IPEX syndrome
IPEX syndrome is short for Immune dysregulation, Polyendocrinopathy, Enteropathy, X-linked syndrome, that primarily affects males and is caused by problems with the immune system 1. The immune system normally protects the body from foreign invaders, such as bacteria and viruses, by recognizing and attacking these invaders and clearing them from the body. However, the immune system can malfunction and attack the body’s own tissues and organs instead, which is known as autoimmunity. IPEX syndrome is characterized by the development of multiple autoimmune disorders in affected individuals. Although IPEX syndrome can affect many different areas of the body, autoimmune disorders involving the intestines, skin, and hormone-producing (endocrine) glands occur most often. IPEX syndrome can be life-threatening in early childhood.
IPEX syndrome is a rare disorder that affects an estimated 1 in 1.6 million people, fewer than 300 affected individuals have been identified worldwide 2.
Almost all individuals with IPEX syndrome develop a disorder of the intestines called autoimmune enteropathy. Autoimmune enteropathy occurs when certain cells in the intestines are destroyed by a person’s immune system. It causes severe diarrhea, which is usually the first symptom of IPEX syndrome. Autoimmune enteropathy typically begins in the first few months of life. It can cause failure to gain weight and grow at the expected rate (failure to thrive) and general wasting and weight loss (cachexia).
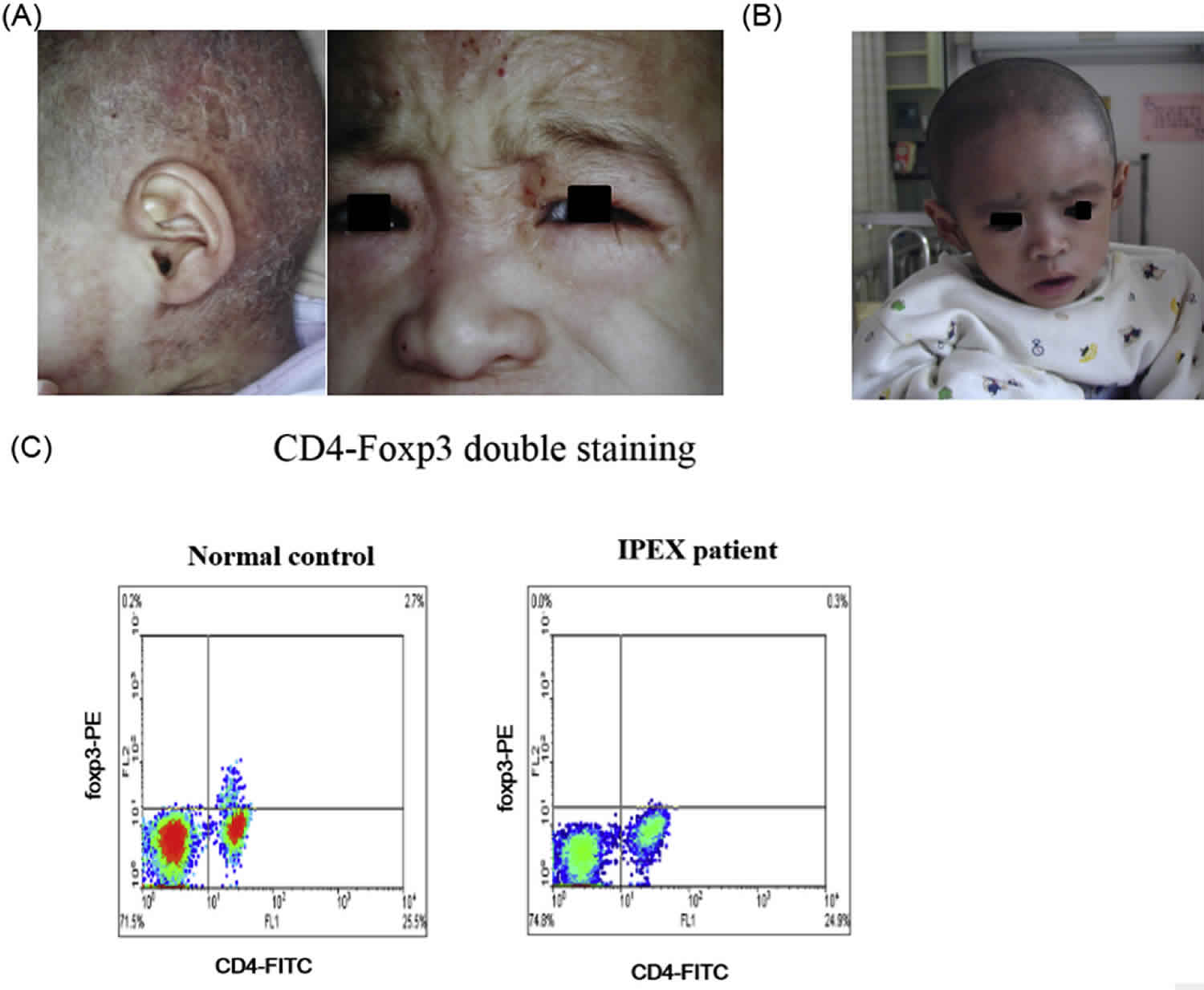
People with IPEX syndrome frequently develop inflammation of the skin, called dermatitis. Eczema is the most common type of dermatitis that occurs in this syndrome, and it causes abnormal patches of red, irritated skin. Other skin disorders that cause similar symptoms are sometimes present in IPEX syndrome.
