Hypophysitis
Hypophysitis is an inflammation of the pituitary gland, and is a rare cause of hypopituitarism 1. Hypophysitis is a generic term that includes a variety of conditions that cause inflammation of the pituitary gland. Hypophysitis is an infiltrative cause of hypopituitarism and can cause symptoms related to sella compression and pituitary hormone deficiencies.
Hypophysitis can be classified according to the anatomic location of pituitary involvement (adenohypophysitis, infundibulo-neurohypophysitis, or panhypophysitis) and the cause either primary hypophysitis (idiopathic) or secondary to sella and parasella lesions, systemic diseases, or drugs (mainly immune checkpoint inhibitors) 2. Primary hypophysitis are characterized by an idiopathic inflammatory process confined to the pituitary gland, while the secondary hypophysitis are triggered by a definite etiology (drugs and intracranial or systemic diseases). Five histologic variants of primary hypophysitis have been described: lymphocytic, granulomatous, xanthomatous, IgG4-related, and necrotizing (Table 1). Lymphocytic hypophysitis is the most common form of hypophysitis and occurs most commonly in women during late pregnancy and the postpartum period. Granulomatous hypophysitis is the second most common variant and possible secondary causes of granulomatous infiltration of the pituitary should be excluded before concluding that a case of granulomatous hypophysitis is idiopathic. Xanthomatous, necrotizing, and IgG4-related hypophysitis are very rare and the latter is often the manifestation of a systemic disease with multi-organ involvement (IgG4-related disease).
Immune checkpoint inhibitors are monoclonal antibodies increasingly used for the treatment of solid and hematological malignancies. They cause a T-lymphocyte activation and proliferation that lead to the anti-tumor response, and may cause autoimmune manifestations known as part of what is called “immune-related adverse events”. A significant number of patients treated with immune checkpoint inhibitors develop immune-related hypophysitis and require prompt diagnosis and treatment. Regardless of the etiology, patients with hypophysitis present with various signs and symptoms caused by the pituitary inflammation that can lead to hypopituitarism and compression of sella and parasella structures. Contrary to other causes of hypopituitarism, adrenocorticotropic hormone and thyroid-stimulating hormone deficiencies are very frequent in the early stages of hypophysitis, and must be identified immediately. The diagnosis of hypophysitis is based on clinical, laboratory, and radiological data; while pituitary biopsy is the gold standard test for diagnosing primary hypophysitis, it should be reserved only for selected cases. Magnetic resonance imaging is the technique of choice for suspected hypophysitis, and the main differential diagnoses are pituitary adenomas in adults, germinomas, and Langerhans cell histiocytosis in adolescents, and metastases in those receiving immune checkpoint inhibitors. The mainstay of treatment of patients with hypophysitis is pituitary hormone replacement. Those with severe signs and symptoms of sella compression should be treated with high-dose glucocorticoids, which usually cause an excellent initial response, although relapse of the pituitary inflammation is common. Pituitary surgery should be considered in patients who do not respond to glucocorticoids and have progressive and debilitating symptoms. Pituitary fibrosis and atrophy often develop in the late stage of the disease, with persistent hypopituitarism.
Primary hypophysitis
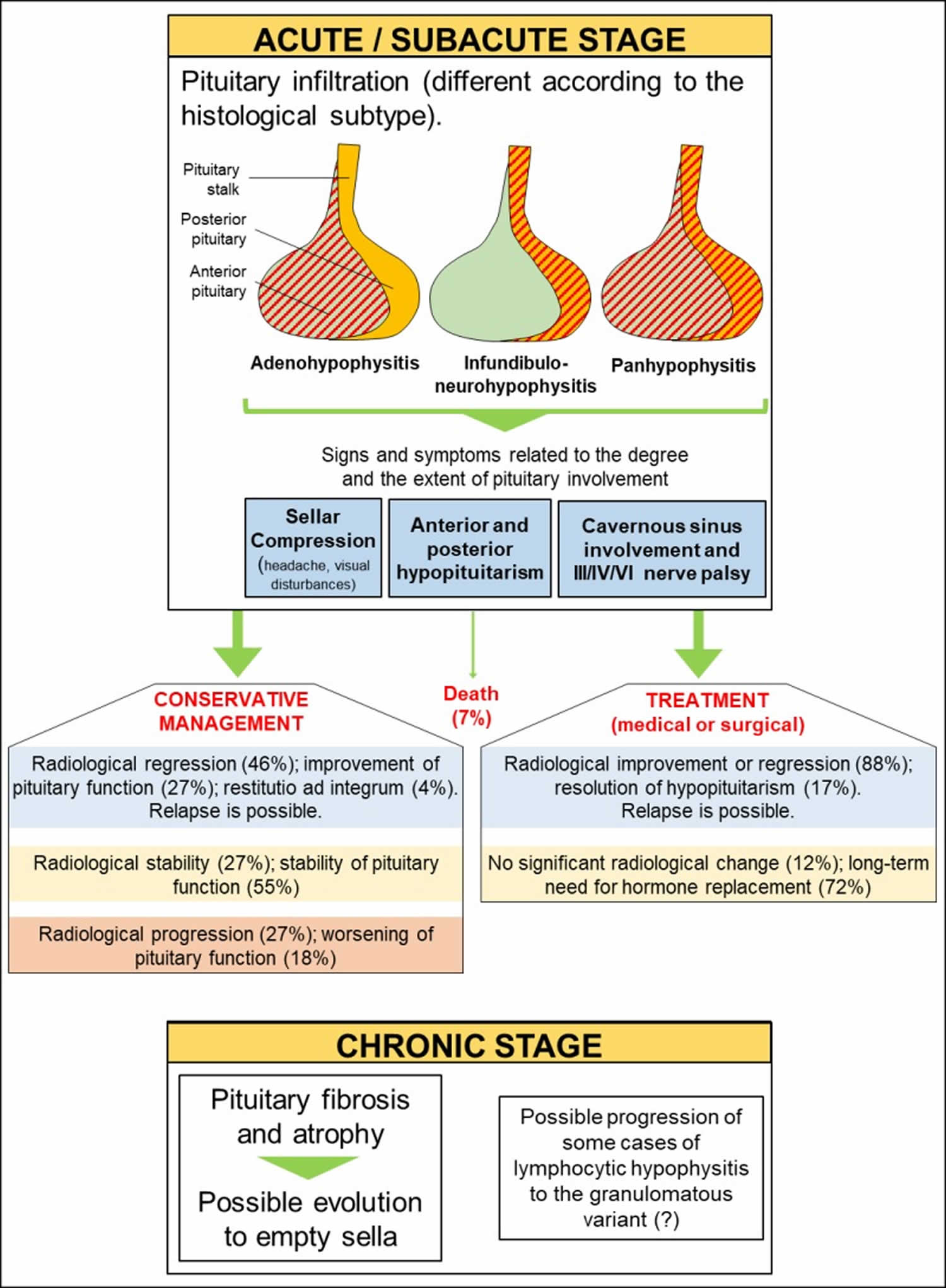
Primary hypophysitis can be self-limiting and spontaneous remission may occur (Figure 1). Considering the low prevalence of the disease, however, robust data regarding the natural history of primary hypophysitis are lacking 3. Moreover, most of the literature regards lymphocytic hypophysitis, while data from other histology subtypes are less robust. A recent German review of 76 cases of primary hypophysitis has shown that patients not receiving any active treatment had improvement, stability or progression of the pituitary involvement at MRI in 46%, 27% and 27% of cases, respectively; pituitary deficiencies improved, remained stable or worsened in 27%, 55% and 18% of patients, respectively 4. A previous study by Khare et al. showed that spontaneous resolution of sella compression symptoms occurred in all patients managed conservatively and that 33% had complete or partial recovery of pituitary function 5. Park et al. also reported similar findings 6.
Primary hypophysitis frequently evolves to fibrosis and pituitary atrophy in the chronic stages of the disease, which often impair pituitary function (Figure 1). The evolution to empty sella has also been shown in a mouse model of autoimmune hypophysitis 7. Caturegli et al. 3 reported that only 4% of patients had spontaneous remission with recovery of pituitary function, while most patients will require long-term replacement of one or more pituitary axes. Whether medical treatment with glucocorticoids has a positive impact on the natural history of primary hypophysitis is still a matter of debate.
Most of the published case series mainly focus on the more frequent lymphocytic hypophysitis. Granulomatous hypophysitis can cause more severe signs and symptoms (headache, chiasmal compression and anterior/posterior hypopituitarism). Xanthomatous hypophysitis seems to cause sella compression and pituitary dysfunction less frequently. IgG4-related hypophysitis can cause various degree of involvement of the anterior pituitary, posterior pituitary and the stalk. Necrotizing hypophysitis is extremely rare and is associated with a high risk of panhypopituitarism and diabetes insipidus. The chronic stage of the disease is most likely related to the extent of damage of the pituitary. Some authors have suggested that some cases of lymphocytic hypophysitis may evolve to the granulomatous form, as mixed forms can rarely be observed. A death rate of 7% has been described in large case series of patients with primary hypophysitis and is probably related to unrecognized acute adrenal insufficiency.
