Hypercortisolism
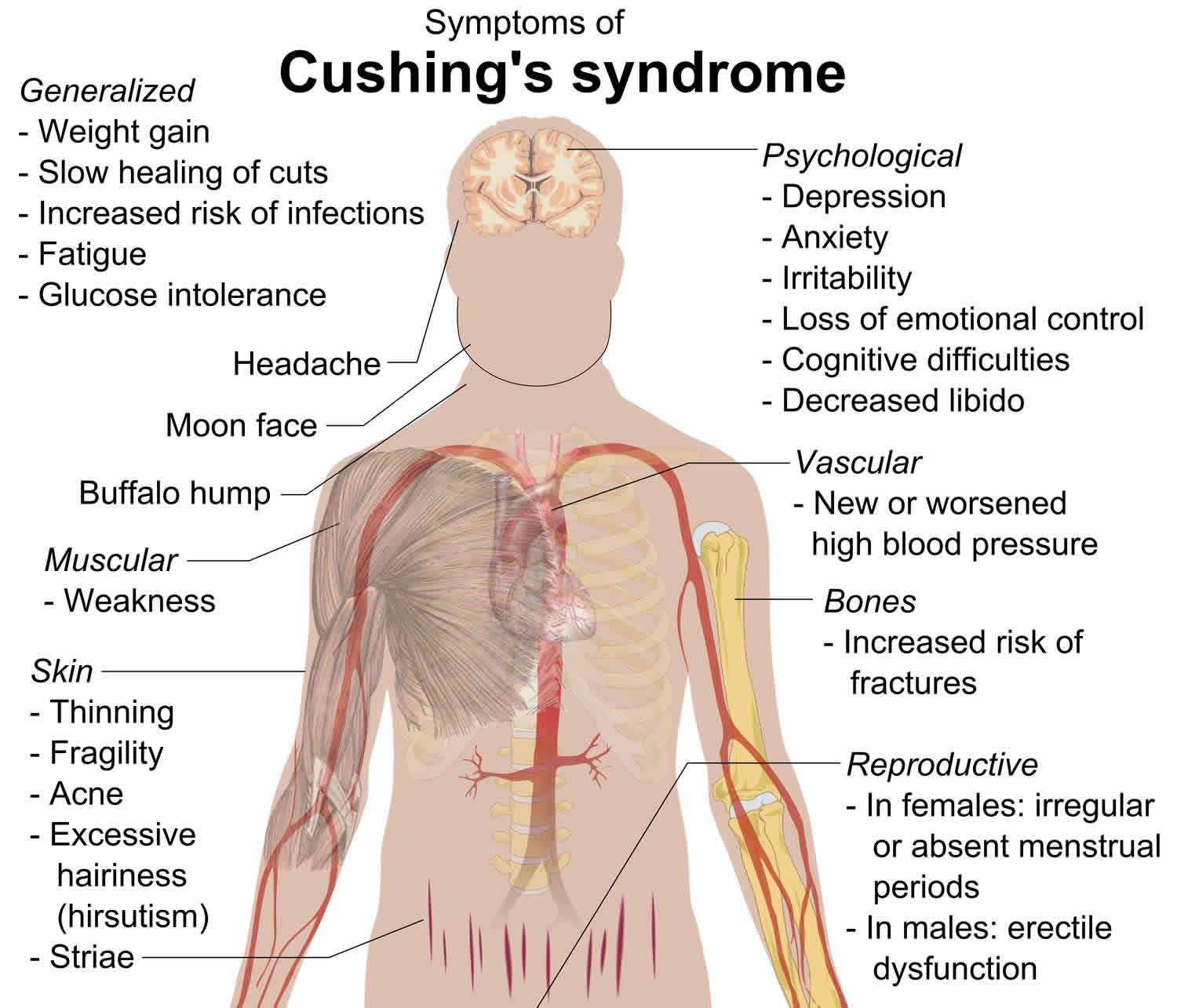
Hypercortisolism also called Cushing’s syndrome, refers to the clinical state caused by chronic exposure of the body’s tissues to elevated levels of a hormone called cortisol – a hormone naturally produced by the adrenal gland and/or other related glucocorticoids 1. Exposure to too much cortisol can occur from long-term use of synthetic glucocorticoid hormones to treat inflammatory illnesses. Pituitary adenomas (benign tumors of the pituitary gland) that secrete increased amounts of ACTH (adrenocorticotropic hormone, a substance that controls the release of cortisol) can also spur overproduction of cortisol. Tumors of the adrenal gland and ectopic ACTH syndrome (a condition in which ACTH is produced by various types of potentially malignant tumors that occur in different parts of the body) can cause similar problems with cortisol balance 2. Common symptoms of Cushing’s syndrome include upper body obesity, severe fatigue and muscle weakness, high blood pressure, backache, elevated blood sugar, easy bruising, and bluish-red stretch marks on the skin. In women, there may be increased growth of facial and body hair, and menstrual periods may become irregular or stop completely. Neurological symptoms include difficulties with memory and neuromuscular disorders.
Hypercortisolism usually occurs in adults between the ages of 20 and 50; however, children may also be affected. The first sign of this condition is usually weight gain around the trunk and in the face. Affected individuals may get stretch marks (striae) on their thighs and abdomen and bruise easily. Individuals with Cushing syndrome can develop a hump on their upper back caused by abnormal deposits of fat. People with hypercortisolism can have muscle weakness, severe tiredness, and progressively thin and brittle bones that are prone to fracture (osteoporosis). They also have a weakened immune system and are at an increased risk of infections. Cushing syndrome can cause mood disorders such as anxiety, irritability, and depression. Hypercortisolism can also affect a person’s concentration and memory. People with Cushing syndrome have an increased chance of developing high blood pressure (hypertension) and diabetes. Women with Cushing syndrome may experience irregular menstruation and have excessive hair growth (hirsutism) on their face, abdomen, and legs. Men with Cushing syndrome may have erectile dysfunction. Children with Cushing syndrome typically experience slow growth.
The difficulty inherent in the recognition, diagnosis, and subsequent management of hypercortisolism is due to the wide range of clinical presentations that it can have. While classic advanced Cushing syndrome has pathognomonic clinical features which are quite obvious with characteristic laboratory findings, that form of hypercortisolism is rather uncommon. Other forms of hypercortisolism including subclinical Cushing syndrome, iatrogenic Cushing syndrome, exogenous Cushing syndrome, factitious, cyclical, intermittent, and pseudo-Cushing syndrome (aka physiologic hypercortisolism) as a group are way more common than classic Cushing syndrome and often more challenging to recognize, diagnose and manage 3.
