What is incontinentia pigmenti
Incontinentia pigmenti also called Bloch-Sulzberger syndrome, is a rare X-linked dominant neurocutaneous syndrome caused by mutations in the IKBKG/NEMO gene on Xq28 (long arm of X chromosome), that can affect many body systems, particularly the skin. hair, teeth, nails, eyes, and central nervous system 1. This means that the abnormal incontinentia pigmenti gene is located on one of the X chromosomes, which determine the sex of a child (XY=male; XX=female). Dominant X-linked disease means that a female with only one copy of the abnormal gene will show the disease, even though they have a normal gene on their other X-chromosome. Males who inherit the abnormal gene do not survive, resulting in miscarriage or stillbirth (X-linked dominant, male-lethal syndrome). Rarely incontinentia pigmenti is reported in males with Klinefelter syndrome (XXY syndrome) or as a result of spontaneous mutations. The IKBKG gene (formerly known as NEMO or NF-kappaB gene) normally encodes a kappa light polypeptide geneenhancer in B cells, kinase gamma, which has a key rolein the modulation of nuclear transcription factor kappa B (NF-B) 2.
In hemizygous males, it is usually lethal, while in females, it has a wide spectrum of clinical manifestations. Incontinentia pigmenti is an uncommon disorder. Incontinentia pigmentihas an estimated incidence of 0.6 to 0.7 cases per 100,000 births 1. Between 900 and 1,200 affected individuals have been reported in the scientific literature. Most of these individuals are female (female:male ratio is 20:1), but several dozen males with incontinentia pigmenti have also been identified 3. Incontinentia pigmenti occurs primarily in females and on occasion in males.
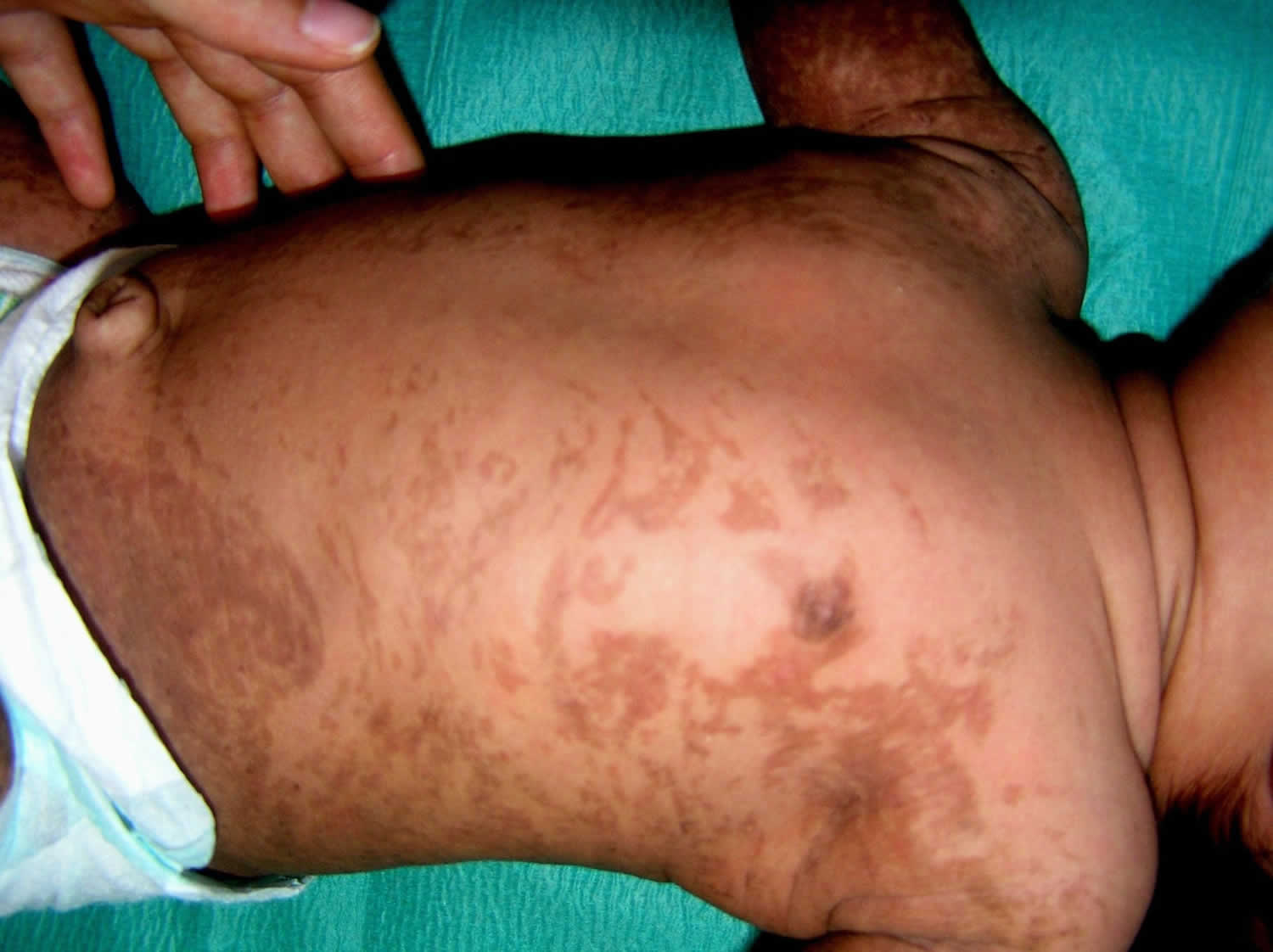
Incontinentia pigmenti is characterized by skin abnormalities that evolve throughout childhood and young adulthood. The characteristic skin lesions of incontinentia pigmenti are present at birth or develop in the first few weeks of life in approximately 90% of patients. Many affected infants have a blistering rash at birth and in early infancy, which heals and is followed by the development of wart-like skin growths. In early childhood, the skin develops grey or brown patches (hyperpigmentation) that occur in a swirled pattern. These patches fade with time, and adults with incontinentia pigmenti usually have lines of unusually light-colored skin (hypopigmentation) on their arms and legs.
Incontinentia pigmenti stages
Characteristic skin lesions evolve through four stages:
- Blistering or vesicobullous stage (birth to age ~4 months). The vesicobullous stage is most frequently observed at birth or within the first two weeks of life, but it is absent in 5-10 percent of cases, in which it is believed to occur in utero 4.
- Most often affects extremities and scalp but can arise on any part of the body