Figure 1. Natural history of Primary Hypophysitis
Hypophysitis causes
Primary hypophysitis
Primary hypophysitis is a rare disease. The incidence is estimated to be ~1 in 9 million/year 8, and hypophysitis accounts for ~0.4% of pituitary surgery cases 9. Five histologic variants of primary hypophysitis have been described, and there are mixed forms as well. Table 1 summarizes the epidemiological and histopathological features of these variants 10. Primary hypophysitis, apart from the rare IgG4-related and necrotizing variants, occurs more frequently in young females. The clinical manifestations of all forms of primary hypophysitis are similar and are linked to the degree of pituitary involvement and the associated hormonal deficiencies.
Table 1. Characteristics of the various forms of Primary Hypophysitis
| Lymphocytic hypophysitis | Granulomatous hypophysitis | Xanthomatous hypophysitis | IgG4-related hypophysitis | Necrotizing hypophysitis | |
| Prevalence | The most common subtype (68%). | The second most common subtype (20%). | Very rare (3%). | Very rare (4%). Higher prevalence in Japan and Korea. | Extremely rare (<1%). |
| Gender predominance | Female, ~3:1 | Female, ~3:1 | Female, ~3:1 | Male, ~2:1 | Male, ~3:1 |
| Association with pregnancy | Yes. ~70% of patients present during pregnancy or postpartum. | No | No | No | No |
| Mean age at presentation | 4th decade (females). 5th decade (males) | 5th decade | 4th decade | 7th decade | Four cases reported (aged 12, 20, 33 and 39) |
| Histopathology | Diffuse lymphocyte infiltration (primarily T cells) of the pituitary gland. Lymphoid follicles can be observed and occasional plasma cells, eosinophils, and fibroblasts may also be present. Pituitary fibrosis and atrophy may occur in later stages of the disease. | Large numbers of multinucleated giant cells and histiocytes with granuloma formation. | Foamy histiocytes (lipid-rich macrophages) without the presence of granulomas. Plasma cells and small round mature lymphocytes are also observed. Pituitary fibrosis may be seen in later stages of the disease. | Extensive gland infiltration by plasma cells with a high degree of IgG4 positivity. Storiform fibrosis is observed *. Pituitary fibrosis and atrophy occur in later stages of the disease, if not treated. | Diffuse non-hemorrhagic necrosis with surrounding lymphocytes, plasma cells and eosinophils. |
Footnote: * Storiform fibrosis: dense, wire-like strands of fibrotic collagen deposition radiating outward from a central point.
Anatomic location of pituitary involvement
- Adenohypophysitis: the inflammation involves the anterior pituitary. It accounts for ~65% of cases of primary hypophysitis;
- Infundibulo-neurohypophysitis: the inflammation involves the posterior pituitary and the stalk. It accounts for ~10% of cases of primary hypophysitis;
- Panhypophysitis: the inflammation involves the entire gland. It accounts for ~25% of cases of primary hypophysitis.
Histopathology forms of primary hypophysitis
- Lymphocytic hypophysitis (68%);
- Granulomatous hypophysitis (20%);
- Xanthomatous hypophysitis (3%);
- IgG4-related (plasmacytic) hypophysitis (4%); The infiltrate focuses around the lesion rather than diffuse in the entire gland. This secondary form of pituitary infiltration is generally a histopathological finding and patient’s signs and symptoms are otherwise related to the primary sella and parasella mass.
- Necrotizing hypophysitis (<1%);
- Mixed forms (lymphogranulomatous; xanthogranulomatous).
Primary hypophysitis causes
- Isolated
- Associated with autoimmune diseases:
- Polyglandular autoimmune syndromes
- Autoimmune thyroiditis (Hashimoto thyroiditis)
- Autoimmune adrenalitis
- Type 1 diabetes mellitus
- Lymphocytic parathyroiditis
- Idiopathic inflammatory myopathy
- Systemic lupus erythematosus
- Sjogren’s syndrome
- Rheumatoid arthritis
- Primary biliary cirrhosis
- Atrophic gastritis
- Optic neuritis
- Myocarditis
- Temporal arteritis
- Bechet’s disease
- Retroperitoneal fibrosis
- Erythema nodosum
- Idiopathic thrombocytopenic purpura
- Dacryoadenitis.
Lymphocytic hypophysitis
Lymphocytic hypophysitis is the most common histologic variant of primary hypophysitis, with over 460 histologically-proven cases reported in the literature 11. Lymphocytic hypophysitis shows a striking temporal association with pregnancy, with ~70% of cases in women presenting during pregnancy or postpartum. Most patients present in the last month of pregnancy or in the first 2 months after delivery 8. Lymphocytic hypophysitis is believed to have an autoimmune etiology. This is supported by the lymphocytic infiltration of the pituitary, the link with pregnancy, the frequent association with other autoimmune diseases, the frequent finding of pituitary antibodies in these patients, the association with particular human leukocyte antigen alleles 2, and the improvement of symptoms in response to immunosuppressive drugs.
Granulomatous hypophysitis
Granulomatous hypophysitis is the second most common subtype of primary hypophysitis and its etiology is unknown. Before concluding that a case of granulomatous hypophysitis is “primary” (i.e. idiopathic), known possible causes of granulomatous infiltration of the pituitary should be excluded. Possible secondary causes of granulomatous hypophysitis include tuberculosis, sarcoidosis, syphilis, Langerhans’ histiocytosis, granulomatosis with polyangitis (formerly known as Wegener’s granulomatosis), and Rathke’s cleft cyst rupture 12.
Xanthomatous hypophysitis
The pituitary shows cystic-like areas of liquefaction infiltrated by lipid-rich macrophages. It has been suggested that many cases of xanthomatous hypophysitis may represent an inflammatory response to components of a ruptured Rathke’s cleft cyst 13.
IgG4-related hypophysitis can be isolated (primary hypophysitis), but is often a manifestation of systemic disease with involvement of multiple organs. Some authors include IgG4-related hypophysitis among the histologic variants of primary hypophysitis, while others report this among the secondary forms of hypophysitis. Considering that the diagnosis and management does not change according to the classification used, we will discuss the features of IgG4-related hypophysitis in this section.
The etiology of IgG4-related hypophysitis is poorly understood and may involve autoimmunity and/or an abnormal tolerance to unspecified allergens and infectious agents 14. IgG4-related disease is diagnosed more frequently in older males and is characterized by a dense lymphoplasmacytic infiltration with a predominance of IgG4-positive plasma cells in the affected tissue and storiform fibrosis in the more advanced stages of the disease (Table 1). One or (more frequently) multiple organs can be affected including lymph nodes, pancreas, liver, salivary and lacrimal glands, retroperitoneum, aorta, pericardium, thyroid, lungs, kidneys, skin, stomach, prostate, ovaries, and the pituitary gland 15. Overall, the prevalence of pituitary involvement in IgG4-related disease is low (2-8%) 16.
IgG4-related disease is considered a rare cause of hypophysitis, although a recent Japanese group reported a strikingly high prevalence of IgG4-related hypophysitis in 170 consecutive patients with hypopituitarism/central diabetes insipidus and a clinical diagnosis of hypophysitis (4% and 30% respectively) 17. Moreover, Bernreuther et al. recently reviewed retrospectively 29 cases of biopsy-proven primary hypophysitis previously diagnosed as “lymphocytic” or “not otherwise specified, non-granulomatous” and found that 41.4% of cases fulfilled the criteria for IgG4-related hypophysitis, suggesting that this entity might be more frequent than previously thought 18.
The diagnosis of IgG4-related hypophysitis is confirmed by characteristic histopathologic findings at pituitary biopsy. However, pituitary biopsy is an invasive procedure and other criteria can be used to establish the diagnosis (Table 2) 19.
It should be considered that patients with IgG4-related hypophysitis have multi-organ involvement in 60-90% of cases. Therefore, they should receive an extensive evaluation for establishing the extent of the disease after the initial diagnosis. The diagnostic work-up should include physical examination, laboratory evaluation and whole-body imaging 20.
Table 2. Diagnostic criteria for IgG4-related hypophysitis
| Criteria | Established diagnosis | |
| Criterion 1 | PITUITARY HISTOPATHOLOGY: Mononuclear infiltration of the pituitary gland, rich in lymphocytes and plasma cells, with >10 IgG4-positive cells/high-power field. * | CRITERION 1 or CRITERIA 2 + 3 or CRITERIA 2 + 4 + 5 |
| Criterion 2 | PITUITARY MRI: Sella mass or thickened pituitary stalk. | |
| Criterion 3 | OTHER INVOLVEMENT: Biopsy-proven involvement in other organs. | |
| Criterion 4 | SEROLOGY: Serum IgG4 level >140 mg/dL (1.4 g/L). | |
| Criterion 5 | RESPONSE TO TREATMENT: Shrinkage of the pituitary mass and symptom improvement with corticosteroids. | |
Footnote: * Low level of infiltration may be seen if the patient is receiving treatment with glucocorticoids 21.