Treatment of hypercortisolism depends on the cause of excess cortisol. If the cause is long-term use of a medication being used to treat another disorder, the physician may reduce the dosage until symptoms are under control. Surgery or radiotherapy may be used to treat pituitary adenomas. Surgery, radiotherapy, chemotherapy, immunotherapy, or a combination of these may be used to treat ectopic ACTH syndrome. The aim of surgical treatment is to cure hypercortisolism by removing the tumor while minimizing the chance of endocrine deficiency or long-term dependence on medications. The U.S. Food and Drug Administration has approved pasireotide diasparate, taken by injection, for individuals who cannot be helped through surgery.
Hypercortisolism causes
Determining the cause of hypercortisolism is a three-tiered process. First, the presence of true hypercortisolism has to be established and confirmed based on clinical manifestations and laboratory test results while excluding differential diagnostic considerations detailed below. This process includes the distinction between true pathologic hypercortisolism and pseudo-Cushing syndrome, also known as physiologic hypercortisolism. The utilization of a screening test can help determine the cause.
After establishing the definitive diagnosis of true pathologic hypercortisolism, the next step in etiologic determination should be whether the hypercortisolism is ACTH dependent or independent. This evaluation should also include a careful determination of whether the hypercortisolism state could have iatrogenic or factitious etiologies.
After resolving the determination of ACTH dependency or independency, the final step of etiologic determination then involves definitive anatomic and pathologic diagnoses.
Iatrogenic Cushing syndrome: Conservative estimates indicate that over 10 million persons in the United States receive pharmacologic dose glucocorticoids annually, and while all these do not develop hypercortisolism, there also evidence that strongly suggests significant under-reporting of both the prevalence and incidence of iatrogenic Cushing syndrome 1. While iatrogenic Cushing syndrome is the dominant etiologic factor in exogenous Cushing syndrome, it is also important to consider the possibility of the less common factitious Cushing syndrome in this subgroup of hypercortisolism 4.
One large cohort that has evaluated retrospectively identified causes of non-iatrogenic, non-exogenous Cushing syndrome in approximately 630 patients at the Vanderbilt University Medical Center identified the following spread of etiologic entities 1:
- 68% due to ACTH dependent Cushing disease
- 12% due to ectopic ACTH Cushing syndrome
- 10% due to functional adrenal adenomas
- 8% due to functional adrenal carcinomas
- Less than 1% due to ectopic corticotropin-releasing hormone (CRH) syndrome
- Less than 1% due to micronodular adrenal hyperplasia
- Less than 1% due to macronodular adrenal hyperplasia and
- Approximately 1.5% due to pseudo-Cushing syndrome states, including major depressive disorder or chronic alcoholism.
Because of the difficulties in the diagnosis and recognition of hypercortisolism, estimates of its exact prevalence and incidence are widely variable. The reported numbers are also dependent on whether the cohorts are population-based, primary care-based, or subspecialty clinic-based, as well as whether the analysis is restricted to adults or include pediatric populations.
It generally accepted that iatrogenic Cushing syndrome is by far the most prevalent form of hypercortisolism. There is some suggestion with more recent data that subclinical Cushing syndrome may be more commonplace than previously presumed. Estimates from one Danish series suggested that during an 11-year period, the incidence of Cushing syndrome was 1.2 to 1.7/million person-years (Cushing disease), 0.6/million person-years (adrenal adenoma) and 0.2/million person-years (adrenal carcinoma) 3. More recent estimates from a US-based population suggested a range of 39.5 to 48.6 /million person-years, which is very different from the Danish cohort 5.
Other cohorts report widely variable incidence rates of Cushing disease (the most prevalent form of endogenous Cushing syndrome) ranging between 1.2 to up to 25 million 5.
There is some suggestion that ectopic Cushing syndrome is more common than reported, but most series suggest a prevalence of approximately 10-15% of all Cushing syndrome patients. Small cell lung cancer is the most common etiologic cause of this form of Cushing syndrome. Only 1% of patients with small-cell lung cancer have ectopic Cushing syndrome suggesting an incidence of approximately 300 new cases annually in the United States 6.
Adrenal mass lesions causing ACTH independent Cushing syndrome is assuming greater importance as far as prevalence and incidence. Adrenal incidentalomas have become more prevalent with the more widespread use of powerful imaging modalities like CT and MRI scans for the evaluation of various non-specific abdominal complaints. Depending on whether radiologic or autopsy based data are used, adrenal incidentalomas have a prevalence of approximately 1.3 to 8.7%.
All the other etiologic causes, including macronodular adrenal hyperplasia, micronodular adrenal hyperplasia (including primary pigmented nodular adrenocortical disease), ectopic corticotropin-releasing hormone (CRH) syndrome are rare, but the clinician needs to be aware of their existence as diagnostic considerations in unusual clinical presentation scenarios.
Overall, Cushing syndrome is more prevalent in women (3 to 8 times more frequent); this applies to both pituitary and adrenal based Cushing syndrome as well as both benign and malignant etiologies. The one subgroup in which men have typically had a greater prevalence is with ectopic Cushing syndrome, and this appears to be due to the greater incidence of tobacco abuse and consequently increased small cell lung cancer in men compared to women. As smoking among women has risen while declining among men, the prevalence disparity of ectopic Cushing syndrome among the sexes has also declined compared to findings from a series of over three decades ago.
Cushing disease most common age bracket of diagnosis is 25 to 45 years while that for ectopic Cushing syndrome is greater than 50 years. Cushing syndrome is less common in pediatric populations, and Cushing disease appears to constitute approximately 30% of Cushing syndrome in pediatric cohorts, which are significantly less than in adult cohorts. Also, small pediatric series suggest that in prepubertal children with Cushing syndrome, boys are more commonly afflicted than girls, while in adolescents, the female preponderance seen in adults is also apparent.
Adrenal neoplastic causes of Cushing syndrome have a bimodal age distribution pattern. There is one peak in the first decade of life and a second more significant peak at approximately age 40 years for adrenal carcinomas and around age 50 years for adrenal adenomas.
Hypercortisolism pathophysiology
Central to the development of hypercortisolism is the development of clinical features resulting from excessive tissue exposure to glucocorticoids (particularly cortisol). When these are presumed to be due to exaggeration of physiologic states known to be associated with hypercortisolemia, pseudo-Cushing syndrome (physiologic hypercortisolism) is diagnosed. This distinction from pathologic hypercortisolism is important as pseudo-Cushing syndrome is generally not sustained, generally resolves with the resolution of the etiologic factor responsible and typically is not associated with the overt cutaneous and/or muscular effects associated with pathologic hypercortisolism 7.
Among the established causes of pseudo-Cushing syndrome are pregnancy, morbid obesity, severe psychologic stress, severe major depressive disorder, poorly controlled diabetes mellitus with associated marked hyperglycemia, and chronic alcoholism. Even more confusing is the fact that patients with these clinical states can also have true endogenous pathologic hypercortisolism.
