What is inclusion body myositis
Inclusion body myositis is the most common subtype of autoimmune inflammatory muscle disease (inflammatory myopathy) in patients older than the age of 50 years. Another word for inflammatory myopathy is myositis. The myo root means muscle, and the itis root means inflammation; so a myositis is an inflammatory muscle disease. Inclusion body myositis is characterized by chronic, progressive muscle inflammation accompanied by muscle weakness. The onset of muscle weakness in inclusion body myositis is generally gradual (over months or years) and affects both proximal (close to the trunk of the body) and distal (further away from the trunk) muscles. Muscle weakness may affect only one side of the body. Falling and tripping are usually the first noticeable symptoms of inclusion body myositis. For some individuals, inclusion body myositis begins with weakness in the wrists and fingers that causes difficulty with pinching, buttoning, and gripping objects. There may be weakness of the wrist and finger muscles and atrophy (thinning or loss of muscle bulk) of the forearm muscles and quadricep muscles in the legs. Difficulty swallowing occurs in approximately half of inclusion body myositis cases. Symptoms of inclusion body myositis usually begin after the age of 50, although inclusion body myositis can occur earlier. Inclusion body myositis occurs more frequently in men than in women. Unlike many other autoimmune diseases, the male-to-female ratio is higher in inclusion body myositis and is approximately 3:1 1.
There is no cure for inclusion body myositis, nor is there a standard course of treatment. The disease is generally unresponsive to corticosteroids and immunosuppressive drugs. Some evidence suggests that intravenous immunoglobulin may have a slight, but short-lasting, beneficial effect in a small number of cases. Physical therapy may be helpful in maintaining mobility. Other therapy is symptomatic and supportive.
Several diagnostic criteria have been proposed for inclusion body myositis based on expert opinion and consensus groups. Their use in clinical practice is however limited due to low sensitivity. The European Neuromuscular Centre 2011 clinically defined diagnostic criteria have a high specificity of greater than 99% to diagnose inclusion body myositis, but like other criteria, its sensitivity is low at 57%.
European Neuromuscular Centre 2011 Inclusion Body Myositis Diagnostic Criteria
Mandatory Features
- Age of onset later than 45 years
- Duration of symptoms more than 12 months
- Serum creatine kinase level, not more than 15 times the upper limit of normal
Clinical Features:
- A weakness of quadriceps more than hip flexors
- A weakness of finger flexors more than shoulder abductors
Pathological Features:
- Endomysial inflammatory infiltrate
- Rimmed vacuoles
- Protein accumulation or 15- to 18-nm filaments
- Upregulation of MHC class I
Classification Criteria:
- Clinicopathologically defined inclusion body myositis: Mandatory criteria + one or both of the clinical criteria plus 1, 2, and 3 of the pathological criteria
- Clinically defined inclusion body myositis: Mandatory criteria plus all clinical criteria plus one or more, but not all the pathological criteria
- Probable inclusion body myositis: Mandatory criteria plus one clinical criterion plus one or more, but not all the pathological criteria
Inclusion body myositis causes
In most cases, the cause of inclusion body myositis is unclear. For some reason, the body’s immune system turns against its own muscles and damages muscle tissue in an autoimmune process.
Viruses might be a trigger for autoimmune myositis. People with the HIV virus, which causes AIDS, can develop a myositis, as can people with a virus called HTLV-1. Some myositis cases have followed infection with the Coxsackie B virus.
There are reports of myositis following exposure to certain drugs. Among the drugs that have been suspected of contributing to myositis are carticaine (a local anesthetic), penicillamine (a drug used to lower copper levels in the body), interferon-alpha (mostly used to treat cancer and hepatitis), cimetidine (used to treat ulcers), carbimazole (to treat thyroid disease), phenytoin (used to treat seizures) and growth hormone. The vaccine for hepatitis B also has been implicated in some cases.
What are inheritance patterns in inclusion body myositis?
There are some genetic forms of inclusion body myositis in which, for the most part, inflammation isn’t a major part of the picture. For this reason, these forms are often called inclusion-body myopathy (muscle disorder), leaving out the “itis” in the disease name to reflect the relative lack of inflammation.
Genetic inclusion-body myopathies can be inherited in either a dominant or a recessive pattern. Dominant genetic disorders require only one genetic flaw to show themselves. Recessive disorders require that both parents pass on a flaw in the same gene before their offspring can show signs of the disease.