Necrotizing hypophysitis
Necrotizing hypophysitis has been reported in four patients (of which only three biopsy-proven) 22. Three patients presented with diabetes insipidus and some degree of anterior pituitary dysfunction was described in all reported cases. Frontal headache at presentation was reported in two patients 22. Three patients were treated surgically and had persistent postoperative panhypopituitarism and central diabetes insipidus 22.
Secondary hypophysitis causes
- Drugs:
- Immune checkpoint inhibitors
- Interferon-α
- Ribavirin
- Ustekinumab
- Sella and parasella diseases:
- Germinoma
- Rathke’s cleft cyst
- Craniopharyngioma
- Pituitary adenoma
- Primary pituitary lymphoma
- Systemic diseases:
- IgG4-related disease: IgG4-related hypophysitis can be isolated, but is often a manifestation of systemic disease with the involvement of multiple organs.
- Sarcoidosis
- Granulomatosis with polyangitis (Wegener’s granulomatosis)
- Langerhans cell histiocytosis
- Erdheim-Chester’s disease
- Rosai-Dorfman disease
- Inflammatory pseudotumor
- Tolosa-Hunt syndrome
- Takayasu’s arteritis
- Cogan’s syndrome
- Crohn’s disease
- Thymoma (anti-Pit-1 antibody syndrome)
- Infections:
- Bacteria (Mycobacterium tuberculosis; Treponema pallidum; Tropheryma whipplei; Borrelia; Brucella)
- Viruses (Cytomegalovirus; Herpes simplex; Varicella-zoster virus; Influenza viruses; Coronavirus; Enterovirus; Coxsackie; Tick-Borne encephalitis virus; Hantavirus)
- Mycoses (Aspergillus; Nocardia; Candida albicans; Pneumocystis jirovecii)
- Parasites (Toxoplasma gondii)
Hypophysitis secondary to sella and parasella disease
Pituitary inflammation can be triggered by sella and parasella disease. The infiltrate is mainly lymphocytic or xanthogranulomatous and focuses around the lesion rather than diffusing to the entire gland 8.
Germinoma
Germinomas are rare brain tumors predominantly affecting prepubertal children. Germinomas are an established cause of secondary hypophysitis 23; moreover, germinomas arising in the sella and parasella region are difficult to differentiate from hypophysitis in children because of similar clinical features (diabetes insipidus + GH deficiency + visual disturbances). This differentiation, nevertheless, is critical for patient care due to different treatments of the two diseases. Biopsy-proven cases of primary hypophysitis are extremely rare in children and adolescents 24; therefore, in children below 10 years a germinoma should be considered the most likely diagnosis.
Tumor markers such as α-fetoprotein, β-human chorionic gonadotropin, or placental alkaline phosphatase in the cerebrospinal fluid may be useful for diagnosing germinoma. However, a pituitary biopsy is the gold standard for differentiating the two conditions, although germinomas can have a marked lymphocytic infiltrate that can outnumber the neoplastic cells making differential diagnosis difficult 25. If germinoma is part of the histologic differential diagnosis, markers for germinomas such as Oct3/4, PLAP and NANOG may be useful.
Finally, it should be noted that the hypopituitarism caused by sella germinomas can precede for years a visible pituitary mass, so that prolonged symptomatic periods prior to diagnosis are common 25.
Rathke’s Cleft Cyst
The rupture of Rathke’s cleft cyst can cause hypophysitis associated with visual disturbances, headache and hypopituitarism including – very frequently – central diabetes insipidus 26. Histopathology can show lymphocytic, granulomatous, xanthomatous or mixed forms of hypophysitis 27. Recent papers have suggested that many cases of xanthomatous hypophysitis may actually be related to rupture of Rathke’s cleft cysts 13.
Other Sella and Parasella Masses
Cases of secondary hypophysitis have been described in association with craniopharyngiomas 28, pituitary adenomas 29 and primary pituitary lymphomas 29.
Hypophysitis secondary to systemic disease
Sarcoidosis
Sarcoidosis is a multisystem inflammatory disease of unknown origin characterized by the formation of non-caseating granulomas that can involve all organ systems. The central nervous system can be affected in 5-15% of patients (neurosarcoidosis) and may be the presenting feature of the disease 30. Granulomas can involve the pituitary, hypothalamus and the stalk in ~0.5% of patients with sarcoidosis, resulting in varying grade of hypopituitarism 31. Approximately 60% of the cases reported in the literature are males presenting in the 3rd and 4th decade. Central diabetes insipidus, FSH/LH deficiency and hyperprolactinemia are among the most frequent hormone abnormalities 32. Patients with sarcoidosis and hypothalamic-pituitary involvement tend to have more frequent multi-organ involvement, as well as neurosarcoidosis and sinonasal involvement 32.
Granulomatosis with Polyangiitis (previously known as Wegener’s Granulomatosis)
Granulomatosis with polyangiitis is an antineutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis of unknown etiology with multisystem involvement and formation of necrotizing granulomas and vasculitis in small- and medium-sized blood vessels. Pituitary involvement is a rare and late manifestation of the disease 31. Secondary hypogonadism and central diabetes insipidus are the most common endocrine abnormalities; diabetes insipidus can recover after adequate treatment of the underlying vasculitis, while anterior pituitary dysfunction is permanent in the majority of patients 33.
Langerhans Cell Histiocytosis
Langerhans cell histiocytosis is a rare disease mainly occurring in childhood, involving clonal proliferation of myeloid Langerhans cells that can infiltrate multiple organs (bones, skin, lymph nodes, lungs, thymus, liver, spleen, bone marrow, and central nervous system including the pituitary). Patients often carry the BRAF V600E mutation in the clonal myeloid cells 34.
The most common endocrine abnormality in patients with Langerhans cell histiocytosis is hypothalamic-pituitary infiltration causing central diabetes insipidus. These patients usually have multi-organ and cranio-facial involvement, although localized disease of the hypothalamic-pituitary region has been reported 35. Up to 40% of patients develop symptoms consistent with diabetes insipidus within the first four years, particularly if there is multisystem involvement and proptosis 36. Anterior pituitary hormone deficiency is also possible at diagnosis and during follow up 35.
Langerhans cell histiocytosis and germinoma are the most common cause of central diabetes insipidus in children and adolescents; therefore, germinoma should always been considered in the differential diagnosis 37.
The definitive diagnosis of Langerhans cell histiocytosis is the biopsy-proven infiltration of the pituitary with Langerhans cells with eosinophils, neutrophils, small lymphocytes, and histiocytes. However, pituitary biopsy is invasive and the diagnosis can be suggested by the presence of the characteristic histopathologic features in other tissues when a multisystem disease is present. For patients with suspected disease isolated to the pituitary, identification of BRAF-V600E in the peripheral blood or cerebrospinal fluid can support the diagnosis and rule out germinoma, although it does not distinguish Langerhans cell histiocytosis from Erdheim-Chester disease 38.
When hypophysitis secondary to Langerhans cell histiocytosis is suspected but pituitary biopsy is not available, it is reasonable to initiate therapy empirically with a plan to follow disease response with MRI. Treatment options include prednisone, alone or in combination with vinblastine, cladribine and vemurafenib, alongside desmopressin and other pituitary hormone replacements to treat hypopituitarism.
Erdheim-Chester disease
Erdheim-Chester’s disease is a rare multisystem histiocytic disorder, most often seen in adults, which may be confused with Langerhans cell histiocytosis. Histiocytic infiltration leads to xanthogranulomatous infiltrates of multiple tissues (bones, skin, lungs, facial, orbital and retro-orbital tissue, retroperitoneum, cardiovascular system and cerebral nervous system including the pituitary). Long bone pain and symmetric osteosclerotic lesions suggest this diagnosis, which is confirmed by tissue biopsies showing histiocytes with non-Langerhans features. Patients often carry the BRAF V600E mutation in the clonal myeloid progenitor cells 34.
Pituitary involvement may manifest as central diabetes insipidus and anterior hypopituitarism, which typically persist even with radiographic regression of the disease. As for Langerhans cell histiocytosis, the definitive diagnosis of Erdheim-Chester’s disease is the finding of the typical histologic features at pituitary biopsy, which can be supported by the finding of the BRAF V600E mutation. Treatment options include vemurafenib, interferon-α, dabrafenib, trametinib, Cobimetinib, cladribine, cyclophosphamide and glucocorticoids.
Rosai-Dorfman disease
Pituitary involvement has been described in Rosai-Dorfman disease, a rare histiocytic disorder. Patients may have both anterior pituitary dysfunction, central diabetes insipidus and visual disturbances 39.
Inflammatory Pseudotumor
The inflammatory pseudotumor is a rare inflammatory disorder commonly involving the lung and orbit. It can be isolated or associated with the IgG4-related disease 40. Pituitary infiltration is a rare manifestation and patients can present with anterior and posterior hypopituitarism. The inflammatory pseudotumor can also spread to the sphenoid sinus, the cavernous sinus and the optic chiasm 41.