Other causes of physiologic hypercortisolism that typically don’t present with the clinical phenotype of pseudo-Cushing syndrome but can have suggestive biochemical anomalies are significant physical stress including surgical and/or hospitalization related stress, severe malnutrition, anorexia nervosa, wasting-cachexia syndrome, intense chronic exercise (including but not restricted to the hyper exercising female athlete syndrome), hypothalamic amenorrhea, various causes of elevated serum cortisol binding globulin (CBG) and glucocorticoid resistance.
Derangement of the hypothalamic-pituitary-adrenal (HPA) axis is central to the development of hypercortisolism. Such disruption can be due to exogenous or endogenous assaults or in some cases, both.
The hypothalamic-pituitary-adrenal axis in the normal physiologic state is primarily for maintenance of baseline continuous cortisol production with provocative secretory responses to various stressful stimuli. Corticotropin-releasing hormone (CRH) is the highest hierarchical control hormone in this axis and is produced from the hypothalamus tonically. It acts on the adenohypophysis (anterior pituitary) upon the basophilic corticotrophs to produce ACTH (corticotrophin), and this then gets secreted from the pituitary into the general circulation from which it then acts on the adrenal cortex to produce cortisol as the dominant endogenous glucocorticoid.
Circadian and stress-related inputs modulate CRH production from the parvicellular neurons of the paraventricular hypothalamic nucleus. Of note is the fact that many of the same neurons concomitantly produce arginine vasopressin, which has a similar though the less dominant modulatory effect on ACTH production from the adenohypophysis 8.
ACTH production occurs via post-translational modification of the precursor molecule pro-opiomelanocortin (POMC). ACTH stimulates the production of cortisol from the adrenal cortex in the free form, most of which is then bound by carrier proteins of which cortisol binding globulin is the dominant (roughly 95% of the total in physiologic settings) one. It is a high specificity low capacity binding protein produced mainly from the liver. There is some cortisol binding also to albumin but significantly fewer degrees. At target tissues and organs, cortisol then dissociates from its carrier protein and has its dominant effects mediated by binding to the glucocorticoid (and to a less degree the mineralocorticoid) receptor which then gets transported to the nuclear ribosomal complex of target cells to alter DNA transcription and protein production to mediate its effects. There is also evidence that cortisol has some non-genomic mediated effects. The details and mechanisms by which these get mediated are still the subjects of ongoing study.
Cortisol in both physiologic and pathologic settings has a negative feedback effect on further cortisol, production by inhibiting both pituitary ACTH production and hypothalamic CRH production in addition to other central nervous system modulatory effects.
The adrenal cortex from which cortisol is produced consists of three distinct anatomic and functional layers: the zona glomerulosa, which predominantly produces aldosterone, the zona fasciculata, which dominantly produces cortisol and the zona reticularis which dominantly produces adrenal androgens. ACTH mediates its stimulatory effects on cortisol production by activating the melanocortin-2 receptor (MC2R) of the zona reticularis cells. It is a typical G protein-coupled receptor with cAMP being the dominant mediatory secondary intracellular messenger.
This intricately orchestrated system has baseline tonic unstressed production that has a well defined circadian rhythm (typical peaks usually soon after early morning waking typically between 6 and 8 AM) with a nadir close to midnight. Superimposed on this circadian baseline rhythm is a pulsatile ultradian rhythm, as well. Various endogenous and exogenous stimuli can perturb this system resulting in either in excess or deficient cortisol production. It is excess of cortisol production and effects at the tissue and organ level of the patient that results in the clinical syndrome of hypercortisolism whether or endogenous or exogenous etiology.
Hypercortisolism symptoms
While the clinical presentation of classic Cushing syndrome is quite distinctive, the vast majority of hypercortisolism patients do not present this way, making a high index of clinical suspicion central to the detection of hypercortisolism. The possible signs and symptoms of hypercortisolism are numerous and individually are not pathognomonic. Many of these findings are non-specific, making hypercortisolism both over-diagnosed and underdiagnosed depending on the unique clinical scenarios involved. The history and physical examination findings though very important, are often not sufficient to diagnose hypercortisolism and generally have to be coupled with appropriate diagnostic laboratory findings. As the clinical presentation of hypercortisolism can be non-specific, a missed diagnosis can have catastrophic consequences. To increase diagnostic sensitivity, the suggestion is to consider screening both with careful problem-focused clinical questioning and examination for distinctive examination findings in certain groups of patients determined to be at higher than the typical risk for possible hypercortisolism. Among these are young adults with osteoporosis, early-onset hypertension and/or hyperglycemia/diabetes, facial plethora, proximal myopathy, presence of distinctive pigmented palpable wide (greater than 1 cm) striae, easy cutaneous bruising especially in young patients, and patients with adrenal incidentalomas 9.
Since exogenous Cushing syndrome is the most common cause of hypercortisolism, the history must include careful questioning regarding exogenous glucocorticoid exposure, which may be from prescriptions including topical and inhalational glucocorticoids (iatrogenic Cushing syndrome) as well as the possibilities of recreational and factitious abuse 10. A careful medication reconciliation history is a critical part of the historical evaluation of potential hypercortisolism patients, as is the attention to the prior history of psychopathology and occupational history that may identify patients with potential occupational access to glucocorticoids, which may be misusing them.
The most common clinical symptom of hypercortisolism is progressive weight gain, which is typically but not invariably centrally dominant. The weight gain in patients with hypercortisolism can, however also be generalized and akin to nonsyndromic obesity 11. Symptoms related to blood pressure elevation like headaches, dizziness, and visual blurring can occur as can symptoms due to worsening hyperglycemia like polyuria, polydipsia, and either polyphagia or anorexia. Reproductive system-related derangements often manifest symptomatically with women presenting with various menstrual irregularities, including but not restricted to oligomenorrhea, amenorrhea, and subfertility or infertility. Pregnancy in the setting of hypercortisolism is thus uncommon and unusual. Among men, reduced libido and symptoms consequent upon secondary hypogonadism such as lower energy levels, fatigue, non-specific weakness, and erectile dysfunction can occur. An easy propensity to bruising is an important symptom to inquire about as it is more distinctive for the syndrome. Muscle weakness, especially in the proximal extremity muscle groups, is also a more specific symptom for hypercortisolism and can manifest as difficulty with climbing stairs, difficulty rising unaided from low set chairs, and inability to do standard squats. Among children and adolescents, the coexistence of reduced or arrested linear growth along with significant excess weight gain is typical and suggestive of hypercortisolism. The discordance between progressive weight gain that crosses centile lines on the child’s weight growth chart as compared to declining or arrested linear growth on the corresponding height charts should raise the suspicion for hypercortisolism and spur appropriate diagnostic testing. Sleep deprivation is common, as are vivid dreams and nightmares. Mood decline and frank depression are also more common in patients with hypercortisolism. Some other patients present with progressive episodes of wide mood swings ranging from wild elation to sudden dysthymia akin to manic-depressive states. Some patients can also present with severe psychotic episodes in the absence of prior background history of established psychiatric pathology. Among women, hirsutism and marked acne are relatively common findings. Frank virilization should raise the suspicion for a malignant neoplastic disease like adrenal carcinoma as the underlying etiology. Clinical presentation on account of osteoporosis associated with hypercortisolism is more common in older patients and those with chronic, long-standing illness. Such patients may present with chronic back pain, loss of height due to vertebral collapse, low impact fragility fractures, and/or local bone pain.
