What is holoprosencephaly
Holoprosencephaly is a complex brain malformation caused by the failure of the prosencephalon (the embryonic forebrain) to sufficiently divide into the double lobes of the cerebral hemispheres, occurring between the 18th and the 28th day of gestation and affecting both the forebrain and the face 1. The result is a single-lobed brain structure and severe skull and facial defects. In most cases of holoprosencephaly, the malformations are so severe that babies die before birth. In less severe cases, babies are born with normal or near-normal brain development and facial deformities that may affect the eyes, nose, and upper lip.
Holoprosencephaly is divided into alobar, semilobar and lobar forms, although there are no clear-cut defining features. A series of facial anomalies are frequently associated, owing to the common origin of the embryonic forebrain and mid-face from the prechordal mesoderm, along with some other anomalies.
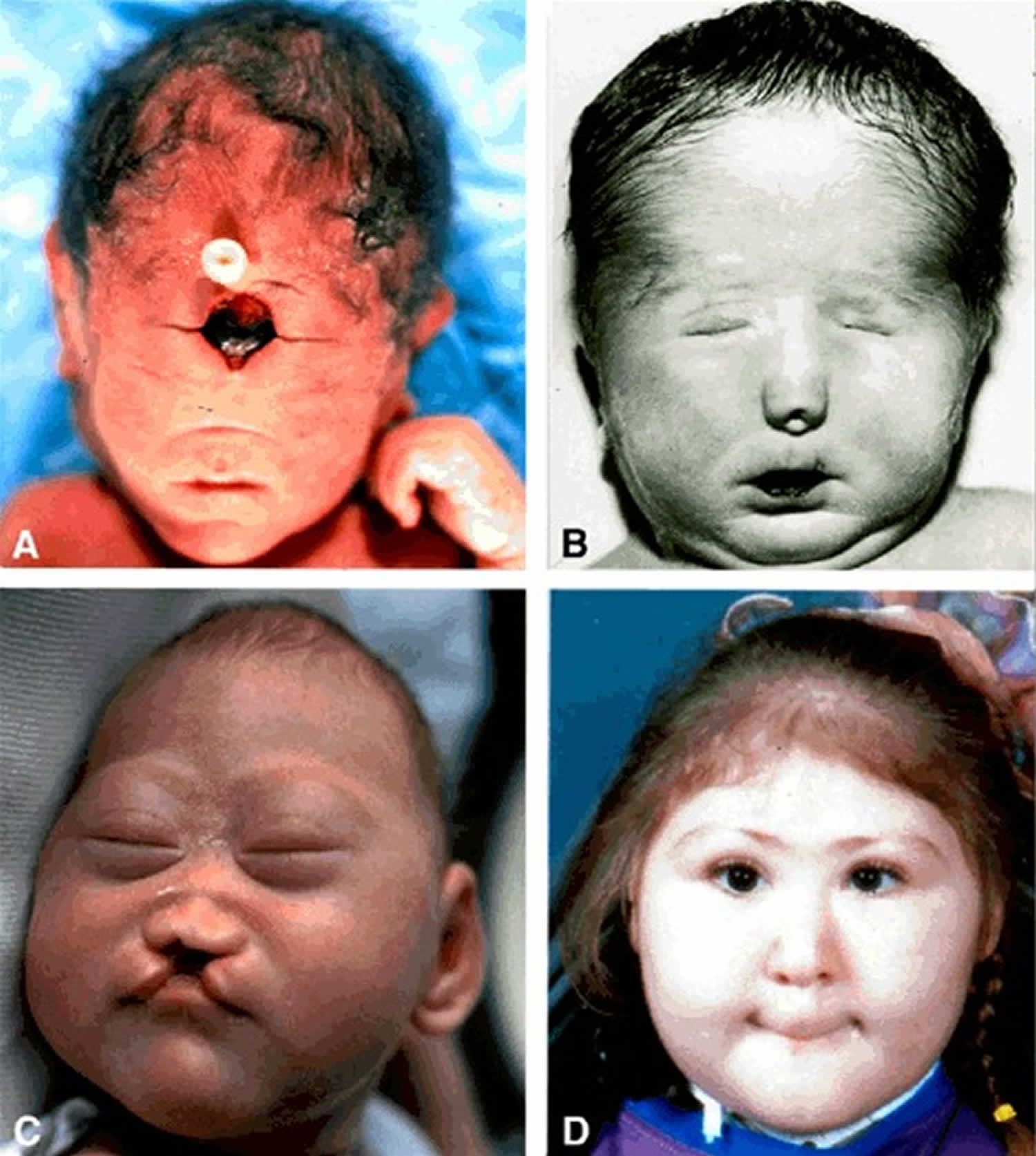
- Alobar holoprosencephaly, in which the brain has not divided at all, is usually associated with severe facial deformities. Cyclopia: single eye or partially divided eye in single orbit with a proboscis above the eye
- Range of findings:
- Cyclopia without proboscis
- Ethmocephaly: extremely closely spaced eyes but separate orbits with proboscis between the eyes
- Cebocephaly: closely spaced eyes with single-nostril nose
- Closely spaced eyes
- Anophthalmia or microophthalmia
- Premaxillary agenesis with median cleft lip, closely spaced eyes, depressed nasal ridge
- Bilateral cleft lip
- Relatively normal facial appearance (especially in persons with pathogenic variants in ZIC2)
- Range of findings:
- Semilobar holoprosencephaly, in which the brain’s hemispheres have somewhat divided, causes an intermediate form of the disorder.
- Range of findings:
- Closely spaced eyes
- Anophthalmia/microophthalmia
- Depressed nasal bridge
- Absent nasal septum
- Flat nasal tip
- Bilateral cleft lip with median process representing the philtrum-premaxilla anlage
- Midline cleft (lip and/or palate)
- Relatively normal facial appearance
- Range of findings:
- Lobar holoprosencephaly, in which there is considerable evidence of separate brain hemispheres, is the least severe form. In some cases of lobar holoprosencephaly the baby’s brain may be nearly normal.
- Range of findings:
- Bilateral cleft lip with median process
- Closely spaced eyes
- Depressed nasal ridge
- Relatively normal facial appearance
- Range of findings:
- Another milder subtype of holoprosencephaly called middle interhemispheric fusion variant (MIHF/MIHV) or syntelencephaly, in which the posterior frontal and parietal lobes fail to separate, with varying lack of cleavage of the basal ganglia and thalami and absence of the body of the corpus callosum but presence of the genu and splenium of the corpus callosum, is also reported 2.
- Range of findings:
- Closely spaced eyes
- Depressed nasal bridge
- Narrow nasal bridge
- Relatively normal facial appearance
- Range of findings:
In most of the cases, facial anomalies are observed in holoprosencephaly, like 1:
- Cyclopia, in which a single, midline, fused eye exists in a single orbit below a proboscis.
- Ethmocephaly, in which ocular hypotelorism is present with an interorbital proboscis.
- Cebocephaly, in which ocular hypotelorism is present with a single-nostril nose.
- Ocular hypotelorism and midline cleft lip/palate.
- Ocular hypotelorism and bilateral cleft lip/palate 3.
- Ocular hypotelorism or solitary median maxillary central incisor in minor forms.
These latter midline defects can occur without the cerebral malformations and then are called microforms.
Other malformations include arhinencephaly (absent olfactory bulbs and tracts), absent thalami, hydrocephalus and neural migration abnormalities. In case 1, ocular hypotelorism and midline clefting were present.
Children with holoprosencephaly have many medical problems:
- Developmental delay and feeding difficulties,
- Epilepsy,
- Instability of temperature, heart rate and respiration.
- Endocrine disorders like:
- diabetes insipidus,
- adrenal hypoplasia,
- hypogonadism,
- thyroid hypoplasia and growth hormone deficiency are frequent.