Tolosa-Hunt syndrome
Tolosa-Hunt syndrome is a painful ophthalmoplegia caused by idiopathic retro-orbital inflammation involving the cavernous sinus or the superior orbital fissure. Histology shows nonspecific granulomatous or nongranulomatous inflammation. Patients with pituitary involvement present with anterior and posterior hypopituitarism, diplopia and retro-orbital pain (often unilateral) 42.
Other Systemic diseases
Cases of secondary hypophysitis have been described in association with Takayasu’s arteritis (granulomatous hypophysitis) 43, Cogan’s syndrome 44 and Crohn’s disease 45. A case of isolated ACTH deficiency in a patient with Crohn’s disease has also been published 46.
Other causes of secondary hypophysitis
Thymoma (Anti-Pit-1 Antibody Syndrome)
Pit-1 is essential for the differentiation, proliferation, and maintenance of somatotrophs, lactotrophs, and thyrotrophs in the pituitary 47. Yamamoto et al. 48 described three cases of acquired combined TSH, GH, and PRL deficiency, with circulating anti-Pit-1 antibodies. Cytotoxic T-cells that react against Pit-1 may be the cause of anti-Pit-1 antibody syndrome 49. Recently, all these patients developed thymomas that express Pit-1. Removal of the thymoma resulted in a decline in antibody titer, suggesting that aberrant expression of Pit-1 in the thymoma plays a causal role in the development of this syndrome 50.
Infections
Infections of the pituitary are a rare cause of hypophysitis and hypopituitarism 51. They can affect either exclusively the pituitary area or as a part of disseminated infections. Risk factors are diabetes mellitus, organ transplantation, human immunodeficiency virus infection, non-Hodgkin lymphoma, chemotherapy and Cushing’s syndrome. They can occur by 31:
- Hematogenous spread in immuno-compromised hosts;
- Contiguous extension from adjacent anatomical sites (meninges, sphenoid sinus, cavernous sinus and skull base);
- Previous infectious diseases of the CNS of different etiologies;
- Iatrogenic inoculation during trans-sphenoidal surgery.
However, in the majority of cases of pituitary abscess an obvious cause cannot be identified.
Tuberculosis can cause granulomatous involvement of the hypothalamus, the pituitary or the stalk. Tubercular meningitis and hypothalamic-pituitary involvement seem to affect mostly anterior pituitary function 52.
Several viruses can cause meningitis, meningoencephalitis and encephalitis that can involve the hypothalamic-pituitary region. Partial or complete hypopituitarism may develop as a result 31. A study by Leow et al. has shown that ~40% of patients with severe acute respiratory syndrome (SARS)-associated with Coronavirus infection can develop reversible central adrenal insufficiency, suggesting a possible inflammation of the pituitary in these patients 53. Hantavirus can also cause viral hypophysitis with pituitary ischemia and hemorrhage as part of the hemorrhagic fever with renal syndrome (HFRS), leading to partial or complete hypopituitarism 54.
Mycoses with hypothalamic-pituitary involvement are extremely rare. Patients frequently present with central diabetes insipidus and anterior pituitary dysfunction (mainly FSH/LH deficiency and hyperprolactinemia) 31.
Hypophysitis symptoms
The signs and symptoms at diagnosis, as well as the pituitary hormone abnormalities depend on the degree of pituitary involvement (Table 3) 11.
Primary hypophysitis more frequently involves the anterior pituitary and patients typically present with severe headaches, visual disturbances due to chiasmal compression and symptoms of adrenal insufficiency. Contrary to other causes of hypopituitarism, impaired adrenocorticotropic hormone (ACTH) and thyroid-stimulating hormone (TSH) secretion is very frequent in the early stages of primary hypophysitis, putting these patients at increased risk of life-threatening adrenal insufficiency. A recent case series has highlighted that secretion of gonadotropins is also impaired very frequently in these patients 55. Growth hormone (GH) deficiency and hyperprolactinemia can also occur.
Less frequently, the inflammation can involve primarily the posterior pituitary and the stalk. Patients with infundibulo-neurohypophysitis typically present with diabetes insipidus and other pituitary hormone deficiencies are less common. As expected, signs of both anterior and posterior pituitary involvement coexist in panhypophysitis (that is, inflammation of the entire gland).
Granulomatous hypophysitis can be associated with more severe symptoms than lymphocytic hypophysitis, with two case series documenting more frequent occurrence of headache, chiasmal compression and hypopituitarism 55. A recent review of the literature found that the most common symptoms of granulomatous hypophysitis at presentation were headache (61%), visual changes (40%), polyuria/polydipsia (27%) and cranial nerve palsies (27%); panhypopituitarism and diabetes insipidus were found in 49% and 27% of cases, respectively 12.
Clinical data regarding xanthomatous and IgG4-related hypophysitis are less robust due to the rarity of these variants. However, Gutenberg et al. 56 found that xanthomatous hypophysitis did not cause chiasmal compression and was associated with a low risk of diabetes insipidus and a less severe anterior pituitary hormone impairment than lymphocytic or granulomatous hypophysitis (FSH/LH and GH deficiencies are more common than TSH and ACTH deficiencies). IgG4-related hypophysitis involves frequently both the pituitary and the stalk (~65%) and causes panhypopituitarism, anterior hypopituitarism and central diabetes insipidus in ~50%, ~25% and ~18% of cases, respectively 57. Cases of intrachiasmal abscess and spreading to the cavernous sinus have also been reported 58.
Primary hypophysitis is rare in children, with less than 100 cases reported in the literature of which only a few were biopsy-proven 59. The clinical presentation, however, seems to differ from adults. A review of the literature showed that the most common presenting symptoms in children are caused by antidiuretic hormone (ADH) deficiency (85%) 59. Growth hormone (GH) deficiency is found in 76% of cases, while FSH/LH, TSH and ACTH deficiencies were less common than in adults (32%, 29% and 20%, respectively). Headaches and visual disturbances were also rarely reported (17% and 8% of cases, respectively) 59. As central diabetes insipidus and growth retardation are the most common presenting symptoms in children with primary hypophysitis, the more frequent intracranial germinoma and Langerhans cell histiocytosis, as well as craniopharyngiomas, have to be considered in the differential diagnosis 60. Moreover, children with a presumptive diagnosis of hypophysitis are at risk of developing germinomas later in life (up to 3 years after the initial diagnosis) and require extended follow-up 61. Germinomas are also a documented cause of secondary hypophysitis.
Table 3. Clinical presentation and prevalence of pituitary hormone abnormalities at diagnosis in patients with Primary Hypophysitis according to the degree of pituitary involvement
| Signs and symptoms at diagnosis | |||
| Adenohypophysitis (~65% of cases) | Infundibulo-neurohypophysitis (~10% of cases) | Panhypophysitis (~25% of cases) | All forms * |
| Headache: 53% Visual disturbances: 43% Adrenal insufficiency: 42% Hyperprolactinemia: 23% Hypothyroidism: 18% Hypogonadism: 12% Lactation failure: 11% Polydipsia/polyuria: 1% | Polydipsia/polyuria: 98% Headache: 13% Adrenal insufficiency: 8% Hyperprolactinemia: 5% Hypogonadism: 3% Visual disturbances: 3% Hypothyroidism: 0% Lactation failure: 0% | Polydipsia/polyuria: 83% Headache: 41% Adrenal insufficiency: 19% Visual disturbances: 18% Hypothyroidism: 17% Hyperprolactinemia: 17% Hypogonadism: 14% Lactation failure: 5% | Headache: 47% Adrenal insufficiency: 35% Polydipsia/polyuria: 35% Visual disturbances: 31% Hypothyroidism: 16% Hypogonadism: 20% Hyperprolactinemia: 20% Lactation failure: 8% |
| Pituitary hormone abnormalities at diagnosis | |||
| Adenohypophysitis (~65% of cases) | Infundibulo-neurohypophysitis (~10% of cases) | Panhypophysitis (~25% of cases) | All forms |
| ACTH deficiency: 56% TSH deficiency: 44% FSH/LH deficiency: 42% GH decreased: 26% Hyperprolactinemia: 25% Hyperprolactinemia: 23% *** ADH deficiency: 0% | ADH deficiency: 98% FSH/LH deficiency: 8% ** Hyperprolactinemia: 5% *** Hyperprolactinemia: 0% ACTH deficiency: 0% TSH deficiency: 0% GH decreased: 0% ** | ADH deficiency: 95% GH decreased: 51% FSH/LH deficiency: 47% ACTH deficiency: 46% Hyperprolactinemia: 40% *** TSH deficiency: 39% Hyperprolactinemia: 16% | ACTH deficiency: 60% FSH/LH deficiency: 55% TSH deficiency: 52% ADH deficiency: 39% GH decreased: 38% Hyperprolactinemia: 37% Hyperprolactinemia: 26% |
Footnotes:
* Other possible symptoms at diagnosis include weight gain (18%) and temperature dysregulation (rare) 55.
** Some case series have reported a high prevalence of GH and FSH/L deficiency in patients with infundibulo-neurohypophysitis 62 .
*** Hyperprolactinemia may be related to stalk compression (disconnection hyperprolactinemia) or to the immune-mediated destruction of prolactin-secreting cells.