The term polyendocrinopathy is used in IPEX syndrome because individuals can develop multiple disorders of the endocrine glands. Type 1 diabetes mellitus is an autoimmune condition involving the pancreas and is the most common endocrine disorder present in people with IPEX syndrome. It usually develops within the first few months of life and prevents the body from properly controlling the amount of sugar in the blood. Autoimmune thyroid disease may also develop in people with IPEX syndrome. The thyroid gland is a butterfly-shaped organ in the lower neck that produces hormones. This gland is commonly underactive (hypothyroidism) in individuals with this disorder, but may become overactive (hyperthyroidism).
Individuals with IPEX syndrome typically develop other types of autoimmune disorders in addition to those that involve the intestines, skin, and endocrine glands. Autoimmune blood disorders are common; about half of affected individuals have low levels of red blood cells (anemia), platelets (thrombocytopenia), or certain white blood cells (neutropenia) because these cells are attacked by the immune system. In some individuals, IPEX syndrome involves the liver and kidneys.
IPEX syndrome diagnosis is established in a male proband with typical clinical findings and by the identification of a hemizygous pathogenic variant in FOXP3. Affected females have not been reported; female carriers of a pathogenic variant do not have clinical findings.
Treatment of IPEX syndrome consists of medications that limit immune system function. Research is ongoing for new and safer treatments 2.
Bone marrow transplantation offers the only potential cure for IPEX syndrome 2. When done at the time of no or mild organ impairment, there is improved resolution of autoimmunity, especially when compared with non-transplanted individuals under chronic immunosuppressive therapy. T cell-directed immune suppression (i.e., sirolimus, cyclosporin A, or tacrolimus), either alone or in combination with steroids, is considered first-line therapy; granulocyte colony stimulating factor (G-CSF) for autoimmune neutropenia; rituximab for pemphigus nodularis and other autoantibody-mediated disease; nutritional support; standard treatment of diabetes mellitus and autoimmune thyroid disease; topical therapies for dermatitis.
How is immunodysregulation polyendocrinopathy enteropathy x-linked syndrome diagnosed in a female?
Immunodysregulation Polyendocrinopathy Enteropathy X-linked (IPEX) syndrome only affects males. Because the FOXP3 gene is found on the X chromosome and males have only one X chromosome, whereas females have two X chromosomes, if there is mutation in a male’s FOXP3 gene, he will have IPEX syndrome. If a male is diagnosed with IPEX syndrome based on clinical symptoms, he may have genetic testing to look for a mutation in the FOXP3 gene. If a mutation is found, his female relatives could then be tested for the same FOXP3 gene mutation. Since females have two FOXP3 genes; if a mutation is found in one of the two FOXP3 genes, she is considered a carrier of IPEX syndrome; she would not be expected to have any symptoms of the disease herself, but she could potentially pass the condition on to a son 2.
IPEX syndrome cause
Mutations in the FOXP3 gene cause IPEX syndrome. The protein produced from this gene is a transcription factor, which means that it attaches (binds) to specific regions of DNA and helps control the activity of particular genes. This protein is essential for the production and normal function of certain immune cells called regulatory T cells. Regulatory T cells play an important role in controlling immune responses and preventing autoimmune disorders. Mutations in the FOXP3 gene impair the normal function of regulatory T cells, making it difficult for the body to turn off immune responses when they are not needed. Normal body tissues and organs are attacked, causing the multiple autoimmune disorders that develop in people with IPEX syndrome.