The least severe of the facial anomalies is the median cleft lip (premaxillary agenesis). The most severe is cyclopia, an abnormality characterized by a single eye located in the area normally occupied by the root of the nose, and a missing nose or a proboscis (a tubular-shaped nose) located above the eye. The least common facial anomaly is ethmocephaly, in which a proboscis separates closely-set eyes. Cebocephaly, another facial anomaly, is characterized by a small, flattened nose with a single nostril situated below incomplete or underdeveloped closely-set eyes.
Holoprosencephaly affects males and females in equal numbers before birth and has been reported in many ethnic groups. The incidence of holoprosencephaly has been estimated at 1 in 250 during early embryonic development, and approximately 1 in 10,000–20,000 live births 4. In the United States, seemingly higher prevalences have been reported in Hispanic, African-American, and Pakistani ethnicities, likely attributable to decreased prenatal diagnosis and termination rates in these groups 5.
Holoprosencephaly can be diagnosed even in the first trimester after 11–12 weeks 4, though mild forms may not be reliably detected prenatally.. So, routine prenatal ultrasound should be carried out in all patients during this period. Ultrasound is a good modality for prenatal diagnosis of holoprosencephaly that is frequently associated with midline face deformity such as cleft lip and palate. In addition to facial anomalies, anomalies of the spine and extremities are frequently associated with it and one must look for them, such as meningomyelocoele and limb abnormalities.
There is no standard course of treatment for holoprosencephaly. Treatment is symptomatic and supportive.
Parts of the human fetal brain and their functions
During fetal development the brain can be divided into five continuous parts (see Figure 1). From top to bottom they are :
- Prosencephalon (the embryonic forebrain):
- The telencephalon (cerebrum) becomes the large cerebral hemispheres. The surface of these hemispheres consists of elevations (gyri) and depressions (sulci) and the hemispheres are partially separated by a deep longitudinal fissure. The cerebrum fills the area of the skull above the tentorium cerebelli and is subdivided into lobes based on position.
- The diencephalon, which is hidden from view in the adult brain by the cerebral hemispheres, consists of the thalamus, hypothalamus, and epithalamus and third ventricle.
- Mesencephalon or the midbrain, gives rise to the midbrain and aqueduct of the midbrain (cerebral aqueduct).
- Rhombencephalon (the hindbrain):
- The metencephalon becomes the pons, cerebellum, and upper part of the fourth ventricle.
- The myelencephalon forms the medulla oblongata and lower part of the fourth ventricle.
At approximately 5 weeks gestation, the prosencephalon cleaves into two secondary vesicles: the telencephalon anteriorly and the diencephalon posteriorly 6. The telencephalon gives rise to the cerebral hemispheres, putamen, and caudate nucleus; the diencephalon forms the thalamus, hypothalamus, globus pallidus, and optic vesicles 7.
The eye field begins as a single midline structure. Under the signaling influence of the prechordal plate, the eye field in vertebrates splits into separate left and right eyes. If this developmental process is not completed correctly, the result is cyclopia 8.
Figure 1. Parts of the human fetal brain
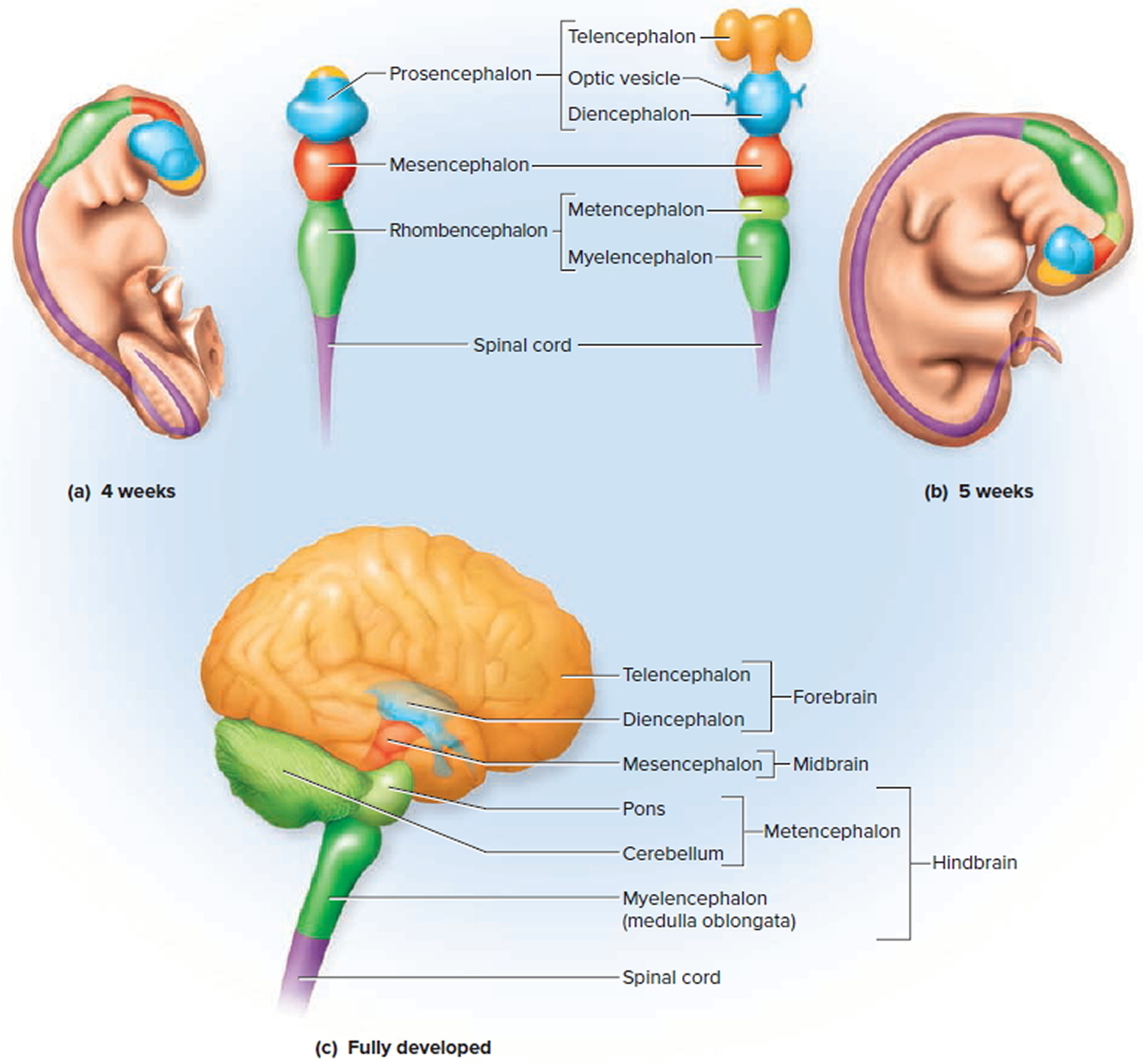
Footnote: (a) The primary vesicles at 4 weeks (fetus). (b) The secondary vesicles at 5 weeks (fetus). (c) The fully developed brain, color-coded to relate its structures to the secondary embryonic vesicles.