- Red, blister-like lesions
- Often appear grouped in lines along the arms and legs (following so-called lines of Blaschko)
- Present at birth or within the first 2 weeks of life in 90% of patients
- May last from a few weeks to a few months and recur throughout the first few months of life
- Verrucous or wart-like rash (for several months)
- Wart-like or pustular lesions
- Thick crusts or scabs form on top of healing blisters
- Lesions may be darker in skin colour (hyperpigmentation)
- May be present at birth but in 70-80% of patients evolves after the first stage
- May last for months, but rarely longer than a year
- Hyperpigmented or swirling macular hyperpigmentation (age ~6 months into adulthood)
- Skin is darkened in a swirled pattern
- Pigmentation ranges from blue-grey or slate to brown
- Present at birth in 5-10% of patients but usually appears within the first few months of life in 90-98% of patients
- Darkened patches may or may not be related to areas affected in stage 1 and 2
- Heavy pigmentation tends to fade slowly with increasing age
- Atrophic/ hypopigmented stage
- Scar-like lesions develop during adolescence and persist into adulthood
- Occur in 30-75% of patients
- Appear as pale, hairless patches or streaks
Other signs and symptoms of incontinentia pigmenti can include hair loss (alopecia) affecting the scalp and other parts of the body, dental abnormalities (such as small teeth or few teeth), eye abnormalities that can lead to vision loss, and lined or pitted fingernails and toenails. Most people with incontinentia pigmenti have normal intelligence; however, this condition may affect the brain. Associated problems can include developmental delays or intellectual disability, seizures, and other neurological problems. Neovascularization of the retina, present in some individuals, predisposes to retinal detachment.
Figure 1. Incontinentia pigmenti vesicular stage

Incontinentia pigmenti causes
Mutations in the IKBKG gene (formerly called NEMO gene) cause incontinentia pigmenti. The IKBKG gene provides instructions for making a protein that helps regulate nuclear factor-kappa-B. Nuclear factor-kappa-B is a group of related proteins that helps protect cells from self-destructing (undergoing apoptosis) in response to certain signals.
About 80 percent of affected individuals have a mutation that deletes some genetic material from the IKBKG gene. This deletion probably leads to the production of an abnormally small, nonfunctional version of the IKBKG protein. Other people with incontinentia pigmenti have mutations that prevent the production of any IKBKG protein. Without this protein, nuclear factor-kappa-B is not regulated properly, and cells are more sensitive to signals that trigger them to self-destruct. Researchers believe that this abnormal cell death leads to the signs and symptoms of incontinentia pigmenti.