Among the typical physical examination, findings are facial rounding (“moon facies”), facial flushing including marked malar telangiectasia, enlarged dorsocervical pad (buffalo hump), and supraclavicular fat pads. The supraclavicular fat pads are more specific for hypercortisolism than the dorsocervical fat pads. The finding of blood pressure elevation on examination is common. Cutaneous atrophy is a typical clinical finding, and in women, scalp hair loss can occur.
Diffuse cutaneous hyperpigmentation in the setting of hypercortisolism should suggest the possibility of ectopic Cushing syndrome.
It is also not uncommon for patients with hypercortisolism to present with clinical features of the comorbidities and complications associated with hypercortisolism. hypercortisolism is a poly systemic clinical state and thus can impact virtually every organ system of the body. Clinical presentations could, therefore, be consequent upon reproductive, dermatologic, orthopedic, metabolic, cardiovascular, neuropsychiatric, infectious, and bariatric comorbidities.
Further modulating the clinical presentation of the individual with hypercortisolism are the duration and degree of severity of the hypercortisolism. hypercortisolism is associated, therefore, with considerable morbidity and mortality risk, which is why its early recognition and appropriate management is so important. Hypercortisolism is associated with high to very high excess cardiovascular risk consequent upon its association with hypertension, which can be severe and resistant at presentation, dysglycemia, or frank diabetes mellitus, which is a well established cardiovascular risk equivalent and increased coagulopathic risk including increased risk for thromboembolic events 12.
Furthermore, hypercortisolism can also be associated with increased risk for heart failure and/or the development of a dilated cardiomyopathy.
Hypercortisolism complications
Hypercortisolism is a clinical syndrome that afflicts virtually every organ system either directly or indirectly 13. The following is a list of the associated comorbidities and complications of hypercortisolism:
- Decreased bone mass, osteopenia, osteoporosis, or osteonecrosis are particularly associated with exogenous rather than endogenous hypercortisolism, with the hips being the most commonly afflicted bones.
- Cardiovascular disease, including hypertension, accelerated atherosclerotic vascular disease (including coronary artery disease and various acute coronary artery syndromes, cerebrovascular disease, and peripheral vascular disease), dilated cardiomyopathy.
- Dyslipidemia including hypertriglyceridemia and hypercholesterolemia
- Development of the dysmetabolic syndrome
- Impaired glucose tolerance or frank diabetes mellitus with all the attendant complications and comorbidities that can accompany that.
- Obesity which is classically described as truncal dominant with relative extremity atrophy (so-called “apple on sticks” phenotype) but can be associated with generalized adiposity
- Linear growth impairment in children
- Proximal myopathy
- Hypogonadism and subfertility or infertility
- Increased predisposition to cutaneous and systemic infections, usually bacterial and fungal.
- Dyspepsia and gastro-esophageal reflux disease
- Nephrolithiasis
- Myriad cutaneous manifestations including cutaneous atrophy, ecchymoses, striae, recurrent cutaneous infections, hyperpigmentation in patients with pituitary Cushing syndrome, ectopic Cushing syndrome, and ectopic CRH syndrome states, alopecia, hirsutism, hypertrichosis, poor wound healing, acanthosis nigricans, excessive acne, etc.
- Ophthalmic complications of which cataracts and open-angle glaucoma are the most prevalent. Exophthalmos has also been described, especially with exogenous glucocorticoid exposure.
- Myriad neuropsychiatric syndromes including but not restricted to so-called “steroid psychosis,” depression, bipolar state, dysthymia, chronic anxiety, etc.
- Cognitive decline, including progressive memory loss and a dementia-like illness in the most severe cases with associated brain cortical atrophy
- A decline in overall quality of life
- Increased coagulopathic risk, including increased risk for deep venous thrombosis and embolism
- Increased risk for secondary or primary adrenal insufficiency and glucocorticoid withdrawal syndrome following the successful treatment of hypercortisolism
- Increased risk for development of Nelson syndrome post bilateral adrenalectomy
- Sleep derangements, including sleep deprivation, chronic insomnia, obstructive sleep apnea, and obesity hypoventilation syndrome.
In the unique and uncommon scenario of hypercortisolism in pregnancy, impaired glucose tolerance or gestational diabetes, hypertensive disorders of pregnancy, preeclampsia, heart failure, psychiatric disorders, and increased fetal loss are the most common consequent complications. While the fetus in such pregnancies tend to be somewhat protected from the maternal hypercortisolism because of the effects of placental 11 beta-hydroxysteroid dehydrogenase which converts approximately 85% of the maternal cortisol reaching the placenta to relatively inert cortisone this protective system can be overwhelmed with increased fetal loss, intrauterine growth retardation, premature delivery, and neonatal adrenal insufficiency described 14.
Hypercortisolism diagnosis
Because the clinical presentation of hypercortisolism is often non-specific, the first step of diagnostic evaluation involves wide-ranging screening, followed by diagnostic confirmation and then finally anatomic localization of the underlying etiology of the hypercortisolism.
It is generally accepted that the diagnosis of hypercortisolism is confirmed when at least two different first-tier screening tests are unequivocally abnormally elevated 15.
The determination of intensity and degree of screening testing is on a case by case basis based on the degree of clinical suspicion (pretest probability) of the diagnosis of hypercortisolism. The first-level screening tests include salivary cortisol tests, which should be obtained as duplicates to evaluate the early morning and late-night levels to evaluate both absolute cortisol levels and the preservation of the normal diurnal rhythm, 24-hour urinary free cortisol estimation, and the 1mg dexamethasone suppression test. The generally accepted threshold for diagnostic fidelity of the overnight 1 mg dexamethasone suppression test is serum cortisol the morning after over 1.8 mcg/dl (50 nmol/l) after confirmation that the patient actually took the dexamethasone tablet and adequately absorbed it (this can only be objectively confirmed by concurrently measure serum dexamethasone levels the morning after). Some centers use other tests for screening, such as the extended low dose dexamethasone suppression test (involves administering 2 mg of dexamethasone daily for two days) 16. These screening tests may need to be repeated multiple times in patients with a high index of clinical suspicion but initially negative findings. Patients with variable discordant results or patients with suspicion of intermittent or cyclical hypercortisolism may also require retesting.
