temporal lobe of a patient dying in status epilepticus, and concluded that neuronal necrosis was the result of impaired blood flow or metabolic disturbances that occurred during the seizures. More recent postmortem studies have also shown significant acute neuronal loss in the hippocampus of patients dying in convulsive status epilepticus 28. DeGiorgio et al 28 compared the hippocampi from five patients dying in status epilepticus, five patients with epilepsy who had a similar degree of physiological compromise (e.g., hypoglycemia, hypotension and hypoxia), and five controls. The neuronal densities were least in those dying with status epilepticus. Interestingly, a later unselected postmortem series identified that there are patients who have had a long history of seizures and even episodes of status epilepticus with no evidence of damage in the hippocampus, suggesting that status epilepticus alone may not be sufficient to cause neuronal damage 29.
From the other perspective, a significant cerebral insult or initial precipitating injury occurring early in life, such as a febrile or prolonged seizure, is often reported (between 30–50% of cases, but up to 80% in one surgical series) in retrospective studies of patients with hippocampal sclerosis 30. The “injury” hypothesis implies that this insult irreversibly damages or alters the hippocampus resulting in a template for the progression to hippocampal sclerosis following a “latent” interval. There appears to be an age-specific sensitivity for this injury, with more severe neuronal loss found when the initial precipitating injury occurs before ages of 4 to 7 years 31. The most direct evidence of the association is the observation with serial neuroimaging that hippocampal sclerosis may follow prolonged febrile convulsions 32. Approximately 10% of children with febrile seizures lasting > 30 minutes (febrile status epilepticus) have an associated high T2-weighted signal in the hippocampus with later evolution to hippocampal sclerosis in most 33. In the other 90%, there was evidence of decreased hippocampal growth, suggesting subtle hippocampal injury 33. Whether there were subtle abnormalities of the hippocampus that may have predisposed to hippocampal sclerosis was unclear. Importantly, in children, the development of hippocampal damage does not seem to be restricted to those who have had febrile status epilepticus, but can occur following any cause of convulsive status epilepticus 34.
Head injury can also result in hippocampal sclerosis. In rat models, fluid percussion injury to the dura results in interneuron loss in the hippocampus 35. The mechanism by which this occurs is unknown. The neuronal loss is accompanied by enhanced excitability of the hippocampus, and eventually spontaneous seizures. Notably, the interneuronal loss is progressive, occurring months after the insult 36. An interesting observation in humans is that traumatic brain injury may be associated with covert status epilepticus that then results in hippocampal injury 37. These experimental and human studies raise a fundamental question: Why is it that only a minority develops hippocampal sclerosis following identical brain insults?
Temporal lobe epilepsy is generally regarded as an acquired disorder with only a small genetic contribution. However, there may be common genetic variants that predispose to hippocampal sclerosis. Recently, such polymorphisms in a region of the genome encoding sodium channel subunits, in particular SCN1A, have been associated with the occurrence of febrile seizures and hippocampal sclerosis 38. This indicates that the risk of hippocampal sclerosis following a brain insult also depends upon the person’s genetic background.
In addition, an underlying maldevelopment of the hippocampus could predispose to hippocampal sclerosis and also to febrile seizures 39; this may partly explain why hippocampal sclerosis is often unilateral. In an MRI study of families with familial febrile convulsions, there were subtle pre-existing hippocampal abnormalities 40 and hippocampal sclerosis has also been reported in patients in association with isolated malformations of the hippocampus 41. In addition, persistence of calretinin positive Cajal–Retzius cells may be found in the sclerosed hippocampus, particularly when associated with febrile seizures 39. These cells secrete reelin, which plays a critical role in neuronal organization in the developing brain; it is possible that their persistence is due to either a developmental abnormality or an “insult” that predated the febrile seizures. A further argument supporting a developmental basis for hippocampal sclerosis comes from the observation that hippocampal sclerosis is often observed in association with subtle cytoarchitectural malformations in the neocortex, such as microdysgenesis 42. There may, therefore, be a more widespread maldevelopmental process involving both mesial and lateral temporal lobe structures.
Hippocampal sclerosis is also well recognized to occur in association with more severe cortical malformations, vascular malformations, and low-grade glioneuronal tumors 43. Here, the hypothesis is that the extrahippocampal lesion generates seizures or subclinical seizure activity that results in neuronal loss in the hippocampus. In such patients (i.e., those with dual pathology), removing both the lesion and the abnormal hippocampus has the best outcome in terms of seizure control, emphasizing the role of the hippocampus in temporal lobe seizures even when there is a second pathology 43.