Figure 2. Holoprosencephaly ultrasound (semilobar holoprosencephaly showing fused thalami)
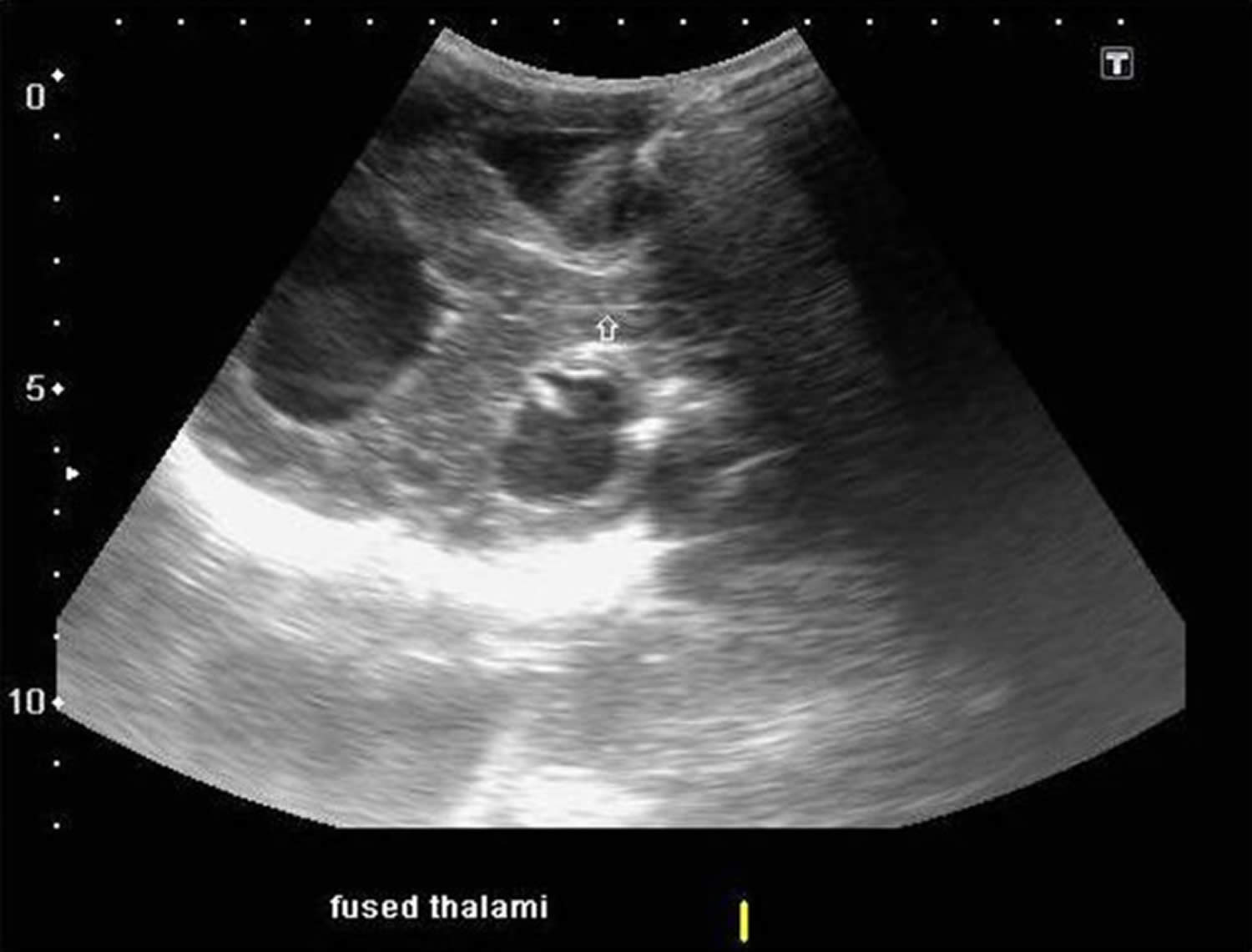
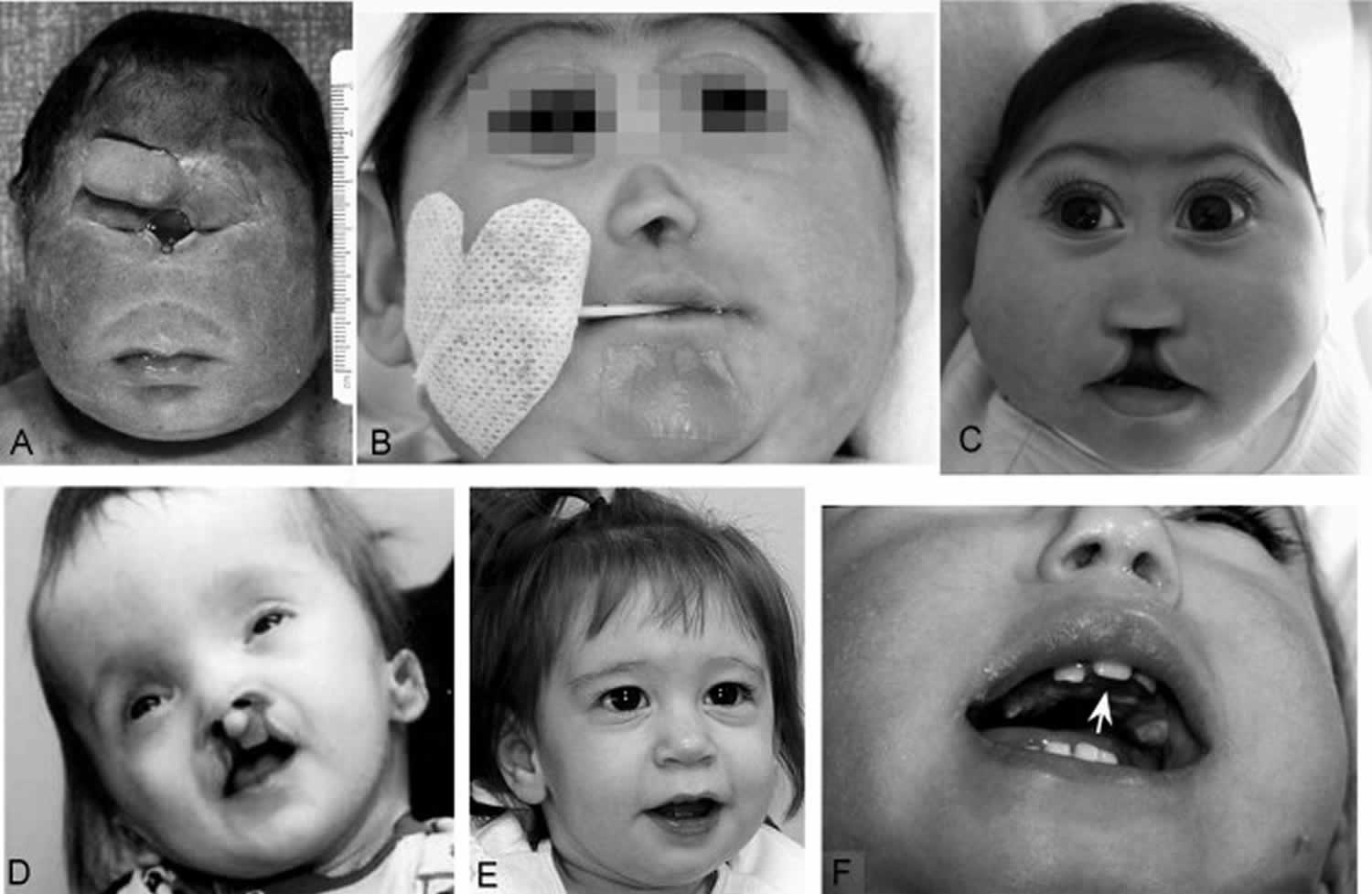
Figure 3. Holoprosencephaly facial features
Footnote:
A. Alobar holoprosencephaly with cyclopia and proboscis above the single eye
B. Alobar holoprosencephaly with cebocephaly and closely spaced eyes
C. Semilobar holoprosencephaly with microcephaly, premaxillary agenesis, and midline cleft lip and palate
D. Semilobar holoprosencephaly with closely spaced eyes, midfaceretrusion, and mild Dysmorphism
Figure 4. Lobar holoprosencephaly
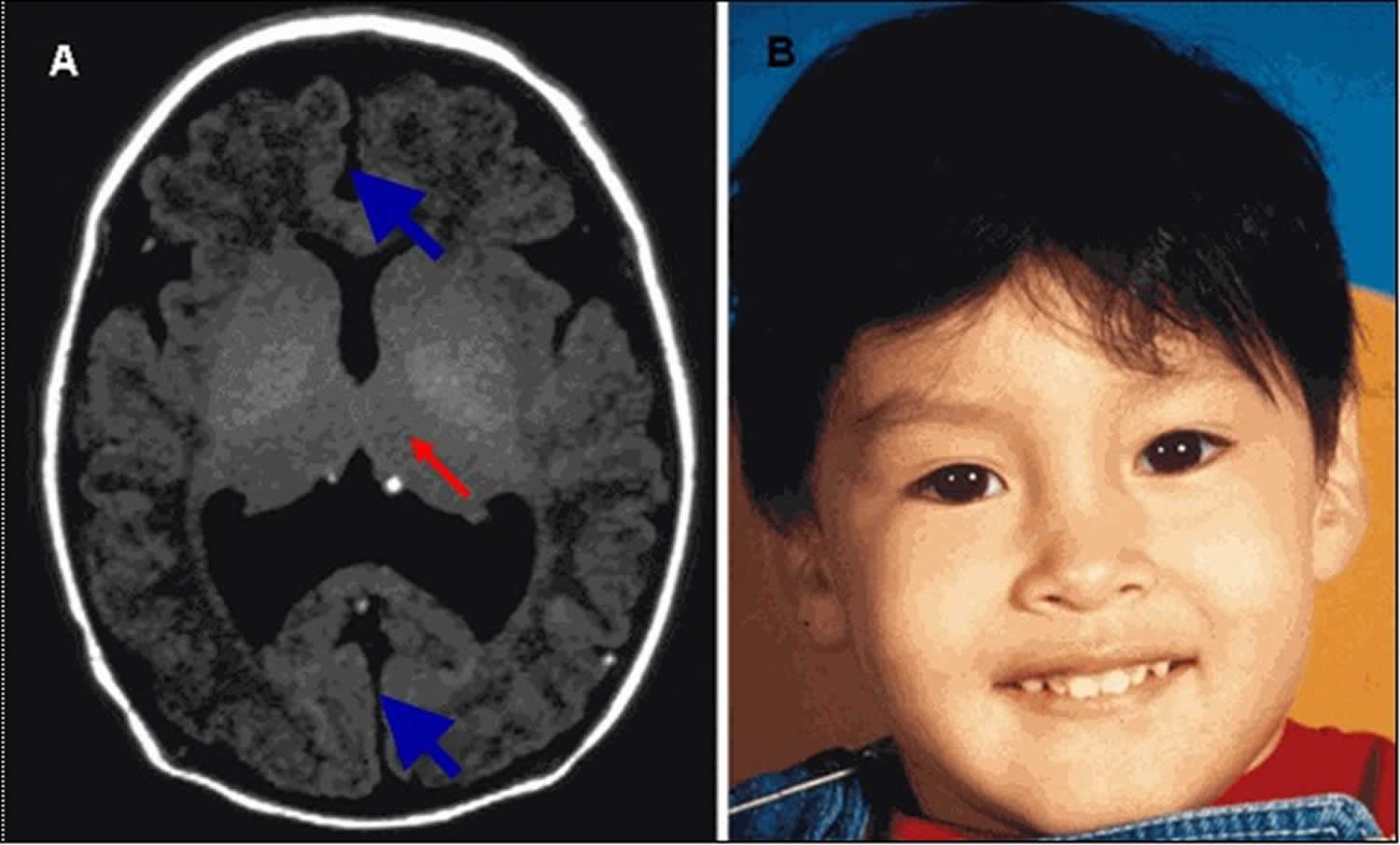
Footnote:
A. MRI in axial plane depicting lobar holoprosencephaly, the least severe of the major types of holoprosencephaly. The cerebral hemispheres are separated (blue/thick arrows); the ventricles are misshapen as a result of absence of the septum pellucidum. The posterior portion of the corpus callosum may be normlly formed. There is varying degree of fusion of the midline gray structures (thalami, basal ganglia, red/thin arrow).