In subjects with abnormal screening tests, confirmatory tests need to be done, and included here is the need to distinguish patients with pseudo-Cushing’s syndrome/physiologic hypercortisolism. Most experts in the field agree regarding the existence of subclinical Cushing syndrome, which is often clinically distinct and not as clinically striking as typical classical Cushing’s syndrome. This form of hypercortisolism is most commonly associated with adrenal etiologies rather than pituitary or ectopic Cushing syndrome though there have been cases of the latter described presenting as subclinical Cushing syndrome. The controversy as regards subclinical Cushing syndrome largely revolves around the exact diagnostic criteria and thresholds for the diagnosis. There are at least five clinical practice guidelines in this area 17. Overall the consensus appears to be that the Endocrine Society and the Italian Association of Medical Endocrinologists guidelines are well accepted. It is also crucial to understand that when there is discordance between screening tests obtained, especially on repeated measures, the possibility of cyclical, intermittent, iatrogenic, and even factitious Cushing’s syndrome merits consideration and excluded. It is also important to understand and exclude the list of other potential variables that can affect the accuracy of these screening tests, including concomitant medication use, variations, and perturbations of dexamethasone metabolism, night shift work, states of cortisol binding globulin excess or deficiency, coexistence sleep disorders, etc 18.
The combined dexamethasone CRH test is generally the best dynamic test for the distinction between pathologic hypercortisolism and physiologic hypercortisolism. Its basis is the premise that patients with the latter tend to have a blunted cortisol response (and less consistently) ACTH response to CRH after dexamethasone suppression (using the standard two-day low dose dexamethasone administration protocol) compared to patients with pathological hypercortisolism. This testing is still an area of controversy, and there are lingering concerns regarding the sensitivity and specificity of the test in distinguishing pseudo-Cushing’s syndrome from true pathologic Cushing’s syndrome.
Based on clinical presentation and the results of multiple screening test results, after deciding that the patient has hypercortisolism the next diagnostic branch point is whether the hypercortisolism is ACTH dependent or independent. As ACTH secretion is pulsatile in fashion and has a well-established diurnal rhythm, this measurement needs to be done on multiple occasions over a period of time to reach a consensus. Persistently low serum ACTH levels less than 5 pg/ml are very strongly predictive of ACTH independent hypercortisolism, which should include exogenous hypercortisolism in the considered etiologic differential diagnosis. Similarly, serum ACTH level consistently greater than 20 pg/ml is typically highly suggestive of ACTH dependent hypercortisolism, which then brings up the clinical decision branch point of distinguishing between pituitary based hypercortisolism (which is typically the cause in 70 to 85% of such cases) versus EcCushing’s syndrome. This distinction is, however, not always easy and may involve the use of other diagnostic testing strategies detailed below.
For patients with persistent serum ACTH levels between 5 to 20 pg/ml, further dynamic testing is necessary to provide clarity. These could include the CRH stimulation test, the vasopressin (or desmopressin, DDAVP) stimulation test, and/or the metyrapone stimulation test. All these tests tend to cause post provocative increase in ACTH secretion in patients with pituitary based hypercortisolism but not in adrenal hypercortisolism nor most EcCushing’s syndrome patients. The exact diagnostic thresholds, sensitivity, and specificity of these tests are, however, contentious, thus limiting their utility in making firm diagnostic distinctions.
Measurement of serum DHEA-S has some diagnostic utility on repeated measures as it tends to be reduced in ACTH independent hypercortisolism but increased or normal in ACTH dependent hypercortisolism 19.
After firmly establishing that a patient has ACTH dependent hypercortisolism, the distinction between pituitary and ectopic Cushing syndrome is the next step in the diagnosis. It is critical to be clear that imaging tests, whether they be adrenal, pituitary, general abdominal, general brain, or another related imaging testing, should only be obtained after a definitive biochemical diagnosis of hypercortisolism is made. Incidentalomas are common enough that prior imaging before making clear biochemical diagnoses can lead to unnecessary testing and possibly incorrect diagnoses with a potentially wrong treatment plan, including unnecessary surgery. Furthermore, the fact that most pituitary based hypercortisolism is the result of microadenomas makes the absence of an identified lesion on pituitary MRI a fairly common clinical scenario. Some reports have indicated up to 40 to 50% of patients with documented biochemical pituitary based ACTH dependent hypercortisolism having reportedly normal pituitary MRI studies 18. The distinction between ectopic Cushing syndrome and pituitary based hypercortisolism is generally better made using dynamic testing, which includes the CRH stimulation test, the vasopressin (or desmopressin, DDAVP) stimulation test and/or less commonly the metyrapone stimulation test. All these tests utilize the fact that pituitary based hypercortisolism tends to preserve the capacity to respond to such secretagogues with a significant increase in ACTH (and less robustly) cortisol levels. Also, high dose dexamethasone tests are an option to provide a distinction between these entities further; this can either use the classic four days extended 8 mg test first described by Liddle and colleagues or the more commonly used overnight 8 mg single dose suppression test. These tests take advantage of the fact that most pituitary based hypercortisolism suppress their associated cortisol production to below 5 mcg/dl (140 nmol/L) in the setting of high dexamethasone exposure unlike most patients with EcCushing’s syndrome.
The specificity and sensitivity of all these tests are not robust enough nor universal enough to avoid scenarios in which doubt as to the location of the source of ACTH excess persists, and in such settings, direct measurement of central and peripheral ACTH production may be necessary. The best-validated strategy for this is the inferior petrosal sinus sampling. The diagnostic utility of the test is heavily dependent on performing the test in the setting of ambient continuous CRH stimulation and requires the availability of high-end imaging for fluoroscopy as well as the availability of highly skilled neuroradiologists. Attempts to obtain similar data using less invasive bilateral internal jugular venous sampling have generally not yielded robust results. Studies of the even more technically difficult cavernous sinus sampling as a method to correctly lateralize the location of tiny pituitary microadenomas causing hypercortisolism have shown that they add little if any utility to the established inferior petrosal sinus sampling and are not currently common practice 20.