Inclusion body myositis pathophysiology
The pathogenesis of inclusion body myositis is not completely understood, but probably consists of an interplay between inflammatory and degenerative pathways 2.
The presence of inflammatory cells, mostly cytotoxic CD8+ T Cells with some macrophages invading and the CD4+T cells and macrophages surrounding the non-necrotic muscle fibers indicates an inflammatory component in the pathogenesis of inclusion body myositis. The activation of T cells is likely an antigen-driven response and is suggested by the presence of abundant antigen presenting cells(APCs) in the muscle fibers. Apart from these, the identification of Anti-Mup 44 antibody targeting a muscle protein cN1A supports a humoral component in the pathogenesis of inclusion body myositis.
Abnormal protein processing associated with aging and subsequent deposition of toxic polymers can cause muscle damage and can also trigger inflammation in the muscle fibers. Beta-amyloid, a degenerative protein identified in the muscle fibers of patients with inclusion body myositis supports the theory of degenerative pathway. The beta-amyloid protein has been demonstrated to stimulate human myoblasts to produce IL-6. This continuous stimulation of IL-6 production could augment the local immune response.
In short, some myofibers appear to be injured by invading cytotoxic T cells, while others have no apparent cause for their morphological abnormalities and have been called degenerative. There is no single, well-supported theory to explain all the features seen in this condition.
Inclusion body myositis histopathology
Histologically, inclusion body myositis is characterized by atrophic muscle fibers, infiltration of non-necrotic myofibers by mononuclear cells in an endomysial and perivascular pattern, rimmed vacuoles, and congophilic inclusions that may be intravacuolar or extravacuolar.
Major histocompatibility complex (MHC)-I is upregulated on immunostaining.
Rimmed vacuoles which are vacuolar degeneration stained positive by Gomori Trichrome stain are the hallmark histological feature of inclusion body myositis. Cytoplasmic inclusions of beta-amyloid are visualized using Congo red and polarised light. Tubulofilamnetous inclusions seen by electron microscopy is also a feature of inclusion body myositis. Increased number of cytochrome c oxidase negative fibers is also observed in a large number of inclusion body myositis patients 2.
Inclusion body myositis symptoms
The initial presenting symptom can vary from patient to patient. Inclusion-body myositis primarily affects men, although women can be affected. Inclusion-body myositis occurs mainly in those older than 50.
Inclusion-body myositis usually begins with the gradual onset of slowly progressive weakness in the muscles of the wrists and fingers, and those at the front of the thigh (quadriceps). The muscles that lift the front of the foot also may be affected. The weakness may not be the same on both sides of the body.
Trouble with gripping, such as a shopping bag or briefcase, and frequent stumbles are common experiences. About a third of people with inclusion body myositis have some weakness of the swallowing muscles.
The heart and lung involvement seen in other inflammatory myopathies like dermatomyositis and polymyositis is not part of the inclusion body myositis picture.
The most common feature is an insidious onset and progressive course of muscle weakness which manifests as the following:
- Difficulty in climbing stairs or arising from a chair (pelvic girdle)
- Decrease in walking speed (hip flexors)
- Frequent falls due to buckling of the knees (quadriceps)
- Foot drop and frequent tripping (ankle dorsiflexion)
- Difficulty in combing hair and in reaching overhead cabinets (shoulder girdle)
- Decrease in grip strength (finger flexors)
Neck muscle involvement can result in difficulty in lifting the head from a pillow.
The distinguishing features of inclusion body myositis from other forms of inflammatory myopathies are the following:
- Asymmetric and distal muscle involvement: The predilection for wrist or finger flexors and foot extensors.
- Insidious onset: The disease course is slow and progressive. The average duration of symptoms before making a diagnosis is 5 years.
- Muscle atrophy: Wasting of finger flexors, wrist flexors, and quadriceps accompany weakness and worsens with progression of the disease. In another inflammatory myositis, muscle atrophy happens as a sequela of the damage caused by the disease, and is, therefore, a late finding in contrast to inclusion body myositis where it can be present during the initial evaluation
- Dysphagia: is seen in approximately 30% to 50% of the patients with inclusion body myositis. It can lead to nasal regurgitation of liquids and pulmonary aspiration. Pharyngeal muscle weakness can also result in dysphonia.