B. Relatively normal facial appearance of a child with lobar holoprosencephaly resulting from a pathogenic variant in ZIC2. Subtle dysmorphisms include narrow forehead (not apparent in this photo), upslanted palpebral fissures, relatively large ears, a relatively depressed nasal bridge, and a broad and well-defined philtrum.
Figure 5. Semilobar holoprosencephaly
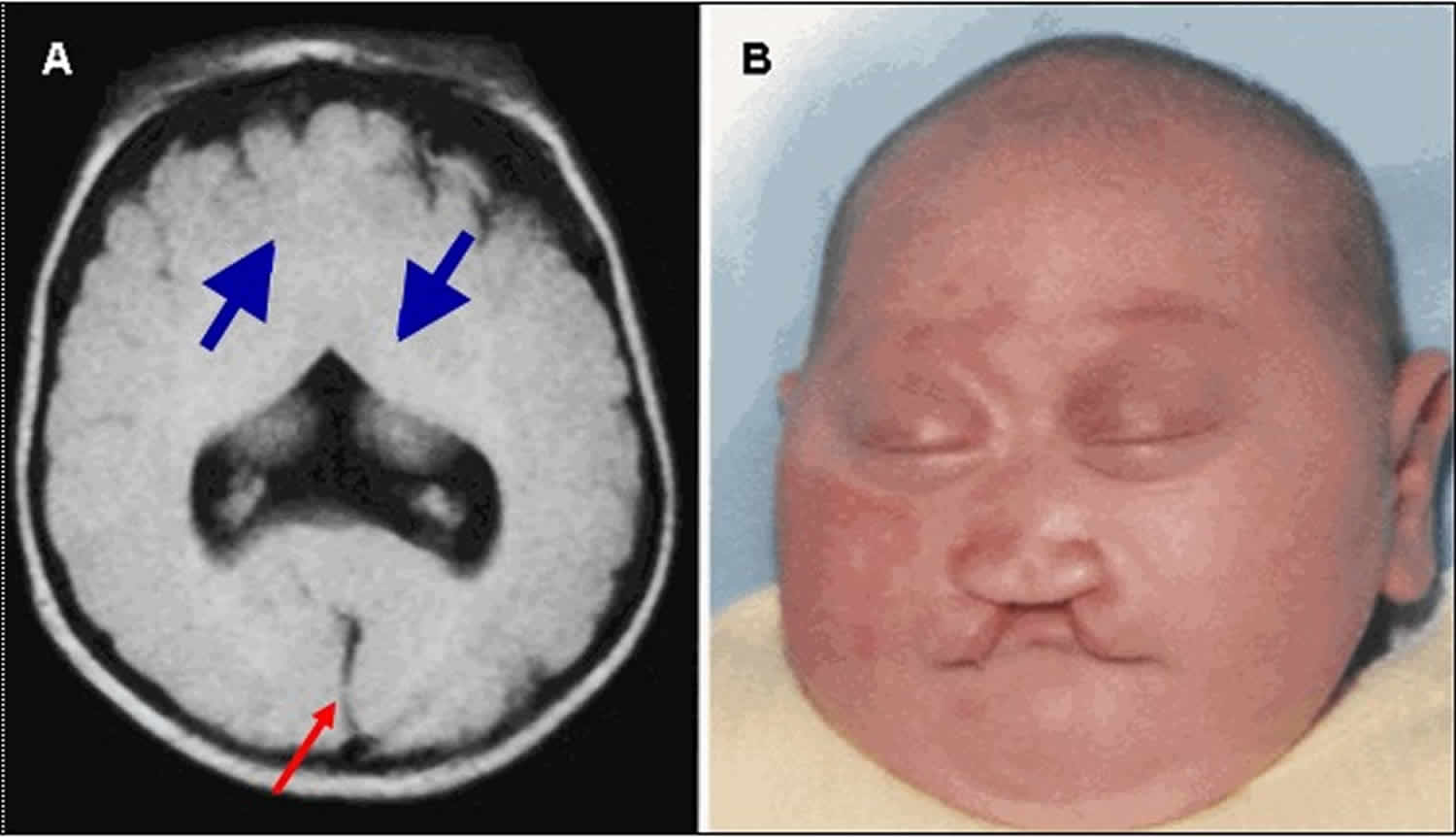
Footnote:
A. MRI showing semilobar holoprosencephaly. Note fusion of the frontal lobes, but presence of some septation posteriorly with presence of a falx and interhemispheric fissure (red/thin arrow). The splenium of the corpus callosum is present but more anterior portions are usually absent. A small, partially formed third ventricle is seen. More significant fusion of anterior brain structures (cortex, basal ganglia, thalamus) persists in this variant (blue/thick arrows). A dorsal cyst may be seen. In mild cases, lack of frontal horn development distinguishes this from the lobar type.