Once the biochemical diagnosis and the anatomic location are determined based on the biochemical and dynamic testing, imaging tests then take center stage. For pituitary and other brain-related causes of hypercortisolism pituitary MRI imaging is the preferred imaging modality for lesion localization. Dynamic contrast-enhanced high-resolution pituitary MRI scans may increase detection, but even the best MRI imaging systems presently available are incapable of detecting all functional pituitary microadenomas causing hypercortisolism while their high degree of sensitivity has resulted in detection of non-functional pituitary microadenomas in 10 to 20% of healthy volunteers routinely imaged 21. In patients with unique limitations, an open MRI or cranial CT with pituitary tomographic imaging may be necessary, but these imaging modalities have significantly lower sensitivity in the detection of tiny pituitary or other brain lesions that may be etiologic in pituitary hypercortisolism hence the primacy of establishing a biochemical diagnosis before imaging studies. In patients in whom the imaging tests are negative despite the positive biochemical diagnosis of pituitary based hypercortisolism because of the considerable morbidity and potential mortality associated with this condition, neurosurgical intervention with pituitary exploration and in some cases, hemi-hypophysectomy is necessary. It is important to note that pituitary imaging has been reported to wrongly suggest a pituitary lesion in approximately 18% of patients who eventually were shown to have EcCushing’s syndrome and so caution in treatment decision making is very important.
For patients whose biochemical and dynamic testing is suggestive of ectopic Cushing syndrome, the process of anatomical localization of the responsible lesion can be particularly challenging. The optimal strategy for imaging such patients is not clearly defined, and the decision is often on a case by case basis based on a careful review of the individual’s clinical presentation. Targeted imaging using CT scans, MRI, PET imaging, and specialized nuclear medicine imaging, including Octreotide scintigraphy and Gallium-68 DOTATATE imaging, is complementary. As most EcCushing’s syndrome lesions are found in the chest, chest based imaging may be the best place to begin in the absence of clinical clues from the history and physical examination. Most malignant etiologic lesions are relatively easy to find by these imaging modalities, but benign carcinoids are notoriously difficult to locate and may require multiple repeated imaging obtained over significant periods of time before finally being discovered.
For patients with ACTH, independent hypercortisolism imaging is generally focused on the adrenals though they have been a few rare cases described of other organ-related tumors (mainly ovarian) associated with ACTH independent cortisol overproduction associated with hypercortisolism. The best imaging study for visualizing the adrenals remains the adrenal dedicated thin slice/section CT scan with and without contrast. Such studies should include clear documentation of the tissue density (Hounsfield units -HU) or CT attenuation value both pre and post-contrast as well as details of the contrast washout properties of identified adrenal lesions. Subsequent decisions regarding diagnosis and management strategies in these patients would depend on whether bilateral or unilateral nodularity is detected, the size of the nodule(s): micronodules vs. macronodules, and the age of the patient. The absence of any obvious nodules in patients with biochemical evidence of ACTH independent hypercortisolism may require bilateral adrenal venous sampling as may the presence of multiple nodules in both adrenals or even the presence of a single nodule in older patients (over 65 years old) where the possibility of such a nodule being a non-functional adenoma rather than the cause of the hypercortisolism can be as great as 25%. Adrenal venous sampling like IPSS requires the availability of high-end fluoroscopy imaging equipment, high fidelity intravenous catheters as well as highly skilled interventional radiologists and so is not widely available. It also requires careful sample collection and labeling and continuous cosyntropin (ACTH) infusion during the sampling. HU scores of greater than 20 raise the possibility of cancerous etiology of identified adrenal lesions as does suboptimal contrast washout (less than 50% over the timed period of observation). In such settings, adrenal MRI imaging may provide additional diagnostic information. Functional imaging of adrenocortical lesions using the iodo-cholesterol scan (NP-59 I-131 Iodo-cholesterol) is hardly ever done anymore as the test is difficult to execute and has demonstrated rather limited diagnostic sensitivity or specificity. In clinical scenarios where the possibility of the adrenal lesion seen could potentially be of adreno-medullary origin (a few cases of adrenal pheochromocytomas associated with ACTH independent hypercortisolism have been reported), obtaining an I-123 MIBG scinti-scan may offer additional diagnostic information. Abdominal sonography has little utility in the evaluation of adrenal lesions involved in ACTH independent hypercortisolism but may have utility in the assessment of the rare clinical scenario of ectopic cortisol producing tumors, which are usually of abdominopelvic origin.
Hypercortisolism treatment
The management of hypercortisolism includes strategies directed at blocking the metabolic and clinical effects of hypercortisolism; strategies directed at the specific causes of the hypercortisolism state (which include medical and surgical interventions) as well as strategies directed at the management and modulation of the complications and co-morbidities associated with hypercortisolism.
In the setting where hypercortisolism is due to exogenous glucocorticoid use, the central management strategy is a carefully monitored reduction in glucocorticoid use with the ultimate aim of total withdrawal of their use. Depending on the initial indication/reason for their use, the duration, and dose of the glucocorticoids and evidence of secondary cortical atrophy, this may or may not be possible. Exogenous hypercortisolism can present the managing clinician with the conundrum of a patient with evidence of hypercortisolism who concomitantly has secondary adrenal insufficiency from cortical atrophy, which can become clinically apparent following the withdrawal of glucocorticoid therapy is entirely withdrawn. Serial monitoring of serum DHEA-S and ACTH levels, as well as serial ACTH (cosyntropin) stimulation tests, may be required in these sorts of patients over time while reducing the glucocorticoid dose to the closet dose to physiologic replacement doses required to prevent symptoms and signs of adrenal insufficiency. For some such patients, the secondary adrenocortical atrophy may be permanent, and such patients may require long term/life term physiologic adrenocortical replacement therapy with hydrocortisone (or other equivalent glucocorticoids) with adjunctive mineralocorticoid repletion (typically with fludrocortisone). For other patients, the medical indication for their glucocorticoid use such as rheumatologic or pulmonary inflammatory states may prevent weaning off these medications, and the strategy in those cases becomes weaning to the lowest required dose of glucocorticoids to maintain therapeutic effect with concurrent management of any consequent associated metabolic and clinical effects of the long term persistent hypercortisolism.
The general principles of proper management of hypercortisolism are to reverse the clinical and metabolic consequences of hypercortisolism by bringing endogenous cortisol production back to normal, removing any neoplastic source of the cortisol excess state, avoiding permanent dependence on medications and avoiding long term secondary hormone deficiency. These goals are not always all attainable in individual patients for various unique patient-specific reasons 22.
In patients where there are delays in making a definitive diagnosis of the underlying cause of hypercortisolism modulation of the hypercortisolism, and its complications (such as management of hyperglycemia, diabetes, hypertension, osteoporosis, hypercoagulopathy, etc.) may have to take center stage until achieving definitive diagnosis to enable more permanent and curative treatment. This approach is central to the prevention and reduction of the comorbidities and complications associated with hypercortisolism 13.