IPEX syndrome inheritance pattern
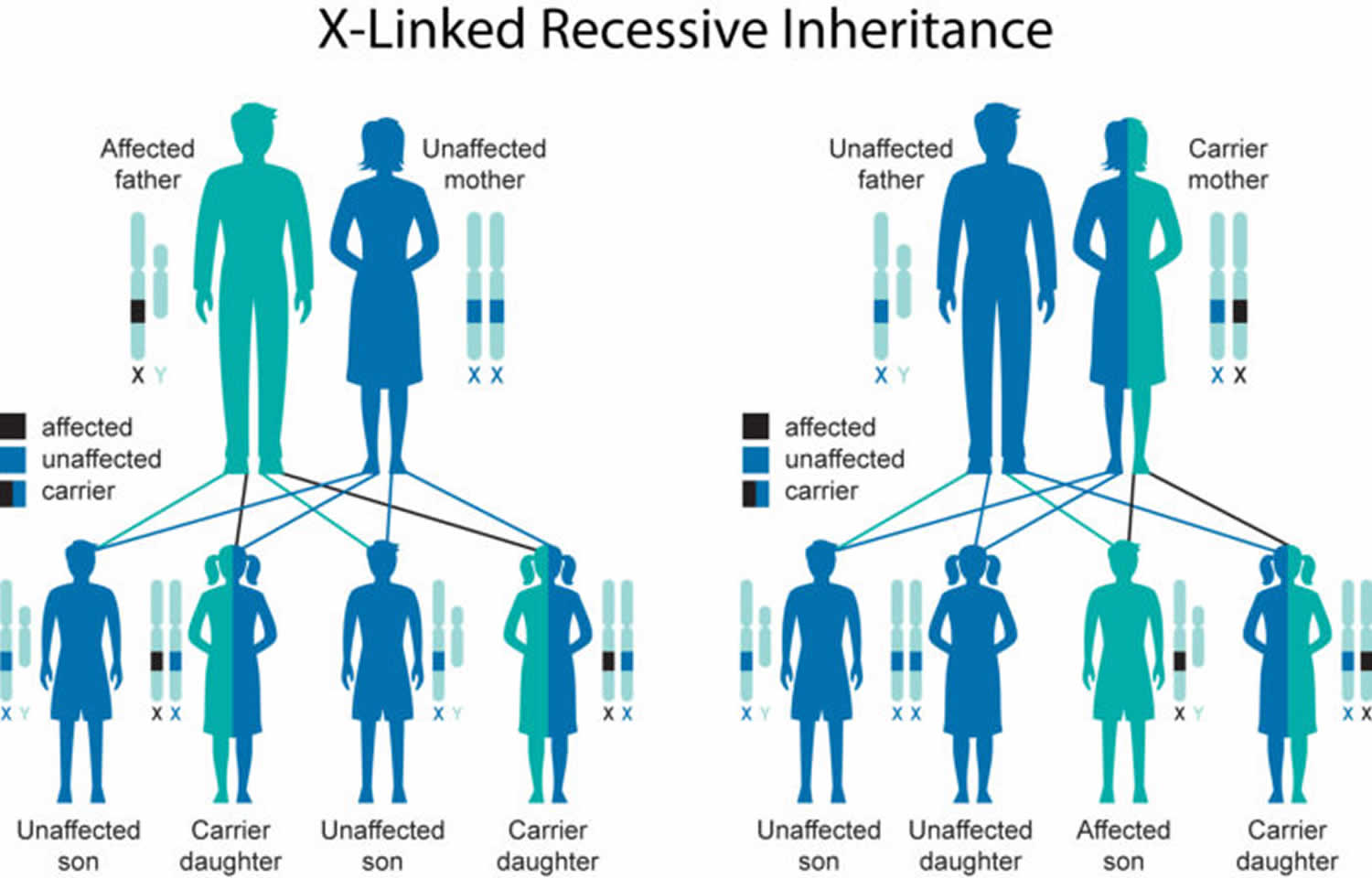
IPEX syndrome is inherited in an X-linked recessive pattern. The FOXP3 gene is located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation must be present in both copies of the gene to cause the disorder. Males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
The risk to sibling of the proband (index case) depends on the carrier status of the mother. If the mother of the proband is a carrier, the chance of transmitting the pathogenic variant in each pregnancy is 50%. Males who inherit the pathogenic variant will be affected; females who inherit the pathogenic variant are carriers and will not be affected. Affected males pass the pathogenic variant to all of their daughters and none of their sons. Carrier testing for at-risk relatives, prenatal testing for pregnancies at risk, and preimplantation diagnosis are possible for families in which the pathogenic variant has been identified.