B. Note microcephaly, closely spaced eyes, depressed nasal ridge with cleft lip.
Figure 6. Alobar holoprosencephaly
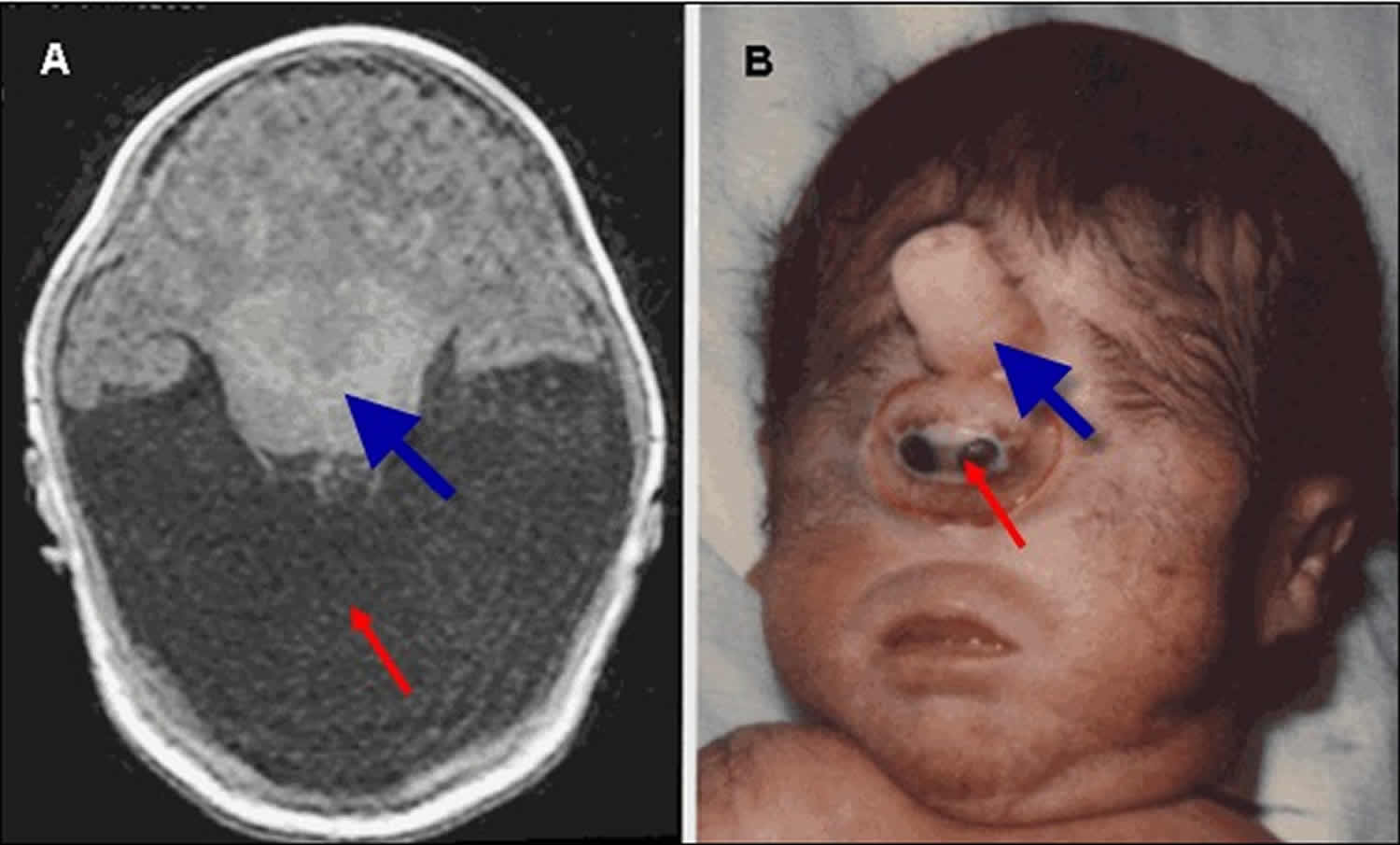
Footnote:
A. MRI of alobar holoprosencephaly (holoprosencephaly), the most severe form of holoprosencephaly, characterized by an enlarged midline monoventricle (holoventricle, red/thin arrow) with fusion of the frontal lobes and the midline gray matter structures (thalami and basal ganglia, blue/thick arrow). Typically, the corpus callosum and the third ventricle are absent.