Medical therapy strategies: While surgical treatment strategies offer the best potential for permanent metabolic resolution and cure many times, this is either delayed or unsuccessful in achieving cure. Among the interventional strategies to include here are the glucocorticoid (and progesterone) receptor antagonist mifepristone (RU-486) and adrenal synthetic enzymes inhibitors like ketoconazole, metyrapone, and mitotane, which is generally restricted for use in adrenocortical carcinoma because of its significant toxicity profile 23. Medical therapy for pituitary adenomas causing hypercortisolism or neuroendocrine tumors causing EcCushing’s syndrome have been described and include cabergoline, pasireotide (Signifor), and other somatostatin analogs including octreotide though the therapeutic effects can be variable 24. In patients for whom oral therapies are impossible or contraindicated, parenteral use of imidazole anesthetic agent Etomidate has some utility in managing hypercortisolism. It blocks 11 beta hydroxylation of deoxycortisol to cortisol 25.
Further studies are ongoing for the potential utility of other medications in the management of resistant hypercortisolism, including fluconazole, levoketoconazole, osilodrostat, and the novel glucocorticoid receptor modulators; CORT-108297 and CORT-125134 26.
For pituitary adenomas causing hypercortisolism (Cushing disease), the vast majority (approximately 60 to 70%) are local microadenomas, but close to 30% may be locally invasive, and 0.2 to 0.2% are due to carcinomas that may be associated with central nervous system (CNS) and/or systemic metastases. In such patients, surgical treatment is rarely curative, and the prognosis is often poor with the need for consideration of systemic adjunctive chemotherapy 27. For Cushing disease, the best potential for cure remains early surgical intervention, usually by trans-sphenoidal surgery. Thus identification of a skilled pituitary neurosurgeon in a clinical facility with appropriate support staff and clinical resources is critical in achieving this goal. Referral to well-established pituitary surgical centers that meet this high standard is a worthwhile consideration. There is also a place for pituitary irradiation therapy in scenarios where, despite well documented and proven biochemical hypercortisolism, radiologic imaging, and possibly even neurosurgical exploration reveals no apparent tumor. It can also serve as adjunctive therapy following debulking non-curative pituitary surgery. This therapy is deliverable by standard conventional brachytherapy or more precisely by stereotactic radiotherapy, radiosurgery, or “gamma knife” therapy, which requires high-level technical expertise and nuclear medicine imaging resources.
In some scenarios where all other efforts ate achieving control of the hypercortisolism fails, bilateral adrenalectomy with subsequent lifelong glucocorticoid and mineralocorticoid repletion therapy may be the last management option to offer. Subsequent follow up of such patients to avoid the development of Nelson syndrome is important.
The management of patients with ectopic Cushing syndrome depends on the localization of the etiologic lesion(s). Surgical resection again offers the best option for optimal therapy and potential cure. Concomitant medical therapy, as detailed above to control the hypercortisolism, is often required until the etiologic lesion is identified and may be required thereafter if curative surgical resection is not feasible.
For patients with adrenal based hypercortisolism due to identified unilateral adrenal adenomas, unilateral adrenalectomy is the best strategy for a cure, and the laparoscopic approach is the preferred management method as long as the adenoma is less than 6 cm in diameter. The role and place of adrenocortical sparing surgery in this setting is still in evolution and is not the standard of care at this time.
While hypercortisolism is typically associated with subfertility/infertility, the uncommon scenario of hypercortisolism in pregnancy occasionally arises. Medical therapy in such settings is often not feasible because of the potential teratogenic effects of most of the available medical treatment options. Metyrapone is the best medical adjunctive treatment option in this setting, but ideally, surgical intervention generally best timed for the second trimester of pregnancy is the preferred therapeutic intervention strategy.
Hypercortisolism prognosis
The prognosis for those with hypercortisolism varies depending on the cause of the disease. Hypercortisolism, when untreated, correlates with marked morbidity and is often fatal. The most common cause of death is cardiovascular events, including acute coronary syndromes, thromboembolic events, and hypertensive complications, including cerebrovascular disease. Less common but also crucial as causative factors in the mortality of these patients are opportunistic bacterial and fungal infections. Older series have indicated a mortality risk as high as 50% within five years after the development of symptoms in the absence of effective treatment. Most cases of pituitary hypercortisolism are curable, and hypercortisolism should be manageable even if it requires the most drastic strategies of bilateral adrenalectomy or mitotane therapy to achieve that goal. Mitotane is the drug most often used for people with adrenal cancer. It blocks hormone production by the adrenal gland and also destroys both adrenal cancer cells and healthy adrenal tissue. This drug can also suppress the usual adrenal steroid hormone production from your other, normal adrenal gland. The underlying cause of the hypercortisolism plays a considerable role in the ultimate prognosis with malignant lesions such as small cell lung carcinoma and adreno-cortical carcinoma having the poorest prognosis while patients with benign adrenal adenomas or benign carcinoids with ectopic Cushing syndrome have the best prognosis.
Most cases of Cushing’s syndrome can be cured. Many individuals with Cushing’s syndrome show significant improvement with treatment, although some may find recovery complicated by various aspects of the causative illness. Some kinds of tumors may recur.
The clinical symptoms and signs of hypercortisolism take time to resolve even with curative therapy and full hormonal and metabolic correction. Resolution typically takes 2 to 12 months, and certain features like associated weight gain, hypertension, and glucose intolerance may never resolve. The loss of bone mass and/or frank osteoporosis typically begins to improve approximately six months after the hypercortisolism gets cured. In such patients, the use of calcium and vitamin supplementation, bisphosphonate therapy, and gonadal steroid repletion therapy can accelerate the recovery process. The health-related quality of life decline associated with hypercortisolism typically only partially resolves upon cure 28. In addition, psychiatric symptoms similarly improve with the cure, but some residual psychopathology typically remains 29.
Children with hypercortisolism often have a residual decline in intelligence quotient and cognitive functional indices even after cure, and this occurs and persists in the absence of any psychopathology and even with reversal of prior noted cerebral atrophy 30. Children tend to demonstrate an improvement in bone density and growth velocity following hypercortisolism treatment, but again, some persistent residual deficit is evident.
References- Uwaifo GI, Hura DE. Hypercortisolism. [Updated 2019 Dec 10]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK551526
- Cushing’s Syndrome Information Page. https://www.ninds.nih.gov/disorders/all-disorders/cushings-syndrome-information-page
- Lindholm J, Juul S, Jørgensen JO, Astrup J, Bjerre P, Feldt-Rasmussen U, Hagen C, Jørgensen J, Kosteljanetz M, Kristensen L, Laurberg P, Schmidt K, Weeke J. Incidence and late prognosis of cushing’s syndrome: a population-based study. J. Clin. Endocrinol. Metab. 2001 Jan;86(1):117-23.
- Hardy RS, Zhou H, Seibel MJ, Cooper MS. Glucocorticoids and Bone: Consequences of Endogenous and Exogenous Excess and Replacement Therapy. Endocr. Rev. 2018 Oct 01;39(5):519-548.