Hippocampal sclerosis may be progressive in some patients 44, suggesting a paradigm in which an epileptogenic hippocampus generates seizures that then cause further damage to the hippocampus and more widespread structures, including the contralateral hippocampus. In support of this hypothesis, early surgical treatment (when the disease is more “confined”) results in better outcomes than late surgical intervention 45.
Lastly, infections can cause the hippocampal sclerosis. There is evidence that it can be associated with neurocysticercosis, although this is likely to be dual pathology rather than a direct impact on the hippocampus 46. Viral encephalitis often targets mesial temporal structures with the evolution to bilateral hippocampal sclerosis and profound memory difficulties emphasizing the importance of aggressive early treatment; hippocampal resection of those with an encephalitic etiology can still render people seizure free 47. Infection with herpes virus may also frequently underlie the occurrence of febrile seizures and later hippocampal sclerosis 48. More recently, autoimmune causes of hippocampal sclerosis have been identified, such as voltage-gated potassium channel antibodies 49. Usually, these autoantibodies result in an acute or subacute encephalitis with enlargement, increased T2-weighted signal, and restricted diffusion of the mesial temporal lobe structures evident on MRI. Later, hippocampal sclerosis can occur, which may be unilateral or bilateral, again emphasizing the importance of aggressive early treatment 50. In some people with established hippocampal sclerosis, autoantibodies can be found, but it is unclear whether they are pathogenic or an epiphenomenon.
Hippocampal sclerosis prevention
Endogenous neuroprotection
There are several endogenous neuroprotection pathways that could be targeted in neuroprotection. One important observation is that preconditioning (i.e., brief seizures protecting against longer seizures) seems to occur in epilepsy in a similar manner to that described in stroke. Such epileptic preconditioning alters gene expression in processes involved with calcium signaling, ion channels, and excitatory neurotransmitter receptors 51. These experimental observations suggest that the impact of status epilepticus may be less severe in people with pre-existing epilepsy.
What about specific pathways involved in neuroprotection? A variety of signaling pathways are involved in neuronal death and endogenous neuroprotection. Among these are the phosphoinositide 3-kinase (PI3-kinase)/Akt and the extracellular signal regulated kinase 1/2 (ERK1/2) pathways that can be activated through several different routes including neurotrophins and calcium entry through NMDA receptors. The PI3 kinase/Akt pathway is intimately related to the mTOR pathway, inhibitors of which are being investigated for antiepileptogenic potential 52. Phosphorylated (activated) Akt inactivates several proapoptotic proteins such as Bad, caspase-9, and transcription factors of the forkhead family 53. Thus, NMDA receptor activity can be neuroprotective. Indeed, NMDA receptor activation has a dichotomous role in neuronal survival/death; low level, chronic activation of synaptic NMDA receptors is neuroprotective, whereas sudden and excessive activation of extrasynaptic NMDA receptors is neurotoxic 54.
Reactive oxygen species can also play an important role in neuronal death. During seizure activity, these are also produced through the excessive activation of NMDA receptors and are mainly generated by cytosolic enzymes (such as NADPH oxidase) 55. There are, however, endogenous mechanisms to protect against these: These endogenous mechanisms are boosted by the transcription factor, nuclear factor erythroid 2-related factor 2 (Nrf2). Nrf2 binds to the cis-acting antioxidant response elements in the nucleus, a specific promoter sequence for genes encoding phase II and antioxidant cytoprotective proteins, including glutathione S-transferase and NADPH:quinone oxidoreductase 56. There is evidence that the ketogenic diet upregulates Nrf2 and may mediate a neuroprotective effect through this mechanism 57. However, reactive oxygen species also show a dichotomous role. Peroxynitrite, which is a potent reactive oxygen species that can result in neuronal death, at very low concentrations activates the Akt pathway and thus may be neuroprotective 58. ERK1/2 can be activated by a variety of extracellular stimuli including neurotrophins 59. ERK1/2 activation may initially counter oxidative stress, but when cellular defenses are exhausted, it serves as a signal to trigger cell death 60.
Together, many of these studies indicate that the binary view of many of these enzymes, receptors, and pathways as neurotoxic or neuroprotective is far too simplistic, and there is often a form of dualism with pathways having both neuroprotective and neurotoxic potential.