Abbreviations: ACTH = adrenocorticotropic hormone; ADH = antidiuretic hormone; FSH = follicle-stimulating hormone; GH = growth hormone; LH = luteinizing hormone; TSH = thyroid-stimulating hormone.
Hypophysitis diagnosis
Pituitary biopsy is the gold standard to confirm the diagnosis of primary hypophysitis. This procedure, however, should be considered only in equivocal cases and when the outcome of the biopsy is expected to change the therapeutic management, and should be performed by a neurosurgeon with extensive expertise in pituitary surgery.
Magnetic resonance imaging (MRI) of the sella region typically shows an enlarged pituitary. In order to avoid unnecessary surgery, primary hypophysitis needs to be differentiated from other sella and parasella masses 63, with pituitary adenomas being the most frequent differential diagnosis in adults.
Primary hypophysitis typically presents as a homogeneous pituitary enlargement with intense and homogeneous enhancement post-gadolinium and no deviation of the stalk; these and other features can help differentiate between primary hypophysitis and pituitary adenomas at MRI 64. Further differential diagnoses, especially for lymphocytic hypophysitis, are the physiologic pituitary enlargement associated with pregnancy and Sheehan’s syndrome, although these patients have no history of obstetric hemorrhage 65. A cautious balance between radiological, clinical and laboratory findings is necessary to reach the correct diagnosis and avoid unnecessary treatment 66.
Several authors have assessed the presence and utility of serum autoantibodies (pituitary and/or hypothalamic antibodies) in patients with primary hypophysitis:
- An autoimmune etiology for lymphocytic hypophysitis was suggested by the presence of pituitary antibodies that may recognize α-enolase, GH, the pituitary gland-specific factors 1a and 2 (PGSF1a and PGSF2), regulatory prohormone-processing enzymes commonly produced in the pituitary gland (PC1/3, PC2, CPE and 7B2), secretogranin II, chromosome 14 open reading frame 166 (C14orf166), the corticotroph-specific transcription factor TPIT and chorionic somatomammotrophin (HCS) 3. Several techniques have been used to detect pituitary antibodies in primary hypophysitis (ELISA, radioligand assay, immunoblotting and immunofluorescence) and the prevalence of antibody-positive hypophysitis is 11-73% depending from the antigen(s) tested and the technique used 67. However, the pathogenic role of these autoantibodies is unclear and they are not specific to hypophysitis. For example, pituitary antibodies were identified by indirect immunofluorescence in ~45% of patients with biopsy-proven hypophysitis, but were also found in the serum of patients with isolated central diabetes insipidus (35%), germinomas (33%), isolated anterior hormone deficiencies (29%), prolactinomas (27%), Rathke’s cleft cysts (25%), craniopharyngiomas (17%) nonfunctioning pituitary tumors (13%), GH-secreting pituitary tumors (12%) and healthy subjects (5%) 67. They can also be found in patients with autoimmune endocrine disorders, especially Hashimoto thyroiditis 68. However, indirect immunofluorescence using human pituitary gland as a substrate and showing a granular cytosolic staining pattern was most commonly found in patients with hypophysitis and isolated hormone deficiencies 67; therefore, the finding of this staining pattern can be useful to clinicians in establishing a diagnosis of hypophysitis;
- The detection of hypothalamic antibodies targeting corticotropin-releasing hormone (CRH)-secreting cells in some patients with GH/ACTH deficiency but with pituitary antibodies targeting only GH-secreting cells indicates that an autoimmune aggression to the hypothalamus can be responsible for some cases of lymphocytic hypophysitis 69. Consequently, not only pituitary but also hypothalamic autoimmunity may contribute to anterior pituitary dysfunction in a subset of patients with primary hypophysitis;
- A search for ADH antibodies in patients with primary hypophysitis may help identifying patients who are prone to developing autoimmune central diabetes insipidus 70. These antibodies alone are not a good diagnostic marker for posterior pituitary involvement, but may serve as a predictive marker of gestational or post-partum diabetes insipidus 71;
- Anti-Rabphilin antibodies have been proposed to be a biomarker for lymphocytic infundibulo-neurohypophysitis 72. Rabphilin is involved in the release of hormones or neurotransmitters and is expressed mainly in the brain, including the posterior pituitary and hypothalamus where ADH is present. Whether anti-Rabphilin antibodies are a cause of central diabetes insipidus or a result of infundibulo-neurohypophysitis is unknown. However, anti-Rabphilin antibodies are detected in 76% of patients with infundibulo-neurohypophysitis and 11% of patients with lymphocytic hypophysitis. In contrast, these antibodies are absent in patients with sella/suprasella masses without lymphocytic hypophysitis, suggesting that this antibody may serve as a biomarker for the diagnosis of infundibulo-neurohypophysitis and may be useful for the differential diagnosis in patients with central diabetes insipidus 63;
- Primary hypophysitis can eventually evolve in pituitary fibrosis and atrophy, documented at imaging as an “empty sella”. Lupi et al. have found pituitary antibodies in 6% of patients with an empty sella not linked to previous head trauma. In this cohort, the presence of pituitary antibodies also correlated with the presence of hypopituitarism 73;
- Antibodies recognizing GH and one peptide from proopiomelanocortin (POMC) have been described in a patient with IgG4-related hypophysitis 74.
Hypophysitis treatment
Due to the rarity of hypophysitis, the management of hypophysitis is controversial 1. An algorithm in line with the more recent literature is reported in Figure 2. Initial evaluation of patients with suspected hypophysitis involves clinical and laboratory assessment. Patients with a suspicion of hypophysitis based on biochemical results should undergo a pituitary MRI, as well as visual assessment to check visual fields and acuity. Secondary causes of hypophysitis and other sella/parasella masses should be considered in the differential diagnosis.
The mainstay of treatment of primary hypophysitis is pituitary hormone replacement 75. As outlined above, ACTH production is frequently impaired at presentation, and most patients will require glucocorticoid replacement. This should be started before thyroxine replacement (if TSH deficiency is present as well) to avoid precipitating acute adrenal insufficiency.
Conservative management is recommended for primary hypophysitis unless symptoms are severe and progressive. The only exception to this rule is IgG4-related hypophysitis that – like other manifestations of the disease – should be promptly treated to revert symptoms and prevent fibrosis 76. The mainstay of treatment are glucocorticoids, which often cause remission of symptoms within a few weeks. A typical starting dose is prednisone 30-40 mg/day (or equivalent), which should be continued for 2-4 weeks, and then tapered gradually over 2-6 months 20. However, some patients may benefit from long-term maintenance glucocorticoid therapy (with or without a steroid-sparing agent), especially in case of extensive multi-organ involvement. Relapse is possible and multiple courses of high-dose glucocorticoids are often necessary. Rituximab has also been used in patients with poor response to glucocorticoids 20. A case of IgG4-related hypophysitis successfully treated with azathioprine has also been reported 77.
High-dose glucocorticoids are the first-line treatment to improve the swelling of the pituitary and improve the symptoms related to significant sella compression. Anterior pituitary function can recover after pulse corticosteroid therapy, although >70% of patients will require long-term replacement with one or more hormones 8; central diabetes insipidus rarely recovers. Honegger et al. documented excellent initial responses to high-dose glucocorticoids, with radiological improvement, stability and progression in 65%, 31% and 4% of cases, respectively 4. However, these patients carried a higher risk of side effects (weight gain, psychiatric symptoms, peripheral edema, diabetes mellitus and glaucoma) and relapse of the pituitary inflammation was documented in 38% of cases. Relapses occurred 2-17 months after starting pulse steroids and the risk or relapse did not correlate with either initial glucocorticoid dose or treatment duration 4.
Hormone deficiencies improved with glucocorticoids only in 15% of patients, while they remained stable or worsened in 70% and 15% of cases, respectively 4. Lupi et al. 78 performed a review of the literature and found somewhat better outcomes with medical therapy, reporting pituitary mass reduction in 84% of cases, improving anterior pituitary function in 45%, and restored posterior pituitary function in 41% after high-dose glucocorticoids and/or azathioprine, with a relatively low risk of relapse (14%). The presence of central diabetes insipidus appears to be an unfavorable prognostic factor for response to glucocorticoids; in fact, patients with concomitant anterior and posterior pituitary dysfunction were found to have poor response to glucocorticoids, which were unable to revert the hypopituitarism 79. Glucocorticoid therapy was also found to be less effective in granulomatous or xanthomatous hypophysitis 56.
In glucocorticoid-resistant cases and when high-dose glucocorticoids cause unacceptable side effects, immunosuppressive drugs such as azathioprine, methotrexate and cyclosporin A have been used successfully. However, the long-term effects are unclear 2. Recently, rituximab has been employed to treat steroid-refractory lymphocytic hypophysitis 80.
Surgery should be considered only in cases with serious and progressive deficits of the visual field, visual acuity, or nerve paralysis not responsive to medical treatment. Surgery generally improves sella compression in the short term; however, Honegger et al. 4 observed progression/relapse of the disease in 25% of patients after a mean follow-up of 3 years. Pituitary function improved only in 8% of patients after surgery, and the rates of resolution of chiasmal compression were also low 4.