Some people have conditions that appear identical to IPEX syndrome, but they do not have mutations in the FOXP3 gene. These conditions do not follow an X-linked inheritance pattern, and females can be affected. Such conditions are classified as IPEX-like syndromes.
Figure 1. IPEX syndrome X-linked recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
IPEX syndrome symptoms
IPEX syndrome should be suspected in males with the following clinical triad, family history, and suggestive laboratory findings.
IPEX syndrome clinical triad
- Enteropathy that manifests as chronic watery diarrhea. Onset is typically in the first months of life; villous atrophy with a mononuclear cell infiltrate (activated T cells) in the lamina propria is the most common finding in biopsy.
- Endocrinopathy, most commonly type 1 diabetes mellitus with onset in the first months or years of life. Autoimmune thyroid disease leading to hypothyroidism or hyperthyroidism has also been observed 3.
- Dermatitis, most commonly eczematous presenting within the first months of life, although prenatal skin desquamation has been reported 4. Erythroderma, exfoliative dermatitis, psoriasis-like lesions, and pemphigus nodularis have also been observed 5.
Males
IPEX syndrome is generally considered to be a syndrome of neonatal enteropathy 6 and neonatal polyendocrinopathy 7 found in males.
The most common presentation of IPEX syndrome is severe watery diarrhea, type 1 insulin-dependent diabetes mellitus, thyroiditis, and dermatitis in males younger than age one year. This disorder is frequently accompanied by other autoimmune phenomena.
- Males with a somewhat milder/atypical disease phenotype can present at older ages 8.
- Fetal presentation of IPEX includes hydrops, echogenic bowel, skin desquamation, intrauterine growth restriction (IUGR), and fetal akinesia. There may be a family history of pregnancy loss 9.
Enteropathy
The enteropathy of IPEX syndrome, often the first sign, is present in virtually all affected individuals. Even in those with milder disease, the diarrhea typically begins in the first six to12 months of life. Watery diarrhea, which may at times also have mucus and blood, leads to malabsorption, failure to thrive, and cachexia, often requiring the use of total parenteral nutrition (TPN) 10. Exocrine pancreatic insufficiency has been observed in some cases 11, which may worsen the diarrhea. Other gastrointestinal manifestations include colitis 12 and gastritis 11. Food allergies are common 13.
Endocrinopathy
Endocrinopathy is present in the majority of affected individuals. Type 1 diabetes mellitus, often with onset in the first months of life, is the most common endocrine manifestation 14. Thyroid disease (thyroiditis with either hypothyroidism [more common] or hyperthyroidism) is also frequently present 14.
Dermatitis
The dermatitis is most frequently eczematous, but psoriasiform and ichthyosiform dermatitis have been reported as well. Other dermatologic manifestations include painful chelitis and skin lesions related to food allergies. Rare cutaneous symptoms include pemphigoid nodularis and epidermolysis bullosa acquisita 15.
Autoimmune disorder
Most affected individuals have other autoimmune phenomena including cytopenias (autoimmune hemolytic anemia, immune thrombocytopenia, autoimmune neutropenia) 16, autoimmune hepatitis 17, and nephropathy (membranous nephropathy, interstitial nephritis, and rarely minimal change nephrotic syndrome) 18. Lymphadenopathy and splenomegaly as a result of lymphoproliferation have been reported [Ochs &Torgerson 2007, Nademi et al 2014, Bacchetta et al 2018, Barzaghi et al 2018]. Alopecia and arthritis have also been observed 16, as well as lung disease related to immune dysregulation 19.