Exogenous neuroprotection
Undoubtedly, the most effective way to prevent seizure-related damage is to halt seizure activity. Indeed, the treatment of status epilepticus in the premonitory phases before status epilepticus has become established may prevent many of the pathological consequences 61. Therefore, early recognition and administration of effective treatment are paramount. If seizures continue, then the excitotoxic cascade is activated. The NMDA receptor and metabotropic glutamate group I receptor activation result in both calcium influx into the neuron and also release of calcium from internal stores 62. Other receptors and ion channels, such as voltage-gated calcium channels and calcium-permeable AMPA receptors, may also contribute to intracellular calcium accumulation. In addition, seizure-induced ion shifts may cause neuronal swelling and necrotic cell death. Inhibition of NMDA receptors before or soon after status epilepticus gives substantial and widespread neuroprotection 63, but it is likely that NMDA receptor antagonists will need to be given early to prevent calcium accumulation.
Calcium accumulation activates many distinct and interconnected downstream mechanisms, including the extrinsic caspase pathway through caspase 8 activation, the intrinsic caspase pathway activated by cytochrome c release from mitochondria, BCL-2 pathways, the formation of reactive oxygen species such as peroxynitrite, disruption of mitochondrial function through mitochondrial calcium accumulation, activation of calpain 1, activation of poly(ADP-ribose) polymerase-1, and so forth 64. There is considerable controversy about the relative roles of these different pathways. This whole area is further confounded by the possibility that different pathways are activated in different seizure models at different times, and that the mechanism of neuronal death may be region/cell specific 65. There are, however, simple measures that could be used to neuroprotect, such as anaplerosis, the replenishment of Krebs’ cycle substrates 66. The ketogenic diet could contribute to neuroprotection through this mechanism.
Independent from neuronal calcium accumulation, inflammatory mechanisms have also been implicated in neuronal death and dysfunction during prolonged seizures, and interventions targeted at brain inflammation in experimental model have shown some success 67.
Clinically, an important question is whether antiepileptic (antiseizure) drugs have any neuroprotective roles beyond their antiseizure effect. There is reasonable preclinical evidence that many of our present antiepileptic drugs have potential neuroprotective properties, but the mechanisms underlying this are not fully elucidated 68. Furthermore, many of the preclinical studies are confounded by an effect of drugs on seizure severity, making it difficult to dissociate the antiseizure from the neuroprotective effect.
Hippocampal sclerosis symptoms
Most patients with hippocampal sclerosis present with complex partial temporal lobe epilepsy.
Common features of temporal lobe epilepsy include the following:
- Memory impairment
- Aura (now called focal aware)
Auras/focal aware may be classified by symptom type, as follows:
- Sensory – auditory, gustatory, hot-cold sensations, olfactory, somatosensory, vestibular, visual
- Autonomic – Heart rate change (asytole, bradycardia, palpitations, tachycardia), flushing, gastrorintestinal, pallor, piloerection, respiratory
- Cognitive/psychic – Déjà vu or jamais vu, dissociation, depersonalization or derealization, forced thinking, aphasia/dysphasia, memory
- Emotional/affective – agitation, aggression, anger, anxiety, fear, paranoia, pleasure, crying (dacrystic) or laughing
Examples of auras include:
- A sudden sense of unprovoked fear or joy
- A deja vu experience — a feeling that what’s happening has happened before
- A sudden or strange odor or taste
- A rising sensation in the abdomen, similar to being on a roller coaster
Sometimes temporal lobe seizures impair your ability to respond to others. This type of temporal lobe seizure usually lasts 30 seconds to two minutes. Characteristic signs and symptoms include:
- Loss of awareness of surroundings
- Staring
- Lip smacking
- Repeated swallowing or chewing
- Unusual finger movements, such as picking motions
Features of temporal lobe complex partial seizure may include the following:
- Aura/focal ware
- Motionless stare, dilated pupils, and behavioral arrest
- Automatism – Oral-facial, eye blinking, alimentary, manual or unilateral dystonic limb posturing, perserveration, vocalization/speech
- Possible evolution to a secondarily generalized tonic-clonic seizure, now called bilateral tonic clonic
- Postictal period that can include confusion, aphasia, or (by definition) amnesia
After a temporal lobe seizure, you may have:
- A period of confusion and difficulty speaking
- Inability to recall what occurred during the seizure
- Unawareness of having had a seizure
- Extreme sleepiness
In extreme cases, what starts as a temporal lobe seizure evolves into a generalized tonic-clonic (grand mal) seizure — featuring convulsions and loss of consciousness.
Hippocampal sclerosis diagnosis
After a seizure, your doctor will thoroughly review your symptoms and medical history. Your doctor may order several tests to determine the cause of your seizure and evaluate how likely it is that you’ll have another one.