B. Facial features seen in the alobar holoprosencephaly spectrum, characterized by a single eye-like structure (cyclopia, red/thin arrow) and an overriding nose-like structure (proboscis, blue/thick arrow)
Figure 7. Middle interhemispheric fusion variant holoprosencephaly
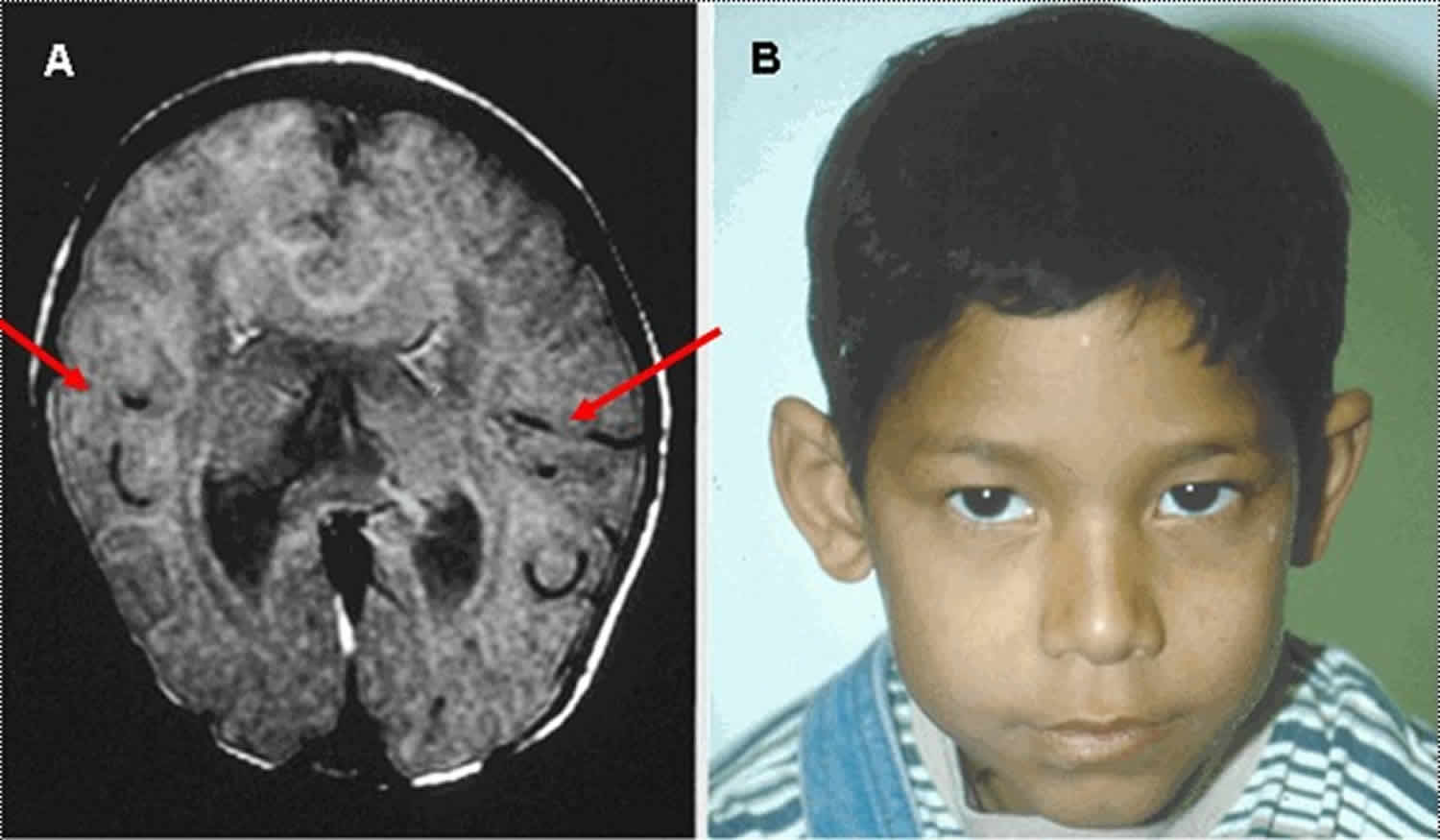
Footnote:
A. MRI in axial plane depicting middle interhemispheric variant of holoprosencephaly in which the anterior portions of the frontal lobes and the occipital lobes are well separated. The sylvian fissures are oriented nearly vertically and are abnormally connected across the midline over the vertex of the brain (red/thin arrows). The genu and splenium of the corpus callosum appear normally formed, but the callosal body is typically absent. The hypothalamus and lentiform nuclei are normally separated; however, the caudate nuclei and the thalami remain incompletely separated.
B. The facial appearance is usually normal.
Holoprosencephaly signs and symptoms
Holoprosencephaly is a malformation sequence with a very variable degree of severity for both the brain and facial abnormalities. Intellectual disability is associated with HPE and seizures are often present.
Children diagnosed with this disorder may have a small head (microcephaly), excessive fluid in the brain (hydrocephalus), facial abnormalities, tooth abnormalities (single central incisor), cleft lip and/or palate, epilepsy, and/or endocrine abnormalities. The most severely affected individuals may have cyclopia, a single central eye that is the most severe eye finding seen in holoprosencephaly, though this is very rare. Abnormalities in the formation of the nose may also occur.
Holoprosencephaly may also affect other systems in the body. Defects in the pituitary gland can cause an abnormally low level of sugar in the blood (hypoglycemia), low levels of sodium in the blood, or genital abnormalities.
Clinical manifestations commonly observed in children with holoprosencephaly include the following 11, 12:
- Developmental delay is present in all individuals with the holoprosencephaly spectrum of CNS (central nervous system) anomalies. The degree of delay is variable, correlating with the severity of the brain malformation, but tends to be severe.
- Seizures are common, and may be difficult to control.
- Hydrocephalus can occur, and may result in macrocephaly, rather than the more commonly-observed microcephaly.
- Neural tube defects occur in a small proportion of individuals.
- Hypothalamic and brain stem dysfunction may lead to swallowing difficulties and instability of temperature, heart rate, and respiration.
- Pituitary dysfunction is manifest by partial or complete panhypopituitarism with abnormal function of any or all of the anterior and/or posterior pituitary hormones, though central diabetes insipidus is by far the most common finding in persons with non-chromosomal, nonsyndromic holoprosencephaly 13.
- Short stature and failure to thrive are common, especially in more severely affected children. Growth hormone deficiency and/or chromosome anomalies may in part be responsible for poor growth in some individuals.
- Feeding difficulties may be a major problem in children with holoprosencephaly. At least part of the difficulty may derive from axial hypotonia, poor suck as a result of neurologic complications, lethargy, seizures and their effects, side effects of medications, and lack of interest. Often gastroesophageal reflux, choking, and gagging occur with feeds. More common problems include slowness in eating, frequent pauses, and frank vomiting with risk of aspiration. Oral-sensory dysfunction may affect feeding especially when associated with textural aversion and labial and lingual weakness. Children with cleft lip and/or palate often have additional difficulties with oral feeding.
- Excessive intestinal gas/colic, irritability, and constipation frequently occur 14.
- Aspiration pneumonia can be a complication of poor coordination of swallowing.
- Erratic sleep patterns can occur.
Holoprosencephaly prognosis
The prognosis for individuals with holoprosencephaly depends on the severity of the brain and facial deformities, the presence of chromosomal abnormalities, the involvement of other organs, and the presence of a multiple anomaly syndrome.
Stashinko et al 15 studied a population of 104 children (mean age, 4 years), only 9% of whom had a karyotype abnormality (i.e, the majority had isolated nonsyndromic holoprosencephaly). This aneuploidy rate is much lower than those in other reports in the literature (38%–55% of cases), likely reflecting selection bias in that the other studies included intrauterine fetal deaths and pregnancy terminations 15. Fifteen percent of the children were between 10 and 19 years old. None of the surviving alobar holoprosencephaly children could sit independently 16 or speak 17. About 50% of children with alobar holoprosencephaly die by 5 months, but as many as 30% live beyond a year 17. More than 50% of children with semilobar or lobar holoprosencephaly were alive at 1 year of age 18. Of those with lobar holoprosencephaly, about 50% are able to walk (some require assistance), have normal to mildly impaired hand function, and can speak single words (some speak in multiword sentences) 16. Children with middle interhemispheric fusion variant (syntelencephaly) may ambulate with assistance and speak and function with mild impairment 15; developmental outcome is similar to that in lobar holoprosencephaly 19.
Hydrocephalus is common, particularly when a dorsal cyst is present. Cerebrospinal fluid shunts are placed in approximately 17% of survivors 16. About half of children with holoprosencephaly have at least one seizure; about 40% require anticonvulsant therapy. Most children with holoprosencephaly have cerebral palsy. Swallowing problems are common in the more severe forms, placing these children at risk for recurrent respiratory problems or chronic lung disease due to aspiration. Poor gastric emptying, gastroesophageal reflux, and constipation are common. Hypothalamic dysfunction leads to homeostasis issues, including sleep problems, disorders of temperature regulation, and thirst and appetite issues—the severity of which seem to be related to the degree of hypothalamic nonseparation. Endocrine disorders include central diabetes insipidus and, less commonly, anterior pituitary dysfunction 16. Diabetes insipidus has not been seen in children with middle interhemispheric fusion variant (syntelencephaly) 19.
Holoprosencephaly life expectancy
A common misperception is that children with holoprosencephaly do not survive beyond early infancy. While this is the case for the most severely affected children, a significant proportion of more mildly affected children (as well as some severely affected children) survive past age 12 months. Among affected individuals with a normal karyotype, an inverse relationship exists between the severity of the facial phenotype and length of survival 10.
- Infants with cyclopia or ethmocephaly generally do not survive beyond age one week 20.
- Approximately 50% of children with alobar holoprosencephaly die before age four to five months and 20% live past the first year of life 14.
- More than 50% of children with isolated semilobar or lobar holoprosencephaly without significant malformations of other organs are alive at age 12 months 21.