Infectious complications
Infections of the gastrointestinal tract, skin, and airways occur in individuals with IPEX syndrome 16, and severe or invasive infections including sepsis, meningitis, pneumonia, and osteomyelitis affect a significant number of subjects 16. Common pathogens identified were Staphylococcus, Enterococcus, cytomegalovirus, and Candida 20. Some infections may be secondary to immunosuppressive therapy; however, many occur prior to the initiation of treatment. Serious infections in individuals with IPEX syndrome are not thought to be due to an intrinsic immune defect but instead are typically related to poor barrier function of the gut and skin 16.
Females
Heterozygous females are generally healthy. However, exceptions have been noted:
- One heterozygous female had an expression level of FOXP3 mRNA intermediate between the very low level observed in her affected son and the normal level in a control 21.
- One heterozygous female has monogenic diabetes mellitus 22.
- X-chromosome inactivation studies performed on one heterozygous female demonstrated that normal and pathogenic variant FOXP3 alleles are equally expressed in peripheral blood mononuclear cells 23. However, subsequent studies of the FOXP3+ regulatory T cell population (CD4+/CD25+) demonstrated a selective advantage for cells utilizing the normal X chromosome, resulting in complete skewing of X chromosome usage in this cell subset 24.
- Recurrent miscarriage of male fetuses has been reported 9.
IPEX syndrome diagnosis
No laboratory findings specifically identify affected individuals. Evidence of immune dysregulation manifested by the following is suggestive of IPEX syndrome:
- Elevated serum concentration of IgE (immunoglobulin E) and in some cases, IgA
- Eosinophilia
- Autoimmune anemia, thrombocytopenia, and/or neutropenia
- Autoantibodies to pancreatic islet antigens, thyroid antigens, small bowel mucosa, and other autoantigens
- Decreased numbers of FOXP3-expressing T cells in peripheral blood determined by flow cytometry – although FOXP3 levels in regulatory T cells (Treg) can be normal in some cases
Establishing the diagnosis
- Male proband. The diagnosis of IPEX syndrome is established in a male proband with the typical clinical findings and by the identification of a hemizygous pathogenic variant in FOXP3 by molecular genetic testing (see Table 1).
- Female proband. Affected females have not been reported. Carrier status is determined by identification of a heterozygous pathogenic variant in FOXP3 by molecular genetic testing.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, exome array, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Because the phenotype of IPEX syndrome is broad, individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with a phenotype indistinguishable from many other inherited disorders with enteropathy, endocrinopathy, and/or immune dysregulation or those in whom the diagnosis of IPEX syndrome has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic and laboratory findings suggest the diagnosis of IPEX syndrome, molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
- Single-gene testing. Sequence analysis of FOXP3 detects small intragenic deletions/insertions and missense, nonsense, and splice site variants. If no pathogenic variant is found, perform gene-targeted deletion/duplication analysis to detect intragenic deletions or duplications.
Note: (1) Lack of amplification by PCR prior to sequence analysis can suggest a putative (multi)exon or whole-gene deletion on the X chromosome in affected males; confirmation requires additional testing by gene-targeted deletion/duplication analysis. (2) Disease-associated variants have been reported in the 5’UTR (c.-7G>T) and the 3’UTR (c.*876A>G and c.*876A>G). Since the 3’UTR variants are further 3′ than typically included in sequencing assays, the assay design may need to be modified to include these variants. - A multigene panel that includes FOXP3 and other genes of interest is most likely to identify the genetic cause of the condition at the most reasonable cost while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Option 2
When the phenotype is indistinguishable from many other inherited disorders characterized by enteropathy, endocrinopathy, or immune dysregulation or when the diagnosis of IPEX syndrome is not considered because an individual has atypical phenotypic features, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. Exome sequencing is most commonly used; genome sequencing is also possible.
Exome array (when clinically available) may be considered if exome sequencing is not diagnostic.