Tests may include:
- Neurological exam. Your doctor may test your behavior, motor abilities and mental function to determine if you have a problem with your brain and nervous system.
- Blood tests. Your doctor may take a blood sample to check for signs of infections, genetic conditions, blood sugar levels or electrolyte imbalances.
- Electroencephalogram (EEG). Electrodes attached to your scalp record the electrical activity of your brain, which shows up as wavy lines on an EEG recording. The EEG may reveal a pattern that tells doctors whether a seizure is likely to occur again, or help rule out other conditions that mimic epilepsy.
- Computerized tomography (CT) scan. A CT scan uses X-rays to obtain cross-sectional images of your brain. CT scans can reveal abnormalities in your brain that might cause a seizure, such as tumors, bleeding and cysts.
- Magnetic resonance imaging (MRI). An MRI uses powerful magnets and radio waves to create a detailed view of your brain. Your doctor may be able to detect lesions or abnormalities in your brain that could lead to seizures.
- Positron emission tomography (PET). PET scans use a small amount of low-dose radioactive material that’s injected into a vein to help visualize active areas of the brain and detect abnormalities.
- Single-photon emission computerized tomography (SPECT). A SPECT test uses a small amount of low-dose radioactive material that’s injected into a vein to create a detailed, 3-D map of the blood flow activity in your brain that happens during a seizure. Doctors may also conduct a form of a SPECT test called subtraction ictal SPECT coregistered to magnetic resonance imaging (SISCOM), which may provide even more-detailed results.
Magnetic resonance imaging (MRI)
MRI is the modality of choice to evaluate the hippocampus, however dedicated temporal lobe epilepsy protocol needs to be performed if good sensitivity and specificity is to be achieved 1. Thin section angled coronal sequences at right angles to the longitudinal axis of the hippocampus are required, to minimize volume averaging.
Coronal volume and coronal high resolution T2WI/FLAIR are best to diagnose hippocampal sclerosis.
Findings include 69:
- reduced hippocampal volume: hippocampal atrophy
- increased T2 signal
- abnormal morphology: loss of internal architecture (interdigitations of hippocampus), stratum radiata, a thin layer of white matter separates the dentate nucleus and Ammon horn
Although comparing left to right side is easiest, it must be remembered that up to 10% of cases are bilateral, and thus if symmetry is the only feature being evaluated, many cases may be misinterpreted as normal.
Often mentioned, but probably one of the least specific findings, is enlargement of the temporal horn of the lateral ventricle 1. If anything, care must be taken not to allow an enlarged horn to trick you into thinking the hippocampus is reduced in size.
When severe and long standing, additional associated findings include 69:
- atrophy of the ipsilateral fornix and mammillary body
- increased signal and or atrophy of the anterior thalamic nucleus
- atrophy of the cingulate gyrus
- increased signal and/or reduction in the volume of the amygdala
- reduction in the volume of the subiculum
- dilatation of temporal horn and temporal lobe atrophy
- collateral white matter and entorhinal cortex atrophy
- thalamic and caudate atrophy
- ipsilateral cerebral hypertrophy
- contralateral cerebellar hemiatrophy
- loss of grey-white matter interface in the anterior temporal lobe 1
- reduced white matter volume in the parahippocampal gyrus 1
Additional 3D volumetric studies can be performed, and although time consuming to post-process may be more sensitive to subtle hippocampal volume loss. Gadolinium is not required 1.
T2 relaxometry may also be useful in detecting cases of hippocampal sclerosis 1.
Diffusion MRI
As a result of neuronal loss, the extracellular space is enlarged and thus diffusion of water molecules is greater on the affected side, resulting in increased values on the affected side (higher signal on ADC).
Conversely, due to neuronal dysfunction and swelling, diffusion is restricted following a seizure, and thus values are lower 1.
MR spectroscopy
MR spectroscopy findings typically represent neuronal dysfunction 1:
- decreased NAA and decreased NAA/Cho and NAA/Cr ratios
- decreased MI in ipsilateral temporal lobe
- increased lipid and lactate soon after as seizure
MR perfusion
MR perfusion demonstrates similar changes to SPECT with blood perfusion depending on when the scan is obtained.
During the peri-ictal phases, perfusion is increased, not only in the mesial temporal lobe but often in large parts of temporal lobe and hemisphere. In interictal periods, conversely, perfusion is reduced 1.
Nuclear medicine
SPECT (Tc-99m HMPAO or ECD) and PET (F18-FDG) imaging are also a useful adjuncts, with both ictal and interictal scans demonstrating abnormalities 70:
- ictal scan: hyperperfusion
- interictal scan: hypoperfusion
Other causes of temporal lobe epilepsy should be considered, especially as small temporal lobe cortical tumors can have similar appearances.