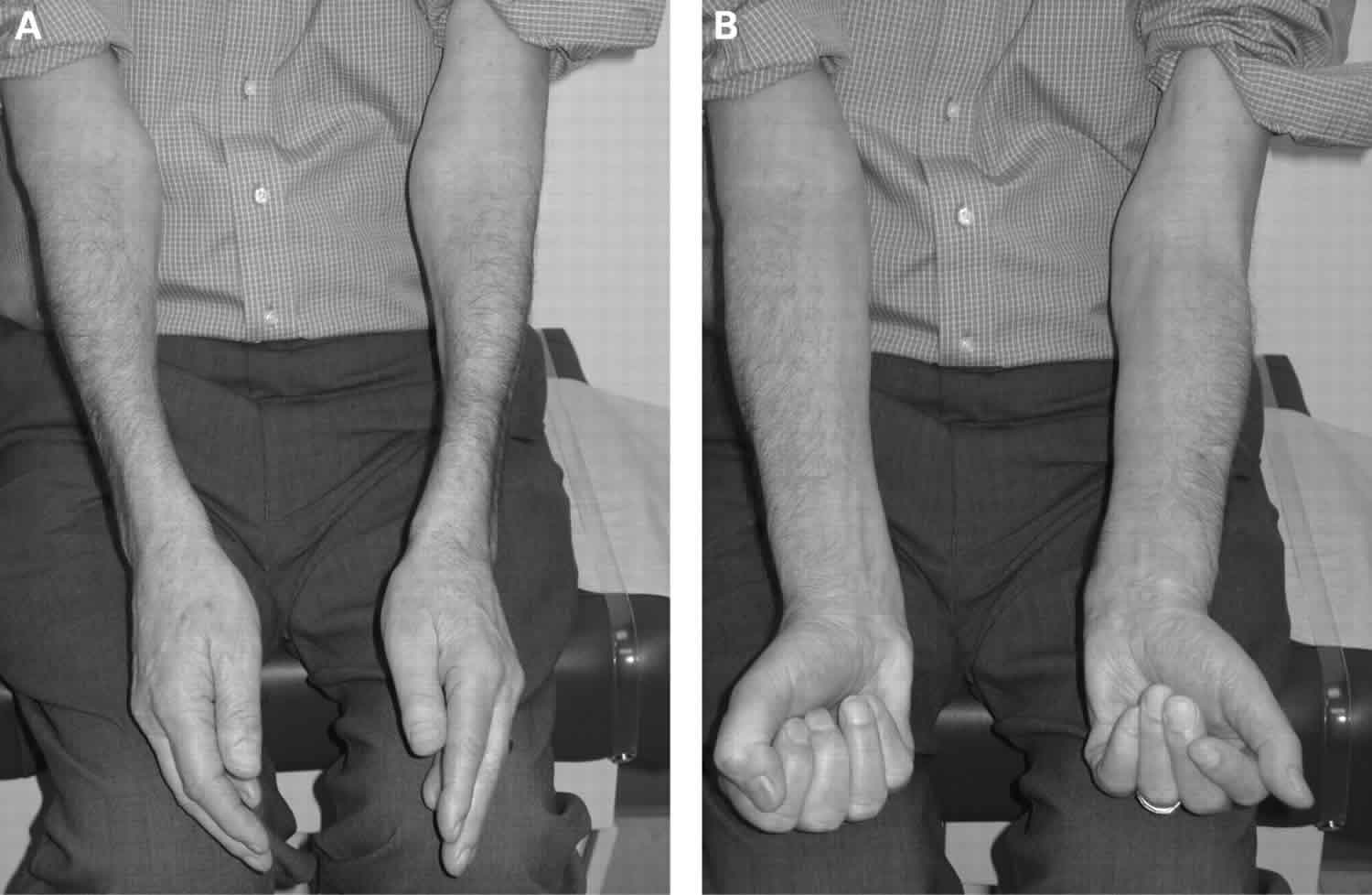
Physical examination will help in objectively assessing the distribution of muscle weakness and atrophy. The clinical hallmarks of inclusion body myositis are weakness and atrophy of the quadriceps and forearm flexors. Weakness in the distal finger flexor is the earliest finding which can be demonstrated by isolating and testing the flexion at the DIP joint of finger flexors.
Inclusion body myositis diagnosis
As with other muscle diseases, a doctor diagnoses inclusion-body myositis by considering the individual’s personal history, family medical history and the results of a careful physical examination. This may be followed by some lab tests, perhaps of the electrical activity inside the muscles. Usually, a muscle biopsy is ordered.