Stereotactic radiotherapy (radiosurgery) has been effectively employed in selected patients who have failed medical treatment or suffer from repeated recurrence of lymphocytic hypophysitis 81.
Figure 2. Diagnosis and management of Primary Hypophysitis
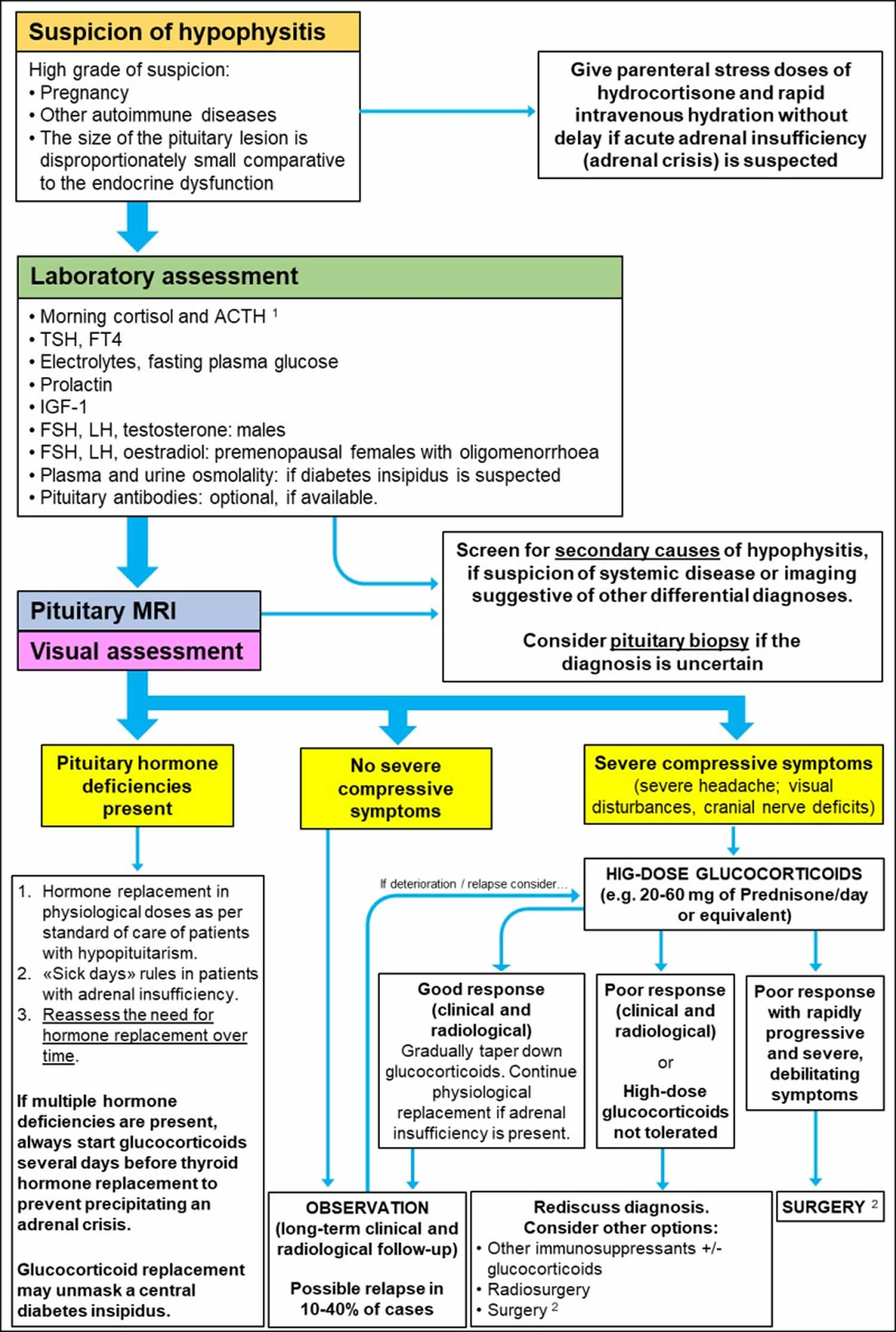
Footnote:
1) Check random ACTH and cortisol if acute adrenal insufficiency is suspected. Consider confirmatory testing with Synacthen if equivocal or borderline results The Synacthen test can give false-positive results in the early stages of central adrenal insufficiency.
2) Pituitary surgery can also provide histology for definitive diagnosis.
[Source 1 ] References- Prete A, Salvatori R. Hypophysitis. 2018 Aug 15. In: Feingold KR, Anawalt B, Boyce A, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK519842
- Bellastella G, Maiorino MI, Bizzarro A, Giugliano D, Esposito K, Bellastella A, De Bellis A. Revisitation of autoimmune hypophysitis: knowledge and uncertainties on pathophysiological and clinical aspects. Pituitary. 2016;19:625–642.
- Caturegli P, Lupi I, Landek-Salgado M, Kimura H, Rose NR. Pituitary autoimmunity: 30 years later. Autoimmun Rev. 2008;7:631–637.
- Honegger J, Buchfelder M, Schlaffer S, Droste M, Werner S, Strasburger C, Stormann S, Schopohl J, Kacheva S, Deutschbein T, Stalla G, Flitsch J, Milian M, Petersenn S, Elbelt U. Pituitary Working Group of the German Society of E. Treatment of Primary Hypophysitis in Germany. J Clin Endocrinol Metab. 2015;100:3460–3469.
- Khare S, Jagtap VS, Budyal SR, Kasaliwal R, Kakade HR, Bukan A, Sankhe S, Lila AR, Bandgar T, Menon PS, Shah NS. Primary (autoimmune) hypophysitis: a single centre experience. Pituitary. 2015;18:16–22.
- Park SM, Bae JC, Joung JY, Cho YY, Kim TH, Jin SM, Suh S, Hur KY, Kim KW. Clinical characteristics, management, and outcome of 22 cases of primary hypophysitis. Endocrinol Metab (Seoul). 2014;29:470–478.
- Lupi I, Zhang J, Gutenberg A, Landek-Salgado M, Tzou SC, Mori S, Caturegli P. From pituitary expansion to empty sella: disease progression in a mouse model of autoimmune hypophysitis. Endocrinology. 2011;152:4190–4198.
- Caturegli P, Newschaffer C, Olivi A, Pomper MG, Burger PC, Rose NR. Autoimmune hypophysitis. Endocr Rev. 2005;26:599–614.
- Faje A. Hypophysitis: Evaluation and Management. Clin Diabetes Endocrinol. 2016;2:15.
- Guaraldi F, Giordano R, Grottoli S, Ghizzoni L, Arvat E, Ghigo E. Pituitary Autoimmunity. Front Horm Res. 2017;48:48–68.
- Caturegli P, Di Dalmazi G, Lombardi M, Grosso F, Larman HB, Larman T, Taverna G, Cosottini M, Lupi I. Hypophysitis Secondary to Cytotoxic T-Lymphocyte-Associated Protein 4 Blockade: Insights into Pathogenesis from an Autopsy Series. Am J Pathol. 2016;186:3225–3235.
- Hunn BH, Martin WG, Simpson S Jr, McLean CA. Idiopathic granulomatous hypophysitis: a systematic review of 82 cases in the literature. Pituitary. 2014;17:357–365.
- Kleinschmidt-DeMasters BK, Lillehei KO, Hankinson TC. Review of xanthomatous lesions of the sella. Brain Pathol. 2017;27:377–395.
- Kuruma S, Kamisawa T, Tabata T, Chiba K, Iwasaki S, Fujiwara T, Kuwata G, Egarashira H, Koizumi K, Koizumi S, Endo Y, Fujiwara J, Arakawa T, Momma K. Allergen-specific IgE antibody serologic assays in patients with autoimmune pancreatitis. Intern Med. 2014;53:541–543.
- Weindorf SC, Frederiksen JK. IgG4-Related Disease: A Reminder for Practicing Pathologists. Arch Pathol Lab Med. 2017;141:1476–1483
- Lin W, Lu S, Chen H, Wu Q, Fei Y, Li M, Zhang X, Tian X, Zheng W, Leng X, Xu D, Wang Q, Shen M, Wang L, Li J, Wu D, Zhao L, Wu C, Yang Y, Peng L, Zhou J, Wang Y, Sha Y, Huang X, Jiao Y, Zeng X, Shi Q, Li P, Zhang S, Hu C, Deng C, Li Y, Zhang S, Liu J, Su J, Hou Y, Jiang Y, You X, Zhang H, Yan L, Zhang W, Zhao Y, Zeng X, Zhang F, Lipsky PE. Clinical characteristics of immunoglobulin G4-related disease: a prospective study of 118 Chinese patients. Rheumatology (Oxford). 2015;54:1982–1990.