IPEX syndrome treatment
To establish the extent of disease in an individual diagnosed with IPEX syndrome, the evaluations summarized in Table 1 (if not performed as part of the evaluation that led to the diagnosis) are recommended:
Table 1. Recommended Evaluations Following Initial Diagnosis in Individuals with IPEX Syndrome
| Organ System | Evaluation | Comment |
| Gastrointestinal | Nutrition assessment | To include serum electrolyte levels, calcium, magnesium, zinc, serum albumin & pre-albumin |
| Hepatic assessment | To include serum AST, ALT, GGT, & total bilirubin & assessment of hepatic autoantibodies | |
| Endocrine |
| |
| Immunologic |
| |
| Dermatologic | Evaluation of any skin lesions | May include histology |
| Hematologic |
| |
| Renal |
| |
| Miscellaneous/Other | Consultation w/clinical geneticist &/or genetic counselor |
Bone marrow transplantation (BMT) offers the only potential cure for IPEX syndrome. Early attempts at BMT using myeloablative conditioning regimens met with only limited success because of transplant-related mortality and other complications related to the underlying disease 25. Approaches using non-myeloablative conditioning regimens have markedly improved outcomes and survival 26. While generally less toxic, these reduced-intensity conditioning regimens still appear to generate long-term, stable engraftment of a donor regulatory T cell population 16 and, if performed in early infancy, may prevent the development of irreversible diabetes mellitus or thyroiditis 27.
A large, multicenter long-term follow-up study suggests that bone marrow transplantation at time of no or mild organ impairment results in improved resolution of autoimmunity, especially when compared with non-transplanted individuals under chronic immunosuppressive therapy 28.
Table 2. Treatment of manifestations in individuals with IPEX Syndrome
| Manifestation | Treatment | Considerations/Other |
|---|---|---|
| Enteropathy of IPEX syndrome | Monitor fluid intake to assure adequate intravascular volume. | |
| Nutritional support including TPN or elemental or low-carbohydrate-containing formula is necessary in almost all individuals. | ||
| T cell-directed immune suppression (i.e., sirolimus, cyclosporin A or tacrolimus), either alone or in combination with steroids | Toxicity, tachyphylaxis, & increased susceptibility to infection related to chronic use of these agents reduce their potential for long-term amelioration of symptoms in most individuals. | |
| Sirolimus (rapamycin) as monotherapy or in combination w/other drugs 4 is considered first-line treatment; calcineurin inhibitors (e.g. tacrolimus) as an alternative 5. | ||
| Endocrinopathy | Standard treatment protocols for diabetes mellitus & autoimmune thyroid disease | |
| Dermatitis |
| For severe dermatitis, wound care specialist can be very helpful |
| Immune dysregulation |
| May be beneficial |
Prevention of secondary complications
Individuals with autoimmune neutropenia or recurrent infections resulting from severe eczema may benefit from prophylactic antibiotic therapy to decrease the risk of severe infectious complications.
Aggressive management of dermatitis with topical steroids and anti-inflammatory agents can help to prevent infections from pathogens that enter as a result of the poor barrier function of the skin.
Surveillance
Monitoring for evidence of autoimmune disease is recommended every three to six months with the following tests: complete blood count, thyroid function, HbA1c and glucose, kidney function (measurement of serum concentration of BUN, creatinine), and liver function (measurement of serum concentration of AST, ALT). Frequent monitoring of growth parameters, nutritional intake, and stooling patterns is important. If under immunosuppression of post-transplantation, monitoring for drug side effects should adhere to standard guidelines.
Agents and circumstances to avoid
Immune activation (e.g., by immunizations or severe infections) has been reported to cause worsening or exacerbation of disease symptoms 32. It is generally best practice to withhold immunizations until after bone marrow transplantation, if possible.
IPEX syndrome prognosis
The outcome of classic IPEX syndrome is males is universally poor. Many children die within the first or second year of life from metabolic derangements, severe malabsorption, or sepsis. Although improvements in immunosuppressive regimens have prolonged survival, long-term immunosuppression did not appear to prevent morbidity due to disease progression and side effects or complications in the majority of patients 16.
Early bone marrow transplantation (BMT) is able to cure IPEX; some survivors are now more than ten years post transplant and doing well. If individuals develop diabetes or thyroiditis prior to BMT, these aspects of the disorder usually persist but the other signs of IPEX resolve. Survival and long-term outcomes are improved if bone marrow transplantation occurs at an earlier age, prior to the individual developing irreversible organ damage related to the extensive, systemic autoimmunity present in virtually all individuals with IPEX 33.
References- Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. https://ghr.nlm.nih.gov/condition/immune-dysregulation-polyendocrinopathy-enteropathy-x-linked-syndrome
- Tan QKG, Louie RJ, Sleasman JW. IPEX Syndrome. 2004 Oct 19 [Updated 2018 Jul 19]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1118
- Gambineri E, Torgerson TR, Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked inheritance (IPEX), a syndrome of systemic autoimmunity caused by mutations of FOXP3, a critical regulator of T-cell homeostasis. Curr Opin Rheumatol. 2003;15:430–5.