Incontinentia pigmenti inheritance pattern
Incontinentia pigmenti is inherited in an X-linked dominant pattern. The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes. In females (who have two X chromosomes), a mutation in one of the two copies of the gene in each cell is sufficient to cause the disorder. Some cells produce a normal amount of IKBKG protein and other cells produce none. The resulting imbalance in cells producing this protein leads to the signs and symptoms of incontinentia pigmenti.
In males (who have only one X chromosome), most IKBKG mutations result in a total loss of the IKBKG protein. A lack of this protein appears to be lethal early in development, so few males are born with incontinentia pigmenti. Affected males who survive may have an IKBKG mutation with relatively mild effects, an IKBKG mutation in only some of the body’s cells (mosaicism), or an extra copy of the X chromosome in each cell.
Some people with incontinentia pigmenti inherit an IKBKG mutation from one affected parent. Other cases result from new mutations in the gene and occur in people with no history of the disorder in their family.
Figure 2. Incontinentia pigmenti X-linked dominant inheritance pattern

People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Incontinentia pigmenti symptoms
Incontinentia pigmenti skin symptoms change with time and begin with a blistering rash in infancy, followed by wart-like skin growths. The growths become swirled grey or brown patches in childhood, and then swirled light patches in adulthood. Other signs and symptoms may include hair loss, small or missing teeth, eye abnormalities that can lead to vision loss, and lined or pitted nails. Most people with incontinentia pigmenti have normal intelligence, but some have developmental delay, intellectual disability, seizures, and/or other neurological problems.
Skin
The skin changes are the most characteristic and common features in incontinentia pigmenti. They are described in four stages. In all the stages, the lesions appear in lines on the arms and legs or a swirled pattern on the trunk. They can be on the face and scalp.
- The first stage or vesicular stage of incontinentia pigmenti may be present at birth or appear during early infancy. This phase consists of redness or inflammation of the skin (erythema), blisters, and boils, most often affecting the extremities and the scalp, that last a few weeks to a few months. It can fade and come back again and again for more than a year, commonly when there is an illness with fever.
- The second stage or verrucous (wart-like) stage may overlap the first and may be present at birth. During this phase, the blisters develop a raised, wart-like (verrucous) appearance, and the lesions look like warts. There can be thick crusts or scabs with healing and areas of darkened skin (increased pigmentation). The extremities are involved almost exclusively in this stage, which may last for several months but rarely as long as year.
- The third or hyperpigmented stage may be present at birth in a small number of affected individuals, but usually appears between the ages of six and 12 months. In this phase, the skin is darkened (hyperpigmented). On the trunk, the dark is sometimes described as a “marble cake” appearance. The hyperpigmentation does not necessarily happen where the stage I and II rashes happen. The heavy pigmentation tends to fade over time and, in some cases, the pigmented areas thin and widen, leaving streaky diminished color of the skin (hypopigmentation).
- In the fourth stage, which is known as the “atrophic” stage, scarring appears that often is present before the hyperpigmentation has faded. Scars are seen in adolescents and adults as pale, hairless patches or streaks. Once the affected individuals reach the late teens and adulthood, the skin changes may have faded and may not be visible to the casual observer.
Teeth
Between 50 to 80 percent of individuals with incontinentia pigmenti have dental abnormalities. These abnormalities include a delay in the eruption of primary teeth; abnormal contours of teeth, giving them a peg-like or cone-shaped appearance; or the congenital absence of both primary and secondary teeth (anodontia); or small teeth, (microdontia).
Nails
Some individuals with incontinentia pigmenti may have ridged, pitted, thickened (onychogryposis), or missing nails on the hands and/or feet. In some cases, painful growths may develop under the nail.
Hair
Approximately 50 percent of individuals with incontinentia pigmenti may also experience abnormal bald patches on the scalp (alopecia). This usually happens where the stage one and two lesions have left scars. The hair generally may be coarse, wiry, and/or lusterless.
Eyes
Nearly one-third of individuals have eye (ocular) abnormalities. The most serious, but least frequent, is a congenitally small, abnormal eye. In any patient there can be an abnormality in the growth of blood vessels in the membrane lining the eyes (retina). If it is going to occur, this typically appears before the age of five. This problem may be treated if detected early. If left untreated, it may cause retinal detachment leading to permanent visual impairment or total blindness.
Nervous System
The majority of individuals with incontinentia pigmenti will have no involvement of the nervous system. Severe neurologic complications can occasionally occur as a consequence of incontinentia pigmenti, the most serious of which is congenital or neonatal strokes. Some affected individuals may experience episodes of uncontrolled electrical disturbances in the brain (seizures). About 30 percent of children with incontinentia pigmenti will have slow motor development, muscle weakness in one or both sides of the body, intellectual disability, and/or seizures.