- Broder MS, Neary MP, Chang E, Cherepanov D, Ludlam WH. Incidence of Cushing’s syndrome and Cushing’s disease in commercially-insured patients <65 years old in the United States. Pituitary. 2015 Jun;18(3):283-9.
- Govindan R, Page N, Morgensztern D, Read W, Tierney R, Vlahiotis A, Spitznagel EL, Piccirillo J. Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: analysis of the surveillance, epidemiologic, and end results database. J. Clin. Oncol. 2006 Oct 01;24(28):4539-44.
- Raff H. Cushing syndrome: update on testing. Endocrinol. Metab. Clin. North Am. 2015 Mar;44(1):43-50.
- Raff H, Carroll T. Cushing’s syndrome: from physiological principles to diagnosis and clinical care. J. Physiol. (Lond.). 2015 Feb 01;593(3):493-506.
- Lacroix A, Feelders RA, Stratakis CA, Nieman LK. Cushing’s syndrome. Lancet. 2015 Aug 29;386(9996):913-27.
- Nieman LK. Diagnosis of Cushing’s Syndrome in the Modern Era. Endocrinol. Metab. Clin. North Am. 2018 Jun;47(2):259-273.
- Nieman LK. Cushing’s syndrome: update on signs, symptoms and biochemical screening. Eur. J. Endocrinol. 2015 Oct;173(4):M33-8.
- van der Pas R, Leebeek FW, Hofland LJ, de Herder WW, Feelders RA. Hypercoagulability in Cushing’s syndrome: prevalence, pathogenesis and treatment. Clin. Endocrinol. (Oxf). 2013 Apr;78(4):481-8.
- Pivonello R, Isidori AM, De Martino MC, Newell-Price J, Biller BM, Colao A. Complications of Cushing’s syndrome: state of the art. Lancet Diabetes Endocrinol. 2016 Jul;4(7):611-29.
- Caimari F, Valassi E, Garbayo P, Steffensen C, Santos A, Corcoy R, Webb SM. Cushing’s syndrome and pregnancy outcomes: a systematic review of published cases. Endocrine. 2017 Feb;55(2):555-563.
- Arnaldi G, Angeli A, Atkinson AB, Bertagna X, Cavagnini F, Chrousos GP, Fava GA, Findling JW, Gaillard RC, Grossman AB, Kola B, Lacroix A, Mancini T, Mantero F, Newell-Price J, Nieman LK, Sonino N, Vance ML, Giustina A, Boscaro M. Diagnosis and complications of Cushing’s syndrome: a consensus statement. J. Clin. Endocrinol. Metab. 2003 Dec;88(12):5593-602.
- Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM, Montori VM. The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2008 May;93(5):1526-40.
- Di Dalmazi G, Pasquali R, Beuschlein F, Reincke M. Subclinical hypercortisolism: a state, a syndrome, or a disease? Eur. J. Endocrinol. 2015 Oct;173(4):M61-71.
- Bansal V, El Asmar N, Selman WR, Arafah BM. Pitfalls in the diagnosis and management of Cushing’s syndrome. Neurosurg Focus. 2015 Feb;38(2):E4.
- Hong AR, Kim JH, Hong ES, Kim IK, Park KS, Ahn CH, Kim SW, Shin CS, Kim SY. Limited Diagnostic Utility of Plasma Adrenocorticotropic Hormone for Differentiation between Adrenal Cushing Syndrome and Cushing Disease. Endocrinol Metab (Seoul). 2015 Sep;30(3):297-304.
- Ilias I, Chang R, Pacak K, Oldfield EH, Wesley R, Doppman J, Nieman LK. Jugular venous sampling: an alternative to petrosal sinus sampling for the diagnostic evaluation of adrenocorticotropic hormone-dependent Cushing’s syndrome. J. Clin. Endocrinol. Metab. 2004 Aug;89(8):3795-800.
- Molitch ME. Nonfunctioning pituitary tumors and pituitary incidentalomas. Endocrinol. Metab. Clin. North Am. 2008 Mar;37(1):151-71, xi.
- Nieman LK, Biller BM, Findling JW, Murad MH, Newell-Price J, Savage MO, Tabarin A., Endocrine Society. Treatment of Cushing’s Syndrome: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2015 Aug;100(8):2807-31.
- Nieman LK. Recent Updates on the Diagnosis and Management of Cushing’s Syndrome. Endocrinol Metab (Seoul). 2018 Jun;33(2):139-146.
- Tritos NA, Biller BMK. Medical Therapy for Cushing’s Syndrome in the Twenty-first Century. Endocrinol. Metab. Clin. North Am. 2018 Jun;47(2):427-440.
- Preda VA, Sen J, Karavitaki N, Grossman AB. Etomidate in the management of hypercortisolaemia in Cushing’s syndrome: a review. Eur. J. Endocrinol. 2012 Aug;167(2):137-43.
- Fleseriu M, Pivonello R, Elenkova A, Salvatori R, Auchus RJ, Feelders RA, Geer EB, Greenman Y, Witek P, Cohen F, Biller BMK. Efficacy and safety of levoketoconazole in the treatment of endogenous Cushing’s syndrome (SONICS): a phase 3, multicentre, open-label, single-arm trial. Lancet Diabetes Endocrinol. 2019 Nov;7(11):855-865.
- Annamalai AK, Dean AF, Kandasamy N, Kovacs K, Burton H, Halsall DJ, Shaw AS, Antoun NM, Cheow HK, Kirollos RW, Pickard JD, Simpson HL, Jefferies SJ, Burnet NG, Gurnell M. Temozolomide responsiveness in aggressive corticotroph tumours: a case report and review of the literature. Pituitary. 2012 Sep;15(3):276-87.
- Lindsay JR, Nansel T, Baid S, Gumowski J, Nieman LK. Long-term impaired quality of life in Cushing’s syndrome despite initial improvement after surgical remission. J. Clin. Endocrinol. Metab. 2006 Feb;91(2):447-53.
- Bourdeau I, Bard C, Noël B, Leclerc I, Cordeau MP, Bélair M, Lesage J, Lafontaine L, Lacroix A. Loss of brain volume in endogenous Cushing’s syndrome and its reversibility after correction of hypercortisolism. J. Clin. Endocrinol. Metab. 2002 May;87(5):1949-54.
- Merke DP, Giedd JN, Keil MF, Mehlinger SL, Wiggs EA, Holzer S, Rawson E, Vaituzis AC, Stratakis CA, Chrousos GP. Children experience cognitive decline despite reversal of brain atrophy one year after resolution of Cushing syndrome. J. Clin. Endocrinol. Metab. 2005 May;90(5):2531-6.





{kind=link}