- Louie RJ, Tan QK, Gilner JB, Rogers RC, Younge N, Wechsler SB, McDonald MT, Gordon B, Saski CA, Jones JR, Chapman SJ, Stevenson RE, Sleasman JW, Friez MJ. Novel pathogenic variants in FOXP3 in fetuses with echogenic bowel and skin desquamation identified by ultrasound. Am J Med Genet A. 2017;173:1219–25.
- McGinness JL, Bivens MM, Greer KE, Patterson JW, Saulsbury FT. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) associated with pemphigoid nodularis: a case report and review of the literature. J Am Acad Dermatol. 2006;55:143–8.
- Ruemmele FM, Brousse N, Goulet O. Autoimmune enteropathy: molecular concepts. Curr Opin Gastroenterol. 2004;20:587–91.
- Dotta F, Vendrame F. Neonatal syndromes of polyendocrinopathy. Endocrinol Metab Clin North Am. 2002;31:283–93.
- Hwang JL, Park SY, Ye H, Sanyoura M, Pastore AN, Carmody D, Del Gaudio D, Wilson JF, Hanis CL, Liu X, Atzmon G, Glaser B, Philipson LH, Greeley SAW. T2D-Genes Consortium. FOXP3 mutations causing early-onset insulin-requiring diabetes but without other features of immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Pediatr Diabetes. 2018;19:388–92.
- Shehab O, Tester DJ, Ackerman NC, Cowchock FS, Ackerman MJ. Whole genome sequencing identifies etiology of recurrent male intrauterine fetal death. Prenatal Diag. 2017;37:1040–5.
- Bacchetta R, Barzaghi F, Roncarolo MG. From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation. Ann N Y Acad Sci. 2018;1417:5–22.
- Scaillon M, Van Biervliet S, Bontems P, Dorchy H, Hanssens L, Ferster A, Segers V, Cadranel S. Severe gastritis in an insulin-dependent child with an IPEX syndrome. J Pediatr Gastroenterol Nutr. 2009;49:368–70.
- Lucas KG, Ungar D, Comito M, Bayerl M, Groh B. Submyeloablative cord blood transplantation corrects clinical defects seen in IPEX syndrome. Bone Marrow Transplant. 2007;39:55–6.
- Torgerson TR, Linane A, Moes N, Anover S, Mateo V, Rieux-Laucat F, Hermine O, Vijay S, Gambineri E, Cerf-Bensussan N, Fischer A, Ochs HD, Goulet O, Ruemmele FM. Severe food allergy as a variant of IPEX syndrome caused by a deletion in a noncoding region of the FOXP3 gene. Gastroenterology. 2007;132:1705–17.
- Rubio-Cabezas O, Minton JA, Caswell R, Shield JP, Deiss D, Sumnik Z, Cayssials A, Herr M, Loew A, Lewis V, Ellard S, Hattersley AT. Clinical heterogeneity in patients with FOXP3 mutations presenting with permanent neonatal diabetes. Diabetes Care. 2009;32:111–6.
- Bis S, Maguiness SM, Gellis SE, Schneider LC, Lee PY, Notarangelo LD, Keles S, Chatila TA, Schmidt BA, Miller DD. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome associated with neonatal epidermolysis bullosa acquisita. Pediatr Dermatol. 2015;32:e74–7.
- Barzaghi F, et al. Primary Immune Deficiency Treatment Consortium (PIDTC) and the Inborn Errors Working Party (IEWP) of the European Society for Blood and Marrow Transplantation (EBMT). Long-term follow-up of IPEX syndrome patients after different therapeutic strategies: An international multicenter retrospective study. J Allergy Clin Immunol. 2018;141:1036–49.e5
- López SI, Ciocca M, Oleastro M, Cuarterolo ML, Rocca A, de Dávila MT, Roy A, Fernández MC, Nievas E, Bosaleh A, Torgerson TR, Ruiz JA. Autoimmune hepatitis type 2 in a child with IPEX syndrome. J Pediatr Gastroenterol Nutr. 2011;53:690–3.