After a careful history and physical exam to document the pattern of weakness in the patient’s muscles, a doctor who suspects myositis likely will order a blood test to check the level of creatine kinase (CK), an enzyme that leaks out of muscle fibers when the fibers are being damaged. Although creatine kinase (CK) levels are usually very high in other inflammatory myopathies, in inclusion body myositis it may be only mildly elevated, or even normal. Other markers of muscle injury like Aldolase, lactate dehydrogenase (LDH), alanine aminotransferase (ALT) and aspartate aminotransferase (AST) can also be elevated. Inflammatory markers like ESR and CRP can be normal. Mup44 antibody against the cytosolic 5’nucleotidase 1A antigen is most commonly present in a patient with inclusion body myositis. However, these antibodies are also detected in about 20% of patients with systemic lupus erythematosus (SLE) and Sjogren syndrome in the absence of muscle disease.
In some cases, the doctor may ask for a blood test for specific antibodies, proteins produced by the immune system in myositis and other autoimmune diseases. Some of these antibodies appear to be specific to autoimmune muscle disease.
Inclusion body myositis can be associated with autoimmune diseases like Sjogren syndrome and sarcoidosis, lymphoproliferative diseases like chronic lymphocytic leukemia and infections like HIV and hepatitis B. Hence, screening tests with antinuclear antibodies (ANA), anti-Ro(SSA), anti-La(SSB), serum immunofixation, human immunodeficiency virus (HIV), and hepatitis C should be considered.
The next step is sometimes an electromyogram (EMG), a test in which tiny needles are inserted into the muscles to test their electrical activity both at rest and when the person tries to contract the muscle. Electromyogram (EMG) can be helpful in distinguishing myopathy from neuropathic causes of weakness. Typical EMG findings of myositis include irritability of the muscle fibers (fibrillation, complex repetitive discharges, and positive sharp waves) at rest and during needle insertion and myopathic motor unit potentials (short duration, low amplitude and polyphasic) during contraction 3. Magnetic resonance imaging (MRI) helps to visualize large areas of muscle and identify edema, inflammation, fatty infiltration, and atrophy. It can be useful in distinguishing active from a chronic inactive disease which is particularly important in the diagnosis of inclusion body myositis.
Both electromyogram (EMG) and magnetic resonance imaging (MRI) are helpful in identifying the muscle appropriate for doing a biopsy. A muscle that does not have clinical signs of advanced or end-stage disease and is at the same time not minimally affected is ideal for doing a biopsy. Biopsy should be done on the contralateral to the one used for EMG testing to avoid inflammation artifact caused during needle insertion. In 90% of cases, degeneration and regeneration of the myofibrils are seen. Typical biopsy finding is perivascular and endomysial inflammatory infiltrates that predominantly consists of CD8+T Cells and invades the non-necrotic muscle fibers that express MHC Class I antigen.
Inflammatory myopathies show a distinctive pattern of electrical activity that can help differentiate them from other types of muscle disease.
A nerve conduction velocity test is sometimes performed. This test measures how fast a nerve impulse travels and how strong it is.
Sometimes these tests are used to rule out disorders that may mimic the symptoms of inclusion body myositis.
A person with a suspected inflammatory myopathy is often asked to undergo a muscle biopsy, a procedure in which a small piece of muscle is removed for examination. This biopsy can enable the physician to pinpoint the diagnosis to a type of myositis.
The biopsy sample from a person with inclusion body myositis is unique because of its inclusion bodies, for which the disease is named.
These “bodies” (which don’t appear in normal cells) contain clumps of discarded cellular material. Inflammatory cells can be seen invading muscle tissue, although some researchers believe this invasion is secondary to the primary events in the muscle tissue, presumably those that cause the inclusion bodies to appear.
Inclusion body myositis treatment
No specific pharmacological therapy is beneficial for sporadic inclusion body myositis. Treatment with drugs that suppress the immune system has been tried in inclusion-body myositis but in general hasn’t been effective. Some physicians may try corticosteroids or other medications that alter the immune response if the patient wishes this treatment, but many feel that side effects outweigh any subtle benefits that might occur with these drugs in inclusion body myositis. There is no data suggesting that supplements such as CoQ10 slow disease progression in inclusion body myositis. Most clinicians treating inclusion body myositis patients do not currently recommend these supplements for inclusion body myositis.