- Bando H, Iguchi G, Fukuoka H, Taniguchi M, Yamamoto M, Matsumoto R, Suda K, Nishizawa H, Takahashi M, Kohmura E, Takahashi Y. The prevalence of IgG4-related hypophysitis in 170 consecutive patients with hypopituitarism and/or central diabetes insipidus and review of the literature. Eur J Endocrinol. 2014;170:161–172.
- Bernreuther C, Illies C, Flitsch J, Buchfelder M, Buslei R, Glatzel M, Saeger W. IgG4-related hypophysitis is highly prevalent among cases of histologically confirmed hypophysitis. Brain Pathol. 2017;27:839–845.
- Leporati P, Landek-Salgado MA, Lupi I, Chiovato L, Caturegli P. IgG4-related hypophysitis: a new addition to the hypophysitis spectrum. J Clin Endocrinol Metab. 2011;96:1971–1980.
- Khosroshahi A, Wallace ZS, Crowe JL, Akamizu T, Azumi A, Carruthers MN, Chari ST, Della-Torre E, Frulloni L, Goto H, Hart PA, Kamisawa T, Kawa S, Kawano M, Kim MH, Kodama Y, Kubota K, Lerch MM, Lohr M, Masaki Y, Matsui S, Mimori T, Nakamura S, Nakazawa T, Ohara H, Okazaki K, Ryu JH, Saeki T, Schleinitz N, Shimatsu A, Shimosegawa T, Takahashi H, Takahira M, Tanaka A, Topazian M, Umehara H, Webster GJ, Witzig TE, Yamamoto M, Zhang W, Chiba T, Stone JH. Second International Symposium on Ig GRD. International Consensus Guidance Statement on the Management and Treatment of IgG4-Related Disease. Arthritis Rheumatol. 2015;67:1688–1699.
- Ohkubo Y, Sekido T, Takeshige K, Ishi H, Takei M, Nishio S, Yamazaki M, Komatsu M, Kawa S, Suzuki S. Occurrence of IgG4-related hypophysitis lacking IgG4-bearing plasma cell infiltration during steroid therapy. Intern Med. 2014;53:753–757.
- Gutenberg A, Caturegli P, Metz I, Martinez R, Mohr A, Bruck W, Rohde V. Necrotizing infundibulo-hypophysitis: an entity too rare to be true? Pituitary. 2012;15:202–208.
- Nishiuchi T, Imachi H, Murao K, Fujiwara M, Sato M, Nishiuchi Y, Kushida Y, Haba R, Shindo A, Tamiya T, Ishida T. Suprasella germinoma masquerading as lymphocytic hypophysitis associated with central diabetes insipidus, delayed sexual development, and subsequent hypopituitarism. Am J Med Sci. 2010;339:195–199.
- Gellner V, Kurschel S, Scarpatetti M, Mokry M. Lymphocytic hypophysitis in the pediatric population. Childs Nerv Syst. 2008;24:785–792.
- Gutenberg A, Bell JJ, Lupi I, Tzou SC, Landek-Salgado MA, Kimura H, Su J, Karaviti LP, Salvatori R, Caturegli P. Pituitary and systemic autoimmunity in a case of intrasella germinoma. Pituitary. 2011;14:388–394.
- Yang C, Wu H, Bao X, Wang R. Lymphocytic Hypophysitis Secondary to Ruptured Rathke Cleft Cyst: Case Report and Literature Review. World Neurosurg. 2018;114:172–177.
- Schittenhelm J, Beschorner R, Psaras T, Capper D, Nagele T, Meyermann R, Saeger W, Honegger J, Mittelbronn M. Rathke’s cleft cyst rupture as potential initial event of a secondary perifocal lymphocytic hypophysitis: proposal of an unusual pathogenetic event and review of the literature. Neurosurg Rev. 2008;31:157–163.
- Puchner MJ, Ludecke DK, Saeger W. The anterior pituitary lobe in patients with cystic craniopharyngiomas: three cases of associated lymphocytic hypophysitis. Acta Neurochir (Wien). 1994;126:38–43.
- Martinez JH, Davila Martinez M, Mercado de Gorgola M, Montalvo LF, Tome JE. The coexistence of an intrasella adenoma, lymphocytic hypophysitis, and primary pituitary lymphoma in a patient with acromegaly. Case Rep Endocrinol. 2011;2011:941738.
- Baughman RP, Teirstein AS, Judson MA, Rossman MD, Yeager H Jr, Bresnitz EA, DePalo L, Hunninghake G, Iannuzzi MC, Johns CJ, McLennan G, Moller DR, Newman LS, Rabin DL, Rose C, Rybicki B, Weinberger SE, Terrin ML, Knatterud GL, Cherniak R. Case Control Etiologic Study of Sarcoidosis research g. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med. 2001;164:1885–1889.
- Pekic S, Popovic V. DIAGNOSIS OF ENDOCRINE DISEASE: Expanding the cause of hypopituitarism. Eur J Endocrinol. 2017;176:R269–R282.
- Langrand C, Bihan H, Raverot G, Varron L, Androdias G, Borson-Chazot F, Brue T, Cathebras P, Pinede L, Muller G, Broussolle C, Cotton F, Valeyre D, Seve P. Hypothalamo-pituitary sarcoidosis: a multicenter study of 24 patients. QJM. 2012;105:981–995.
- Kapoor E, Cartin-Ceba R, Specks U, Leavitt J, Erickson B, Erickson D. Pituitary dysfunction in granulomatosis with polyangiitis: the Mayo Clinic experience. J Clin Endocrinol Metab. 2014;99:3988–3994.
- Milne P, Bigley V, Bacon CM, Neel A, McGovern N, Bomken S, Haniffa M, Diamond EL, Durham BH, Visser J, Hunt D, Gunawardena H, Macheta M, McClain KL, Allen C, Abdel-Wahab O, Collin M. Hematopoietic origin of Langerhans cell histiocytosis and Erdheim-Chester disease in adults. Blood. 2017;130:167–175.
- Radojkovic D, Pesic M, Dimic D, Radjenovic Petkovic T, Radenkovic S, Velojic-Golubovic M, Novak V, Ilic I, Radojkovic M. Localised Langerhans cell histiocytosis of the hypothalamic-pituitary region: case report and literature review. Hormones (Athens). 2018;17:119–125.
- Grois N, Fahrner B, Arceci RJ, Henter JI, McClain K, Lassmann H, Nanduri V, Prosch H, Prayer D, Histiocyte Society CNSLCHSG. Central nervous system disease in Langerhans cell histiocytosis. J Pediatr 2010; 156:873-881, 881 e871.
- Prosch H, Grois N, Bokkerink J, Prayer D, Leuschner I, Minkov M, Gadner H. Central diabetes insipidus: Is it Langerhans cell histiocytosis of the pituitary stalk? A diagnostic pitfall. Pediatr Blood Cancer. 2006;46:363–366.
- Allen CE, Ladisch S, McClain KL. How I treat Langerhans cell histiocytosis. Blood. 2015;126:26–35.
- Rotondo F, Munoz DG, Hegele RG, Gray B, Khatun N, Bonert M, Kovacs K. Rosai-Dorfman disease involving the neurohypophysis. Pituitary. 2010;13:256–259.
- Patnana M, Sevrukov AB, Elsayes KM, Viswanathan C, Lubner M, Menias CO. Inflammatory pseudotumor: the great mimicker. AJR Am J Roentgenol. 2012;198:W217-227.
- Al-Shraim M, Syro LV, Kovacs K, Estrada H, Uribe H, Al-Gahtany M. Inflammatory pseudotumor of the pituitary: case report. Surg Neurol. 2004;62:264–267.
- Kita D, Tachibana O, Nagai Y, Sano H, Yamashita J. Granulomatous pachymeningitis around the sella turcica (Tolosa-Hunt syndrome) involving the hypophysis–case report. Neurol Med Chir (Tokyo). 2007;47:85–88.
- Toth M, Szabo P, Racz K, Szende B, Balogh I, Czirjak S, Slowik F, Glaz E. Granulomatous hypophysitis associated with Takayasu’s disease. Clin Endocrinol (Oxf). 1996;45:499–503.
- Kanatani M, Nakamura R, Kurokawa K, Taoda M, Nemoto Y, Kamakura K, Kugai N, Nagata N, Takatani O, Tsuchiya K. Hypopituitarism associated with Cogan’s syndrome; high-dose glucocorticoid therapy reverses pituitary swelling. Jpn J Med. 1991;30:164–169.
- Freeman HJ, Maguire J. Sella inflammatory mass with inflammatory bowel disease. Can J Gastroenterol. 2010;24:58–60.
- Kalambokis G, Vassiliou V, Vergos T, Christou L, Tsatsoulis A, Tsianos EV. Isolated ACTH deficiency associated with Crohn’s disease. J Endocrinol Invest. 2004;27:961–964.
- Larkin S, Ansorge O. Development And Microscopic Anatomy Of The Pituitary Gland. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A, eds. Endotext. South Dartmouth (MA)2000.