Hippocampal sclerosis treatment
Temporal lobe epilepsy is initially managed medically with anti-epileptic agents. In patients who are refractory to medical management temporal lobectomy or selective amygdalohippocampectomy may be performed. Anterior temporal lobectomy is successful in 75-90% of patients with hippocampal sclerosis.
Following a diet that’s high in fat and low in carbohydrates, known as a ketogenic diet, can improve seizure control. Variations on a high-fat, low-carbohydrate diet, such as the low glycemic index and modified Atkins diets, may be less effective. However, they aren’t as restrictive as the ketogenic diet and might provide some benefit.
Medications
Many medications are available to treat temporal lobe seizures. However, many people don’t achieve seizure control with medications alone, and side effects, including fatigue, weight gain and dizziness, are common.
Older antiepileptic drugs used for seizure control in temporal lobe epilepsy have some long-term side effects and require lab monitoring:
- Phenytoin
- Carbamazepine
- Valproate
- Phenobarbital
Newer antiepileptic drugs appear to be comparably effective but with fewer side effects and don’t requre lab monitoring for the most part:
- Gabapentin
- Pregabalin
- Topiramate
- Lamotrigine
- Levetiracetam
- Oxcarbazepine
- Zonisamide
- Lacosamide
- Briviacetam
- Clobazam
- Rufinamide
- Perampanel
- Vigabatrin (for intractable)
- Felbamate (for intractable)
Discuss possible side effects with your doctor when deciding about treatment options. Also ask what effect your seizure medications and other medications you take, such as oral contraceptives, may have on each other.
Surgical or other procedures
When anti-seizure medications aren’t effective, other treatments may be an option:
- Surgery. The goal of surgery is to stop seizures from happening. This is often done through a traditional surgery, where surgeons operate to remove the area of the brain where seizures begin. In certain people, surgeons may be able to use MRI-guided laser therapy as a less invasive way to destroy the area of damaged tissue that causes seizures. Surgery works best for people who have seizures that always originate in the same place in their brains. Surgery generally isn’t an option if your seizures come from more than one area of the brain, your seizure focus can’t be identified or your seizures come from a region of the brain that performs vital functions.
- Temporal lobectomy is the definitive treatment for medically intractable temporal lobe epilepsy with high seizure-free rate.
- Vagus nerve stimulation (VNS). A device implanted underneath the skin of your chest stimulates the vagus nerve in your neck, sending signals to your brain that inhibit seizures. With vagus nerve stimulation, you may still need to take medication, but you may be able to lower the dose.
- Responsive neurostimulation. During responsive neurostimulation, a device implanted on the surface of your brain or within brain tissue can detect seizure activity and deliver an electrical stimulation to the detected area to stop the seizure.
- Deep brain stimulation (DBS) is awating FDA approval but is available in other countries.
- Camacho DL, Castillo M. MR imaging of temporal lobe epilepsy. Semin Ultrasound CT MR. 2007;28(6):424-436. doi:10.1053/j.sult.2007.09.005
- Walker MC. Hippocampal Sclerosis: Causes and Prevention. Semin Neurol. 2015;35(3):193-200. doi:10.1055/s-0035-1552618 https://www.thieme-connect.com/products/ejournals/html/10.1055/s-0035-1552618
- Blumcke I, Spreafico R, Haaker G, et al. Histopathological Findings in Brain Tissue Obtained during Epilepsy Surgery. N Engl J Med. 2017;377(17):1648-1656. doi:10.1056/NEJMoa1703784
- Jackson GD, Chambers BR, Berkovic SF. Hippocampal sclerosis: development in adult life. Dev Neurosci. 1999;21(3-5):207-214. https://doi.org/10.1159/000017400
- Van Paesschen W, Duncan JS, Stevens JM, Connelly A. Longitudinal quantitative hippocampal magnetic resonance imaging study of adults with newly diagnosed partial seizures: one-year follow-up results. Epilepsia 1998; 39 (6) 633-639
- Blümcke I, Spreafico R. Cause matters: a neuropathological challenge to human epilepsies. Brain Pathol 2012; 22 (3) 347-349
- Walker M, Chan D, Thom M, Hippocampus and human disease. In: Andersen P, Morris R, Amaral D, Bliss T, O’Keefe J. , eds. The Hippocampus Book. Oxford: Oxford University Press; 2007: 769-812
- Blümcke I, Coras R, Miyata H, Ozkara C. Defining clinico-neuropathological subtypes of mesial temporal lobe epilepsy with hippocampal sclerosis. Brain Pathol. 2012 May;22(3):402-11.