The treatments include glucocorticoids, methotrexate, cyclophosphamide, azathioprine, IVIG 4 and alemtuzumab. Alemtuzumab has shown a reduction in key biomarkers such as IL 1 beta and Class I MHC complex in a pilot study. In a small randomized controlled trial, bimagrumab, an antibody against type II activin receptors showed an increase in the muscle volume at the end of 8 weeks 5. An IL-1 receptor blocker, anakinra has shown positive results in small case series 6 and case reports 7. There are ongoing trials with follistatin gene therapy and agents like arimoclomol, natalizumab among others.
The subset of patients likely to respond to treatment is that with serum CK greater than 5 times the upper limit of normal, those with evidence of active inflammation in imaging or biopsy, the presence of myositis autoantibodies and those that overlap with other connective tissue diseases.
For severe dysphagia not responding to immunosuppressants and intravenous immunoglobulin (IVIg), surgical methods like cricopharyngeal dilation or myotomy can be considered, and in very severe cases, a gastrostomy tube may be needed.
Even though not evidence-based, some physicians have patients supplement daily supplementation with creatine monohydrate.
Anakinra, an IL-1 receptor antagonist, was tested in case reports and in a small case series, with positive results in some patients.
Inclusion body myositis physical therapy
Physical therapy and rehabilitation is a critical aspect of the treatment. Exercise can help in improving the muscle strength and quality of life of the patients 8. Graduated rehabilitation under an experienced physiatrist should be started as soon as possible for optimal results. Occupational therapy will be helpful in learning techniques to accommodate social and professional life. In many cases, both muscle strengthening and aerobic exercises are appropriate for patients with inclusion body myositis. However, the appropriate exercises may depend on other health issues and the severity of the weakness. The best approach is to work with a physical therapist who has experience taking care of patients with inclusion body myositis or genetic muscle diseases (as with inclusion body myositis, these are progressive).
Inclusion body myositis prognosis
Inclusion body myositis is generally resistant to all therapies and its rate of progression appears to be unaffected by currently available treatments.
Inclusion body myositis life expectancy
Inclusion-body myositis is generally a slowly progressive disease, and life expectancy isn’t significantly affected. Most people with inclusion body myositis remain able to walk, although they may require a cane or wheelchair for long distances. Some are more profoundly affected, becoming gradually more disabled and require a wheelchair full time within 10 or 15 years of the first symptoms. Causes of death are usually due to respiratory, aspiration, dysphagia and cachexia.
References- Panginikkod S, Musa R. Inclusion Body Myositis. [Updated 2019 Jun 4]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK538200
- Naddaf E, Barohn RJ, Dimachkie MM. Inclusion Body Myositis: Update on Pathogenesis and Treatment. Neurotherapeutics. 2018 Oct;15(4):995-1005
- Nojszewska M, Gawel M, Kierdaszuk B, Sierdziński J, Szmidt-Sałkowska E, Seroka A, Kamińska AM, Kostera-Pruszczyk A. Electromyographic findings in sporadic inclusion body myositis. J Electromyogr Kinesiol. 2018 Apr;39:114-119
- Anh-Tu Hoa S, Hudson M. Critical review of the role of intravenous immunoglobulins in idiopathic inflammatory myopathies. Semin. Arthritis Rheum. 2017 Feb;46(4):488-508.
- Amato AA, Sivakumar K, Goyal N, David WS, Salajegheh M, Praestgaard J, Lach-Trifilieff E, Trendelenburg AU, Laurent D, Glass DJ, Roubenoff R, Tseng BS, Greenberg SA. Treatment of sporadic inclusion body myositis with bimagrumab. Neurology. 2014 Dec 09;83(24):2239-46.
- Zong M, Dorph C, Dastmalchi M, Alexanderson H, Pieper J, Amoudruz P, Barbasso Helmers S, Nennesmo I, Malmström V, Lundberg IE. Anakinra treatment in patients with refractory inflammatory myopathies and possible predictive response biomarkers: a mechanistic study with 12 months follow-up. Ann. Rheum. Dis. 2014 May;73(5):913-20
- Kosmidis ML, Alexopoulos H, Tzioufas AG, Dalakas MC. The effect of anakinra, an IL1 receptor antagonist, in patients with sporadic inclusion body myositis (sIBM): a small pilot study. J. Neurol. Sci. 2013 Nov 15;334(1-2):123-5
- Alexanderson H. Exercise in Myositis. Curr Treatm Opt Rheumatol. 2018;4(4):289–298. doi:10.1007/s40674-018-0113-3 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6299050





{kind=link}