Other
Abnormalities in the development of the breast, ranging from extra nipples to complete absence of the breast, are sometimes seen in individuals with incontinentia pigmenti.
Incontinentia pigmenti diagnosis
The diagnosis of incontinentia pigmenti is based on clinical evaluation, detailed patient history, and molecular genetic testing for mutation in the IKBKG gene. IKBKG is the only gene known to be associated with incontinentia pigmenti. 65 percent of patients have a specific deletion within the gene. Another 20 percent or so have mutations found by gene sequencing. A skin biopsy to confirm the diagnosis in a female is now rarely needed given the widespread availability and sensitivity of molecular genetic testing. Nonetheless, skin biopsy may be helpful in confirming the diagnosis in a female with borderline or questionable findings in whom molecular genetic testing has not identified a disease-causing mutation.
Clinical testing and work-up
It is very important for babies born with incontinentia pigmenti to have an eye examination by a pediatric ophthalmologist. This should be done monthly until age four months, then every three months from age four months to one year, every six months from age one to three years, and annually after age three years. The eye problems associated with incontinentia pigmenti can be severe, but may be effectively managed if recognized early.
The following evaluations may be done to determine the severity of disease in those affected with incontinentia pigmenti. physical examination with particular emphasis on the skin, hair, nails, and neurologic system, electroencephalography (EEG) and magnetic resonance imaging (MRI) if seizures, other neurologic abnormalities, or retinal abnormalities are present, magnetic resonance angiography to look for cerebrovascular lesions, and developmental screening.
Because incontinentia pigmenti can be very mild, even in infancy, an affected woman may not know that she has it. It is important that a full evaluation of the mother be done by a geneticist, dermatologist, or other physician after the birth of a child with incontinentia pigmenti.
Incontinentia pigmenti treatment
There is no specific treatment for incontinentia pigmenti 6. Skin abnormalities characteristic of incontinentia pigmenti usually disappear by adolescence or adulthood without any treatment.
Cryotherapy and laser photocoagulation may be used to treat affected individuals with retinal neovascularization that predisposes to retinal detachment.
Dental abnormalities can often be treated effectively by dentists who may provide dental implants in childhood as needed. Also if dental abnormalities interfere with chewing and/or speech, assistance from a speech pathologist and/or pediatric nutritionist may be necessary.
Hair problems may require the attention of a dermatologist in some cases, although they are usually not severe. Neurological symptoms such as seizures, muscle spasms or mild paralysis may be controlled with various drugs and/or medical devices.
Genetic counseling is recommended for affected individuals and their families. Other treatment is symptomatic and supportive.
Incontinentia pigmenti prognosis
Incontinentia pigmenti prognosis is variable and depends on the degree of involvement of organ systems other than the skin, in particular the presence of neurodevelopmental complications. There are generally no significant complications secondary to the cutaneous manifestations. Morbidity and mortality primarily result from neurologic and ophthalmologic complications, including mental retardation, seizures, and vision loss. Patients with structural brain abnormalities and neonatal seizures are at greater risk for motor and intellectual impairment 7.
References- Cammarata-Scalisi, Francisco & Fusco, F & Ursini, Matilde. (2019). Incontinentia Pigmenti. Actas Dermo-Sifiliográficas (English Edition). 110. 10.1016/j.adengl.2019.03.009.
- Danescu S, Has C, Baican C, Müller T, Baican A. A novel IKBKGmutation in a patient with incontinentia pigmenti and fea-tures of hepatic ciliopathy. Australas J Dermatol. 2018:10,1111/ajd.12805
- Incontinentia pigmenti. https://ghr.nlm.nih.gov/condition/incontinentia-pigmenti
- Hadj-Rabia S, Froidevaux D, Bodak N, Hamel-Teillac D, Smah A, Touil Y, Fraitag S, Prost Y, Bodemer C. Clinical Study of 40 cases of incontinentia Pigmenti. Arch Dermatol. 2003 Sep;139(9):1163-70
- Osório, F., Magina, S., Nogueira, A., & Azevedo, F. (2010). Incontinentia Pigmenti with vesicular stage. Dermatology Online Journal, 16(10). Retrieved from https://escholarship.org/uc/item/9dz2p5bk
- Incontinentia pigmenti. https://www.dermnetnz.org/topics/incontinentia-pigmenti/
- Incontinentia pigmenti. https://emedicine.medscape.com/article/1114205-overview





{kind=link}