- Liu TT, Ye XL, Zhang JP, Yu TT, Cheng SS, Zou XC, Xu Y, Chen GQ, Yin ZY. Increased adult neurogenesis associated with reactive astrocytosis occurs prior to neuron loss in a mouse model of neurodegenerative disease. CNS Neurosci Ther. 2017 Nov;23(11):885-893.
- Becker AJ, Chen J, Zien A, Sochivko D, Normann S, Schramm J, Elger CE, Wiestler OD, Blümcke I. Correlated stage- and subfield-associated hippocampal gene expression patterns in experimental and human temporal lobe epilepsy. Eur. J. Neurosci. 2003 Nov;18(10):2792-802.
- Houser CR. Granule cell dispersion in the dentate gyrus of humans with temporal lobe epilepsy. Brain Res. 1990 Dec 10;535(2):195-204.
- Vespa PM, McArthur DL, Xu Y, et al. Nonconvulsive seizures after traumatic brain injury are associated with hippocampal atrophy. Neurology 2010;75(9):792–798.
- Blümcke I, Thom M, Aronica E, et al. International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: a Task Force report from the ILAE Commission on diagnostic methods. Epilepsia 2013;54(7):1315–1329.
- Blümcke I, Thom M, Aronica E , et al. International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: a Task Force report from the ILAE Commission on diagnostic methods. Epilepsia 2013; 54 (7) 1315-1329
- Thom M, Liagkouras I, Elliot KJ , et al. Reliability of patterns of hippocampal sclerosis as predictors of postsurgical outcome. Epilepsia 2010; 51 (9) 1801-1808
- Watson C, Nielsen SL, Cobb C, Burgerman R, Williamson B. Pathological grading system for hippocampal sclerosis: correlation with magnetic resonance imaging-based volume measurements of the hippocampus. J Epilepsy 1996; 9 (1) 56-64
- Schoene-Bake J-C, Keller SS, Niehusmann P , et al. In vivo mapping of hippocampal subfields in mesial temporal lobe epilepsy: relation to histopathology. Hum Brain Mapp 2014; 35 (9) 4718-4728
- Borelli P, Shorvon SD, Stevens JM, Smith SJ, Scott CA, Walker MC. Extratemporal ictal clinical features in hippocampal sclerosis: their relationship to the degree of hippocampal volume loss and to the outcome of temporal lobectomy. Epilepsia 2008; 49 (8) 1333-1339
- Bernasconi N, Bernasconi A, Caramanos Z, Antel SB, Andermann F, Arnold DL. Mesial temporal damage in temporal lobe epilepsy: a volumetric MRI study of the hippocampus, amygdala and parahippocampal region. Brain 2003; 126 (Pt 2) 462-469
- Bernasconi N, Bernasconi A, Caramanos Z , et al. Entorhinal cortex atrophy in epilepsy patients exhibiting normal hippocampal volumes. Neurology 2001; 56 (10) 1335-1339
- Jones RS. Ictal epileptiform events induced by removal of extracellular magnesium in slices of entorhinal cortex are blocked by baclofen. Exp Neurol 1989; 104 (2) 155-161
- Sinjab B, Martinian L, Sisodiya SM, Thom M. Regional thalamic neuropathology in patients with hippocampal sclerosis and epilepsy: a postmortem study. Epilepsia 2013; 54 (12) 2125-2133
- Barron DS, Fox PM, Laird AR, Robinson JL, Fox PT. Thalamic medial dorsal nucleus atrophy in medial temporal lobe epilepsy: a VBM meta-analysis. Neuroimage Clin 2012; 2: 25-32
- Coan AC, Campos BM, Yasuda CL , et al. Frequent seizures are associated with a network of gray matter atrophy in temporal lobe epilepsy with or without hippocampal sclerosis. PLoS ONE 2014; 9 (1) e85843
- Keller SS, Richardson MP, Schoene-Bake J-C , et al. Thalamotemporal alteration and postoperative seizures in temporal lobe epilepsy. Ann Neurol 2015; ; Epub ahead of print
- Tarkka R, Pääkkö E, Pyhtinen J, Uhari M, Rantala H. Febrile seizures and mesial temporal sclerosis: No association in a long-term follow-up study. Neurology. 2003;60(2):215-218. doi:10.1212/01.wnl.0000037482.55894.b1
- Pfleger L. Beobachtungen uber schrumpfung und sclerose des ammonshornes bei epilepsie. Allg Zeitschrift für Psychiatr 1880; 36: 359-365
- DeGiorgio CM, Tomiyasu U, Gott PS, Treiman DM. Hippocampal pyramidal cell loss in human status epilepticus. Epilepsia 1992; 33 (1) 23-27