Almost all survivors have apparently normal vision and hearing; they smile and demonstrate memory 14.
Holoprosencephaly causes
Holoprosencephaly is a birth defect that arises during the first few weeks of the pregnancy. Diabetes in the mother during the pregnancy can increase the risk of holoprosencephaly in the fetus by 200 fold 22. Furthermore, ethyl alcohol, cigarette smoking, and retinoic acid have all been listed as possible causes of holoprosencephaly 23, although the validity of several of these associations has been doubted in a 2012 study 24. However, for most children, no known intrauterine exposure is identified that is causally related to holoprosencephaly in that child.
Some children will have an identifiable genetic cause of holoprosencephaly. Approximately one-third of children born with holoprosencephaly have an abnormality of the chromosomes, which contain the genetic material (DNA). The most common chromosomal abnormality associated with holoprosencephaly is when there are 3 copies of chromosome 13 (trisomy 13 or Patau syndrome), although a number of other chromosomal changes can also cause holoprosencephaly.
In other children, holoprosencephaly is due to a change in a specific gene. These changes cause the genes and their proteins to function abnormally, and this affects the development of the brain, resulting in holoprosencephaly. Some of these genes are SHH, SIX3, TGIF1, ZIC2, PTCH1, FOXH1, NODAL, CDON, FGF8, and GLI2. Holoprosencephaly can also occur in certain genetic syndromes in which there are other medical issues besides those mentioned in this report that affect organs in addition to the brain and face (e.g., Smith-Lemli-Opitz syndrome).
Despite the proven usefulness of genetic mutation screening, , the exact cause of the condition is not identified for many individuals. Nearly 75% of holoprosencephaly cases with normal chromosomes do not have identified mutations; thus, holoprosencephaly pathogenesis is likely due to many additional uncharacterized causes, including additional unidentified genetic factors and environmental agents other than those already known 8; this is Muenke’s “multiple-hit hypothesis” 25. Nonchromosomal, nonsyndromic holoprosencephaly is considered autosomal dominant, with incomplete penetrance and markedly variable expressivity 26. Multiple autosomal recessive entities are associated with holoprosencephaly; these include Smith-Lemli-Opitz syndrome, Meckel syndrome and hydrolethalus syndrome 27.
Holoprosencephaly diagnosis and testing
Imaging of the brain by CT scan or (preferably) MRI confirms the diagnosis of holoprosencephaly, may define the anatomic subtype, and identifies associated CNS anomalies 28. Holoprosencephaly is most frequently diagnosed during the newborn period when abnormal facial findings and/or neurologic presentation prompt further evaluation. Often holoprosencephaly is first identified on prenatal ultrasound examination. Infants with normal facies or only mildly abnormal facies and either mild or intermediate brain anomalies may not be diagnosed until the first year of life when neuroimaging studies obtained during evaluation for developmental delay and/or failure to thrive reveal holoprosencephaly.
Approximately 25%-50% of individuals with holoprosencephaly have a numeric or structural chromosome abnormality detectable by chromosome analysis. Approximately 18%-25% of individuals with monogenic holoprosencephaly have a recognizable syndrome and the remainder have nonsyndromic holoprosencephaly. Molecular genetic testing is possible for many of the genes associated with nonsyndromic holoprosencephaly. Approximately 10% of individuals with holoprosencephaly have defects in cholesterol biosynthesis.
Holoprosencephaly treatment
Treatment and care for a baby with holoprosencephaly are supportive and based on the specific medical issues present for an individual child.
An endocrinology evaluation should be performed to assess for pituitary abnormalities. A neurologist should also be involved in the child’s care and can guide treatment for seizures if they are present. Plastic reconstructive surgery of cleft lip and palate or other facial features may be needed if indicated. A developmental pediatrician can help direct developmental therapies. Other treatments can be instituted as appropriate.
A clinical genetics evaluation and genetic counseling should be obtained for patients and their families once the diagnosis is made. Relatives of a child with holoprosencephaly may have an increased risk of having a child with holoprosencephaly, and this should be assessed and discussed by the child’s physicians, especially the neurologist and/or clinical geneticist. There are specific features that suggest an increased risk for having another child with holoprosencephaly (e.g., a single central upper incisor), and these should be carefully assessed in parents and family members. A chromosome analysis and gene testing is often performed.
Pediatricians, neurologists, dentists, special education teachers, surgeons, therapists, psychologists, developmental pediatricians, and others must systematically and comprehensively plan the child’s treatment for holoprosencephaly.
Evaluations Following Initial Diagnosis
To establish the extent of disease in a child diagnosed with holoprosencephaly (holoprosencephaly), evaluation for the following is recommended at a minimum (other organ systems may also be investigated depending on specific clinical findings):
- Cleft lip and/or palate
- Hydrocephalus and/or features of holoprosencephaly or other cortical anomalies. All children with midline facial anomalies should undergo brain MRI; attention should also be paid to the pituitary region, which often requires high-resolution thin sections.
- Growth deficiency. Height, weight, and head circumference should be measured. It is important to compare weight to height in addition to plotting absolute measurements. Evaluation should include thyroid function tests, bone age, complete blood count, blood chemistries, sedimentation rate, insulin-like growth factor 1, and insulin-like growth factor binding protein 3. If growth hormone deficiency is found, panhypopituitarism should be assessed by specific hormone testing and brain MRI.
- Pituitary dysfunction. Sagittal MRI can be used to determine pituitary absence or ectopia and anatomic information. CNS anomalies and absent corpus callosum and/or septum pellucidum may accompany endocrine dysfunction. Serum analysis for specific hormones can be performed to evaluate pituitary function.
- Oral feeding and swallowing. Evaluation should include assessment of caloric intake, swallowing abilities, oral motor skills, and presence of gastroesophageal reflux. Occupational and speech evaluations are warranted to evaluate and address feeding concerns. Studies for diagnosis of gastroesophageal reflux including esophageal pH probe, milk scan, barium swallow, and/or endoscopy may be considered.
Treatment of Manifestations
Treatment for holoprosencephaly varies according to the brain malformations and associated anomalies 11. Most affected children benefit from a multidisciplinary team approach with clinicians very familiar with holoprosencephaly.
- Hormone replacement therapy has been successful in some children with pituitary dysfunction.
- Antiepileptic drugs can help decrease the frequency and intensity of seizures.
- Feeding difficulties and failure to thrive may be managed with gastrostomy tube placement and Nissen fundoplication if gastroesophageal reflux and vomiting are issues. Thickening of feeds and upright positioning after feeding may be helpful to alleviate gastroesophageal reflux. To achieve the best growth in the child with holoprosencephaly, the quality of the feeds is more important than the quantity.
- Accommodations for oral feeding with cleft lip and/or palate may require specific nipples, cups, and parental training. Early surgical repair may improve feeding.
- Placement of a ventriculo-peritoneal shunt may be necessary in children with holoprosencephaly and hydrocephalus.
- In older children, surgical repair of cleft lip and/or palate may be indicated.
- For children with cleft lip and/or palate, referral to a specialized cleft or craniofacial clinic is recommended.
- Onset of new neurologic findings or deterioration warrant evaluation for seizures and/or hydrocephalus and/or shunt malfunction. Such evaluation would include vital sign monitoring, neurologic examination, EEG, and MRI.
- A major aspect of treatment is support and counseling of the parents 29.
Prevention of Secondary Complications
Children with hormonal disturbances should receive prompt evaluation during times of stress (e.g., illness, surgery).
Consultation with subspecialists regarding fluid and electrolyte management should be sought if elective surgery is planned.
Children with diabetes insipidus need careful monitoring of fluid and electrolyte intake.
Surveillance
Height, weight, and head circumference should be measured during health maintenance evaluations.