- Yamamoto M, Iguchi G, Takeno R, Okimura Y, Sano T, Takahashi M, Nishizawa H, Handayaningshi AE, Fukuoka H, Tobita M, Saitoh T, Tojo K, Mokubo A, Morinobu A, Iida K, Kaji H, Seino S, Chihara K, Takahashi Y. Adult combined GH, prolactin, and TSH deficiency associated with circulating PIT-1 antibody in humans. J Clin Invest. 2011;121:113–119.
- Bando H, Iguchi G, Fukuoka H, Yamamoto M, Hidaka-Takeno R, Okimura Y, Matsumoto R, Suda K, Nishizawa H, Takahashi M, Tojo K, Takahashi Y. Involvement of PIT-1-reactive cytotoxic T lymphocytes in anti-PIT-1 antibody syndrome. J Clin Endocrinol Metab. 2014;99:E1744–1749.
- Bando H, Iguchi G, Okimura Y, Odake Y, Yoshida K, Matsumoto R, Suda K, Nishizawa H, Fukuoka H, Mokubo A, Tojo K, Maniwa Y, Ogawa W, Takahashi Y. A novel thymoma-associated autoimmune disease: Anti-PIT-1 antibody syndrome. Sci Rep. 2017;7:43060.
- Chung TT, Monson JP. Hypopituitarism. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A, eds. Endotext. South Dartmouth (MA)2000.
- Dhanwal DK, Vyas A, Sharma A, Saxena A. Hypothalamic pituitary abnormalities in tubercular meningitis at the time of diagnosis. Pituitary. 2010;13:304–310.
- Leow MK, Kwek DS, Ng AW, Ong KC, Kaw GJ, Lee LS. Hypocortisolism in survivors of severe acute respiratory syndrome (SARS). Clin Endocrinol (Oxf). 2005;63:197–202.
- Tarvainen M, Makela S, Mustonen J, Jaatinen P. Autoimmune polyendocrinopathy and hypophysitis after Puumala hantavirus infection. Endocrinol Diabetes Metab Case Rep 2016
- Honegger J, Schlaffer S, Menzel C, Droste M, Werner S, Elbelt U, Strasburger C, Stormann S, Kuppers A, Streetz-van der Werf C, Deutschbein T, Stieg M, Rotermund R, Milian M, Petersenn S. Pituitary Working Group of the German Society of E. Diagnosis of Primary Hypophysitis in Germany. J Clin Endocrinol Metab. 2015;100:3841–3849.
- Gutenberg A, Hans V, Puchner MJ, Kreutzer J, Bruck W, Caturegli P, Buchfelder M. Primary hypophysitis: clinical-pathological correlations. Eur J Endocrinol. 2006;155:101–107.
- Shikuma J, Kan K, Ito R, Hara K, Sakai H, Miwa T, Kanazawa A, Odawara M. Critical review of IgG4-related hypophysitis. Pituitary. 2017;20:282–291.
- Hadjigeorgiou GF, Lund EL, Poulsgaard L, Feldt-Rasmussen U, Rasmussen AK, Wegener M, Fugleholm K. Intrachiasmatic abscess caused by IgG4-related hypophysitis. Acta Neurochir (Wien). 2017;159:2229–2233.
- Kalra AA, Riel-Romero RM, Gonzalez-Toledo E. Lymphocytic hypophysitis in children: a novel presentation and literature review. J Child Neurol. 2011;26:87–94.
- Gan HW, Bulwer C, Spoudeas H. Pituitary and Hypothalamic Tumor Syndromes in Childhood. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A, eds. Endotext. South Dartmouth (MA)2000.
- Di Iorgi N, Morana G, Maghnie M. Pituitary stalk thickening on MRI: when is the best time to re-scan and how long should we continue re-scanning for? Clin Endocrinol (Oxf). 2015;83:449–455.
- Chiloiro S, Tartaglione T, Angelini F, Bianchi A, Arena V, Giampietro A, Mormando M, Sciandra M, Laino ME, De Marinis L. An Overview of Diagnosis of Primary Autoimmune Hypophysitis in a Prospective Single-Center Experience. Neuroendocrinology. 2017;104:280–290.
- Allix I, Rohmer V. Hypophysitis in 2014. Ann Endocrinol (Paris). 2015;76:585–594.
- Evanson J. Radiology of the Pituitary. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A, eds. Endotext. South Dartmouth (MA)2000.
- Molitch ME. Pituitary and Adrenal Disorders of Pregnancy. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A, eds. Endotext. South Dartmouth (MA)2000.
- Gutenberg A, Larsen J, Lupi I, Rohde V, Caturegli P. A radiologic score to distinguish autoimmune hypophysitis from nonsecreting pituitary adenoma preoperatively. AJNR Am J Neuroradiol. 2009;30:1766–1772.
- Ricciuti A, De Remigis A, Landek-Salgado MA, De Vincentiis L, Guaraldi F, Lupi I, Iwama S, Wand GS, Salvatori R, Caturegli P. Detection of pituitary antibodies by immunofluorescence: approach and results in patients with pituitary diseases. J Clin Endocrinol Metab. 2014;99:1758–1766.
- Guaraldi F, Caturegli P, Salvatori R. Prevalence of antipituitary antibodies in acromegaly. Pituitary. 2012;15:490–494.
- De Bellis A, Sinisi AA, Pane E, Dello Iacovo A, Bellastella G, Di Scala G, Falorni A, Giavoli C, Gasco V, Giordano R, Ambrosio MR, Colao A, Bizzarro A, Bellastella A. Italian Autoimmune Hypophysitis Network G. Involvement of hypothalamus autoimmunity in patients with autoimmune hypopituitarism: role of antibodies to hypothalamic cells. J Clin Endocrinol Metab. 2012;97:3684–3690.
- De Bellis A, Colao A, Di Salle F, Muccitelli VI, Iorio S, Perrino S, Pivonello R, Coronella C, Bizzarro A, Lombardi G, Bellastella A. A longitudinal study of vasopressin cell antibodies, posterior pituitary function, and magnetic resonance imaging evaluations in subclinical autoimmune central diabetes insipidus. J Clin Endocrinol Metab. 1999;84:3047–3051.
- Bellastella G, Bizzarro A, Aitella E, Barrasso M, Cozzolino D, Di Martino S, Esposito K, De Bellis A. Pregnancy may favour the development of severe autoimmune central diabetes insipidus in women with vasopressin cell antibodies: description of two cases. Eur J Endocrinol. 2015;172:K11–17.
- Iwama S, Sugimura Y, Kiyota A, Kato T, Enomoto A, Suzuki H, Iwata N, Takeuchi S, Nakashima K, Takagi H, Izumida H, Ochiai H, Fujisawa H, Suga H, Arima H, Shimoyama Y, Takahashi M, Nishioka H, Ishikawa SE, Shimatsu A, Caturegli P, Oiso Y. Rabphilin-3A as a Targeted Autoantigen in Lymphocytic Infundibulo-neurohypophysitis. J Clin Endocrinol Metab. 2015;100:E946–954.
- Lupi I, Manetti L, Raffaelli V, Grasso L, Sardella C, Cosottini M, Iannelli A, Gasperi M, Bogazzi F, Caturegli P, Martino E. Pituitary autoimmunity is associated with hypopituitarism in patients with primary empty sella. J Endocrinol Invest. 2011;34:e240–244.
- Landek-Salgado MA, Leporati P, Lupi I, Geis A, Caturegli P. Growth hormone and proopiomelanocortin are targeted by autoantibodies in a patient with biopsy-proven IgG4-related hypophysitis. Pituitary. 2012;15:412–419.
- Nicolaides NC, Chrousos GP, Charmandari E. Adrenal Insufficiency. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A, eds. Endotext. South Dartmouth (MA)2000.
- Huguet I, Clayton R. Pituitary-Hypothalamic Tumor Syndromes: Adults. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A, eds. Endotext. South Dartmouth (MA)2000.
- Caputo C, Bazargan A, McKelvie PA, Sutherland T, Su CS, Inder WJ. Hypophysitis due to IgG4-related disease responding to treatment with azathioprine: an alternative to corticosteroid therapy. Pituitary. 2014;17:251–256.
- Lupi I, Manetti L, Raffaelli V, Lombardi M, Cosottini M, Iannelli A, Basolo F, Proietti A, Bogazzi F, Caturegli P, Martino E. Diagnosis and treatment of autoimmune hypophysitis: a short review. J Endocrinol Invest. 2011;34:e245–252.
- Lupi I, Cosottini M, Caturegli P, Manetti L, Urbani C, Cappellani D, Scattina I, Martino E, Marcocci C, Bogazzi F. Diabetes insipidus is an unfavorable prognostic factor for response to glucocorticoids in patients with autoimmune hypophysitis. Eur J Endocrinol. 2017;177:127–135.
- Schreckinger M, Francis T, Rajah G, Jagannathan J, Guthikonda M, Mittal S. Novel strategy to treat a case of recurrent lymphocytic hypophysitis using rituximab. J Neurosurg. 2012;116:1318–1323.
- Ray DK, Yen CP, Vance ML, Laws ER, Lopes B, Sheehan JP. Gamma knife surgery for lymphocytic hypophysitis. J Neurosurg. 2010;112:118–121.





{kind=link}