- Sheikine Y, Woda CB, Lee PY, Chatila TA, Keles S, Charbonnier LM, Schmidt B, Rosen S, Rodig NM. Renal involvement in the immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) disorder. Pediatr Nephrol. 2015;30:1197–202.
- Baris S, Schulze I, Ozen A, Karakoc Aydiner E, Altuncu E, Karasu GT, Ozturk N, Lorenz M, Schwarz K, Vraetz T, Ehl S, Barlan IB. Clinical heterogeneity of immunodysregulation, polyendocrinopathy, enteropathy, X-linked: pulmonary involvement as a non-classical disease manifestation. J Clin Immunol. 2014;34:601–6.
- Barzaghi F, Passerini L, Bacchetta R. Immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome: a paradigm of immunodeficiency with autoimmunity. Front Immunol. 2012;3:211
- Bennett CL, Brunkow ME, Ramsdell F, O’Briant KC, Zhu Q, Fuleihan RL, Shigeoka AO, Ochs HD, Chance PF. A rare polyadenylation signal mutation of the FOXP3 gene (AAUAAA–>AAUGAA) leads to the IPEX syndrome. Immunogenetics. 2001;53:435–9.
- Alkorta-Aranburu G, Carmody D, Cheng YW, Nelakuditi V, Ma L, Dickens JT, Das S, Greeley SAW, Del Gaudio D. Phenotypic heterogeneity in monogenic diabetes: the clinical and diagnostic utility of a gene panel-based next-generation sequencing approach. Mol Genet Metab. 2014;113:315–20.
- Tommasini A, Ferrari S, Moratto D, Badolato R, Boniotto M, Pirulli D, Notarangelo LD, Andolina M. X-chromosome inactivation analysis in a female carrier of FOXP3 mutation. Clin Exp Immunol. 2002;130:127–30.
- Di Nunzio S, Cecconi M, Passerini L, McMurchy AN, Baron U, Turbachova I, Vignola S, Valencic E, Tommasini A, Junker A, Cazzola G, Olek S, Levings MK, Perroni L, Roncarolo MG, Bacchetta R. Wild-type FOXP3 is selectively active in CD4+CD25hi regulatory T cells of healthy female carriers of different FOXP3 mutations. Blood. 2009;114:4138–41.
- Baud O, Goulet O, Canioni D, Le Deist F, Radford I, Rieu D, Dupuis-Girod S, Cerf-Bensussan N, Cavazzana-Calvo M, Brousse N, Fischer A, Casanova JL. Treatment of the immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) by allogeneic bone marrow transplantation. N Engl J Med. 2001;344:1758–62.
- Rao A, Kamani N, Filipovich A, Lee SM, Davies SM, Dalal J, Shenoy S. Successful bone marrow transplantation for IPEX syndrome after reduced-intensity conditioning. Blood. 2007;109:383–5.
- Burroughs LM, Torgerson TR, Storb R, Carpenter PA, Rawlings DJ, Sanders MD, Scharenberg AM, Skoda-Smith S, Englund J, Ochs HD, Woolfrey AE. Stable hematopoietic cell engraftment after low-intensity nonmyeloablative conditioning in patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. J Allergy Clin Immunol. 2010;126:1000–5.
- Barzaghi F, Passerini L, Bacchetta R. Immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome: a paradigm of immunodeficiency with autoimmunity. Front Immunol. 2012;3:211.
- Yong PL, Russo P, Sullivan KE. Use of sirolimus in IPEX and IPEX-like children. J Clin Immunol. 2008;28:581–7.
- Strauss L, Whiteside TL, Knights A, Bergmann C, Knuth A, Zippelius A. Selective survival of naturally occurring human CD4+CD25+Foxp3+ regulatory T cells cultured with rapamycin. J Immunol. 2007;178:320–9.
- McGinness JL, Bivens MM, Greer KE, Patterson JW, Saulsbury FT. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) associated with pemphigoid nodularis: a case report and review of the literature. J Am Acad Dermatol. 2006;55:143–8
- Powell BR, Buist NR, Stenzel P. An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy. J Pediatr. 1982;100:731–7.
- Kucuk ZY, Bleesing JJ, Marsh R, Zhang K, Davies S, Filipovich AH. A challenging undertaking: Stem cell transplantation for immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. J Allergy Clin Immunol. 2016;137:953–5.e4





{kind=link}