Evaluation for endocrine deficiencies should be undertaken at appropriate intervals and during health maintenance visits.
Evaluation of Relatives at Risk
A careful family history by a clinical geneticist familiar with holoprosencephaly is critical, as genetic changes associated with holoprosencephaly, even in a mildly affected individual, would be considered a risk factor for manifestations such as developmental delay.
References- Dubourg C, Bendavid C, Pasquier L, Henry C, Odent S, David V. Holoprosencephaly. Orphanet Journal of Rare Diseases. 2007;2:8. doi:10.1186/1750-1172-2-8. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1802747/
- Barkovich AJ, Quint DJ. Middle interhemispheric fusion: an unusual variant of holoprosencephaly. AJNR Am J Neuroradiol. 1993;14:431–40. https://www.ncbi.nlm.nih.gov/pubmed/8456724
- Cohen HL, Sivit CJ. Holoprosencephaly. In: Cohen HL, Sivit CJ, editors. , eds. Fetal and pediatric ultrasound: a casebook approach. New York: McGraw-Hill, 2001:12–16.
- Chaudhari HD, Thakkar G, Darji P, Khokhani P. Prenatal ultrasound diagnosis of holoprosencephaly and associated anomalies. BMJ Case Reports. 2012;2012:bcr0320126129. doi:10.1136/bcr-03-2012-6129. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4542616/
- Epidemiology of holoprosencephaly: Prevalence and risk factors. Orioli IM, Castilla EE. Am J Med Genet C Semin Med Genet. 2010 Feb 15; 154C(1):13-21. https://www.ncbi.nlm.nih.gov/pubmed/20104599/
- Bardo D. Pediatric neuroradiology. I. Embryologic basis for brain malformation. Appl Radiol 2009 (Jul–Aug): 29–40.
- Winter T. Semilobar holoprosencephaly. In: Woodward PJ, ed. Diagnostic imaging: obstetrics. 2nd ed. Altona, Manitoba, Canada: Amirsys, 2011
- Roessler E, Muenke M. The molecular genetics of holoprosencephaly. Am J Med Genet C Semin Med Genet 2010;154C (1):52–61.
- Solomon BD, Gropman A, Muenke M. Holoprosencephaly Overview. 2000 Dec 27 [Updated 2013 Aug 29]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Figure 6. [Facial findings in holoprosencephaly (HPE)]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1530/figure/hpe-overview.F6
- Solomon BD, Gropman A, Muenke M. Holoprosencephaly Overview. 2000 Dec 27 [Updated 2013 Aug 29]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1530/
- Levey EB, Stashinko E, Clegg NJ, Delgado MR. Management of children with holoprosencephaly. Am J Med Genet C Semin Med Genet. 2010;154C:183–90.
- Solomon BD, Lacbawan F, Mercier S, Clegg NJ, Delgado MR, Rosenbaum K, Dubourg C, David V, Olney AH, Wehner LE, Hehr U, Bale S, Paulussen A, Smeets HJ, Hardisty E, Tylki-Szymanska A, Pronicka E, Clemens M, McPherson E, Hennekam RC, Hahn J, Stashinko E, Levey E, Wieczorek D, Roeder E, Schell-Apacik CC, Booth CW, Thomas RL, Kenwrick S, Keaton A, Balog JZ, Hadley D, Zhou N, Long R, Velez JI, Pineda-Alvarez DE, Odent S, Roessler E, Muenke M. Mutations in ZIC2 in Human Holoprosencephaly: description of a novel ZIC2-specific phenotype and comprehensive analysis of 157 individuals. J Med Genet. 2010a;47:513–24.
- Lacbawan F, Solomon BD, Roessler E, El-Jaick K, Domené S, Vélez JI, Zhou N, Hadley D, Balog JZ, Long R, Fryer A, Smith W, Omar S, McLean SD, Clarkson K, Lichty A, Clegg NJ, Delgado MR, Levey E, Stashinko E, Potocki L, Vanallen MI, Clayton-Smith J, Donnai D, Bianchi DW, Juliusson PB, Njølstad PR, Brunner HG, Carey JC, Hehr U, Müsebeck J, Wieacker PF, Postra A, Hennekam RC, van den Boogaard MJ, van Haeringen A, Paulussen A, Herbergs J, Schrander-Stumpel CT, Janecke AR, Chitayat D, Hahn J, McDonald-McGinn DM, Zackai EH, Dobyns WB, Muenke M. Clinical spectrum of SIX3-associated mutations in holoprosencephaly: correlation between genotype, phenotype and function. J Med Genet. 2009;46:389–98.
- Barr M Jr, Cohen MM Jr. Holoprosencephaly survival and performance. Am J Med Genet. 1999;89:116–20.
- Stashinko EE, Clegg NJ, Kammann HA et al. A retrospective survey of perinatal risk factors of 104 living children with holoprosencephaly. Am J Med Genet A 2004;128A(2): 114–119.
- Levey EB, Stashinko E, Clegg NJ, Delgado MR. Management of children with holoprosencephaly. Am J Med Genet C Semin Med Genet 2010;154C(1):183–190.
- Bullen PJ, Rankin JM, Robson SC. Investigation of the epidemiology and prenatal diagnosis of holoprosencephaly in the North of England. Am J Obstet Gynecol 2001;184(6):1256–1262.
- Volpe P, Campobasso G, De Robertis V, Rembouskos G. Disorders of prosencephalic development. Prenat Diagn 2009;29(4):340–354.
- Lewis AJ, Simon EM, Barkovich AJ et al. Middle interhemispheric variant of holoprosencephaly: a distinct cliniconeuroradiologic subtype. Neurology 2002;59(12):1860–1865.
- Croen LA, Shaw GM, Lammer EJ. Holoprosencephaly: epidemiologic and clinical characteristics of a California population. Am J Med Genet. 1996;64:465–72.
- Olsen CL, Hughes JP, Youngblood LG, Sharpe-Stimac M. Epidemiology of holoprosencephaly and phenotypic characteristics of affected children: New York State, 1984-1989. Am J Med Genet. 1997;73:217–26.
- Holoprosencephaly. Online Mendelian Inheritance in Man, OMIM. Johns Hopkins University, Baltimore, Md. MIM number: 236100. http://omim.org/entry/236100
- Stashinko EE, Clegg NJ, Kammann HA et al. A retrospective survey of perinatal risk factors of 104 living children with holoprosencephaly. Am J Med Genet A 2004;128A(2): 114–119
- Vaz SS, Chodirker B, Prasad C, Seabrook JA, Chudley AE, Prasad AN. Risk factors for nonsyndromic holoprosencephaly: a Manitoba case-control study. Am J Med Genet A 2012;158A(4):751–758.
- Volpe P, Campobasso G, De Robertis V, Rembouskos G. Disorders of prosencephalic development. Prenat Diagn 2009;29(4):340–354
- Solomon BD, Mercier S, Vélez JI et al. Analysis of genotype-phenotype correlations in human holoprosencephaly. Am J Med Genet C Semin Med Genet 2010;154C(1):133–141
- Mercier S, Dubourg C, Belleguic M et al. Genetic counseling and “molecular” prenatal diagnosis of holoprosencephaly (HPE). Am J Med Genet C Semin Med Genet 2010; 154C(1):191–196.
- Hahn JS, Barnes PD. Neuroimaging advances in holoprosencephaly: refining the spectrum of the midline malformation. Am J Med Genet Part C Semin Med Genet. 2010;154C:120–32.
- Mercier S, Dubourg C, Belleguic M, Pasquier L, Loget P, Lucas J, Bendavid C, Odent S. Genetic counseling and “molecular” prenatal diagnosis of holoprosencephaly (HPE). Am J Med Genet C Semin Med Genet. 2010;154C:191–6.





{kind=link}