- Thom M, Zhou J, Martinian L, Sisodiya S. Quantitative post-mortem study of the hippocampus in chronic epilepsy: seizures do not inevitably cause neuronal loss. Brain 2005; 128 (Pt 6) 1344-1357
- French JA, Williamson PD, Thadani VM , et al. Characteristics of medial temporal lobe epilepsy: I. Results of history and physical examination. Ann Neurol 1993; 34 (6) 774-780
- Davies KG, Hermann BP, Dohan Jr FC, Foley KT, Bush AJ, Wyler AR. Relationship of hippocampal sclerosis to duration and age of onset of epilepsy, and childhood febrile seizures in temporal lobectomy patients. Epilepsy Res 1996; 24 (2) 119-126
- Lewis DV, Barboriak DP, MacFall JR, Provenzale JM, Mitchell TV, VanLandingham KE. Do prolonged febrile seizures produce medial temporal sclerosis? Hypotheses, MRI evidence and unanswered questions. Prog Brain Res 2002; 135: 263-278
- Lewis DV, Shinnar S, Hesdorffer DC , et al; FEBSTAT Study Team. Hippocampal sclerosis after febrile status epilepticus: the FEBSTAT study. Ann Neurol 2014; 75 (2) 178-185
- Yoong M, Martinos MM, Chin RF, Clark CA, Scott RC. Hippocampal volume loss following childhood convulsive status epilepticus is not limited to prolonged febrile seizures. Epilepsia 2013; 54 (12) 2108-2115
- Lowenstein DH, Thomas MJ, Smith DH, McIntosh TK. Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci 1992; 12 (12) 4846-4853
- Pavlov I, Huusko N, Drexel M , et al. Progressive loss of phasic, but not tonic, GABAA receptor-mediated inhibition in dentate granule cells in a model of post-traumatic epilepsy in rats. Neuroscience 2011; 194: 208-219
- Vespa PM, McArthur DL, Xu Y , et al. Nonconvulsive seizures after traumatic brain injury are associated with hippocampal atrophy. Neurology 2010; 75 (9) 792-798
- Kasperaviciute D, Catarino CB, Matarin M , et al; UK Brain Expression Consortium. Epilepsy, hippocampal sclerosis and febrile seizures linked by common genetic variation around SCN1A. Brain 2013; 136 (Pt 10) 3140-3150
- Blümcke I, Thom M, Wiestler OD. Ammon’s horn sclerosis: a maldevelopmental disorder associated with temporal lobe epilepsy. Brain Pathol 2002; 12 (2) 199-211
- Fernández G, Effenberger O, Vinz B , et al. Hippocampal malformation as a cause of familial febrile convulsions and subsequent hippocampal sclerosis. Neurology 1998; 50 (4) 909-917
- Baulac M, De Grissac N, Hasboun D , et al. Hippocampal developmental changes in patients with partial epilepsy: magnetic resonance imaging and clinical aspects. Ann Neurol 1998; 44 (2) 223-233
- Hardiman O, Burke T, Phillips J , et al. Microdysgenesis in resected temporal neocortex: incidence and clinical significance in focal epilepsy. Neurology 1988; 38 (7) 1041-1047
- Li LM, Cendes F, Andermann F , et al. Surgical outcome in patients with epilepsy and dual pathology. Brain 1999; 122 (Pt 5) 799-805
- Briellmann RS, Berkovic SF, Syngeniotis A, King MA, Jackson GD. Seizure-associated hippocampal volume loss: a longitudinal magnetic resonance study of temporal lobe epilepsy. Ann Neurol 2002; 51 (5) 641-644
- Janszky J, Janszky I, Schulz R , et al. Temporal lobe epilepsy with hippocampal sclerosis: predictors for long-term surgical outcome. Brain 2005; 128 (Pt 2) 395-404
- Bianchin MM, Velasco TR, Santos AC, Sakamoto AC. On the relationship between neurocysticercosis and mesial temporal lobe epilepsy associated with hippocampal sclerosis: coincidence or a pathogenic relationship?. Pathog Glob Health 2012; 106 (5) 280-285
- Davies KG, Hermann BP, Wyler AR. Surgery for intractable epilepsy secondary to viral encephalitis. Br J Neurosurg 1995; 9 (6) 759-762
- Epstein LG, Shinnar S, Hesdorffer DC , et al; FEBSTAT study team. Human herpesvirus 6 and 7 in febrile status epilepticus: the FEBSTAT study. Epilepsia 2012; 53 (9) 1481-1488
- Vincent A, Buckley C, Schott JM , et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain 2004; 127 (Pt 3) 701-712
- Wagner J, Witt J-A, Helmstaedter C, Malter MP, Weber B, Elger CE. Automated volumetry of the mesiotemporal structures in antibody-associated limbic encephalitis. J Neurol Neurosurg Psychiatry 2014; ; (e-pub ahead of print).
- Jimenez-Mateos EM, Hatazaki S, Johnson MB , et al. Hippocampal transcriptome after status epilepticus in mice rendered seizure damage-tolerant by epileptic preconditioning features suppressed calcium and neuronal excitability pathways. Neurobiol Dis 2008; 32 (3) 442-453
- Ostendorf AP, Wong M. mTOR Inhibition in Epilepsy: Rationale and Clinical Perspectives. CNS Drugs 2015; 29 (2) 91-99
- Brunet A, Bonni A, Zigmond MJ , et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999; 96 (6) 857-868
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 2010; 11 (10) 682-696
- Kovac S, Domijan A-M, Walker MC, Abramov AY. Seizure activity results in calcium- and mitochondria-independent ROS production via NADPH and xanthine oxidase activation. Cell Death Dis 2014; 5: e1442
- Baird L, Dinkova-Kostova AT. The cytoprotective role of the Keap1-Nrf2 pathway. Arch Toxicol 2011; 85 (4) 241-272
- Milder JB, Liang L-P, Patel M. Acute oxidative stress and systemic Nrf2 activation by the ketogenic diet. Neurobiol Dis 2010; 40 (1) 238-244
- Delgado-Esteban M, Martin-Zanca D, Andres-Martin L, Almeida A, Bolaños JP. Inhibition of PTEN by peroxynitrite activates the phosphoinositide-3-kinase/Akt neuroprotective signaling pathway. J Neurochem 2007; 102 (1) 194-205
- Hetman M, Gozdz A. Role of extracellular signal regulated kinases 1 and 2 in neuronal survival. Eur J Biochem 2004; 271 (11) 2050-2055
- Luo Y, DeFranco DB. Opposing roles for ERK1/2 in neuronal oxidative toxicity: distinct mechanisms of ERK1/2 action at early versus late phases of oxidative stress. J Biol Chem 2006; 281 (24) 16436-16442
- Treiman DM, Walker MC. Treatment of seizure emergencies: convulsive and non-convulsive status epilepticus. Epilepsy Res 2006; 68 (Suppl. 01) S77-S82
- Meldrum BS. Implications for neuroprotective treatments. Prog Brain Res 2002; 135: 487-495
- Fujikawa DG, Daniels AH, Kim JS. The competitive NMDA receptor antagonist CGP 40116 protects against status epilepticus-induced neuronal damage. Epilepsy Res 1994; 17 (3) 207-219
- Rowley S, Patel M. Mitochondrial involvement and oxidative stress in temporal lobe epilepsy. Free Radic Biol Med 2013; 62: 121-131
- Narkilahti S, Pirttilä TJ, Lukasiuk K, Tuunanen J, Pitkänen A. Expression and activation of caspase 3 following status epilepticus in the rat. Eur J Neurosci 2003; 18 (6) 1486-1496
- Kovac S, Abramov AY, Walker MC. Energy depletion in seizures: anaplerosis as a strategy for future therapies. Neuropharmacology 2013; 69: 96-104
- Vezzani A, Aronica E, Mazarati A, Pittman QJ. Epilepsy and brain inflammation. Exp Neurol 2013; 244: 11-21
- Pitkänen A, Kubova H. Antiepileptic drugs in neuroprotection. Expert Opin Pharmacother 2004; 5 (4) 777-798
- Chan S, Erickson JK, Yoon SS. Limbic system abnormalities associated with mesial temporal sclerosis: a model of chronic cerebral changes due to seizures. Radiographics. 1997;17(5):1095-1110. doi:10.1148/radiographics.17.5.9308104
- Juni JE1, Waxman AD, Devous MD Sr, Tikofsky RS, Ichise M, Van Heertum RL, Holman BL, Carretta RF, Chen CC. Procedure guideline for brain perfusion SPECT using technetium-99m radiopharmaceuticals. Society of Nuclear Medicine. J Nucl Med. 1998 May;39(5):923-6





{kind=link}