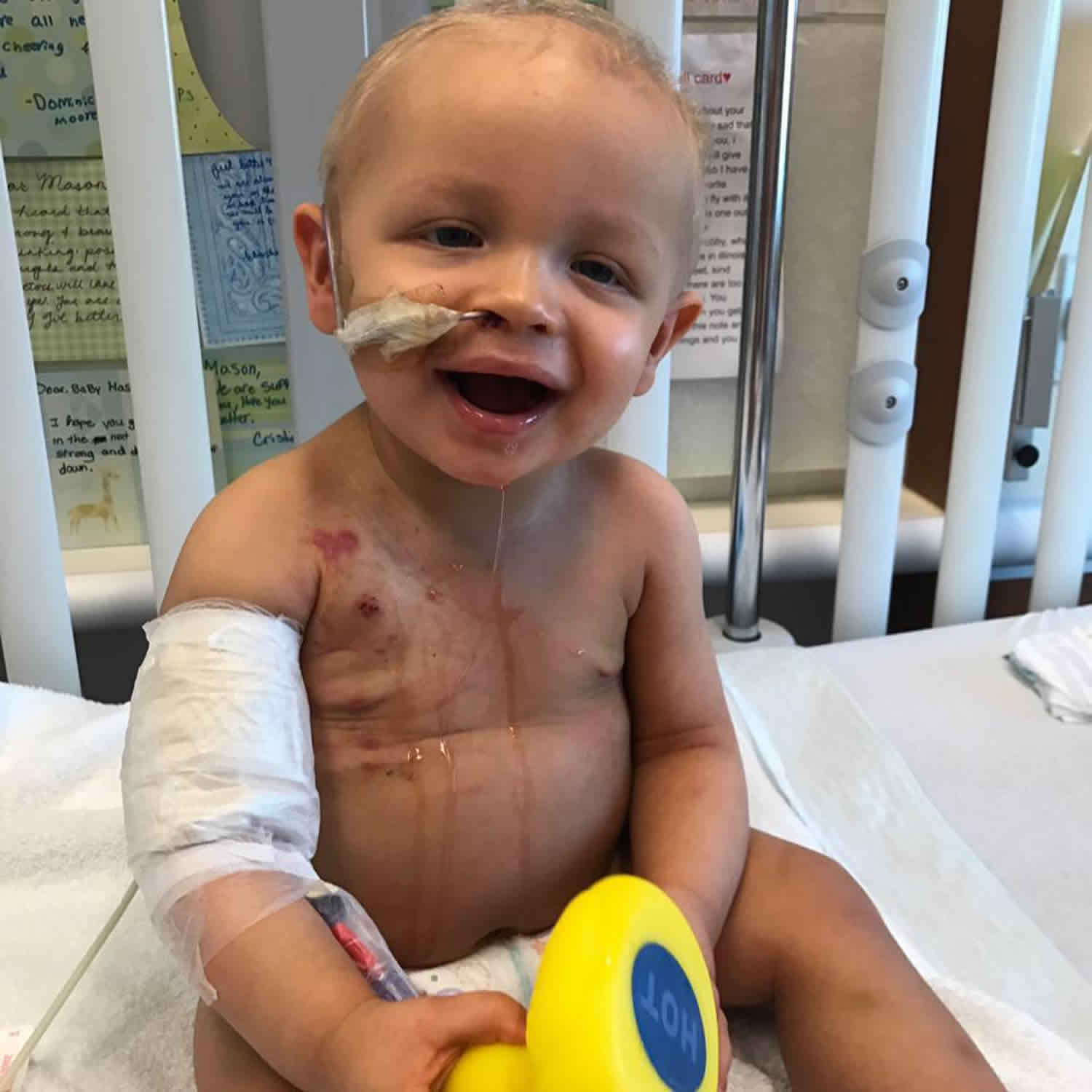
Kostmann syndrome
Kostmann syndrome also called “severe congenital neutropenia”, Kostmann disease, Kostmann neutropenia, genetic infantile agranulocytosis or infantile genetic agranulocytosis, is a rare inherited disorder in which there is a shortage of neutrophils (a type of white blood cell that is important in fighting infections and inflammation) with absolute neutrophil counts less than 500 cells/mm³. Mild neutropenia is classified as absolute neutrophil counts less than 1500 granulocytes/mm³, moderate is less than 1000 granulocytes/mm³, severe is less than 500 granulocytes/mm³, and very severe is less than 200 granulocytes/mm³. Severe congenital neutropenia usually presents in infancy with an absolute neutrophil count of less than 200 cells/mm³ 1. Infants with Kostmann disease get recurrent infections caused by bacteria (e.g. otitis media, pneumonia, sinusitis, urinary tract infections, abscesses of skin and/or liver) and are at an increased risk of acute myelogenous leukemia (AML) or myelodysplasia (a bone marrow disorder). Periodontal disease, as well as neurological symptoms, such as cognitive impairment, severe neurodegeneration and epilepsy, have been reported in some patients 2.
The deficiency of neutrophils, called neutropenia, is apparent at birth or soon afterward. It leads to recurrent infections beginning in infancy, including infections of the sinuses, lungs, and liver. Affected individuals can also develop fevers and inflammation of the gums (gingivitis) and skin. Approximately 40 percent of affected people have decreased bone density (osteopenia) and may develop osteoporosis, a condition that makes bones progressively more brittle and prone to fracture. In people with Kostmann syndrome, these bone disorders can begin at any time from infancy through adulthood.
Approximately 20 percent of people with Kostmann syndrome develop certain cancerous conditions of the blood, particularly myelodysplastic syndrome or leukemia during adolescence.
Some people with Kostmann’s syndrome have additional health problems such as seizures, developmental delay, or heart and genital abnormalities.
The incidence of Kostmann syndrome is estimated to be 1 in 200,000 individuals 3.
Kostmann syndrome causes
Kostmann syndrome or severe congenital neutropenia can result from mutations in one of many different genes (i.e. ELANE, HAX1, WASp, G6PC3, and CSF3R). These genes play a role in the maturation and function of neutrophils, which are cells produced by the bone marrow. Neutrophils secrete immune molecules and ingest and break down foreign invaders.
Gene mutations that cause Kostmann disease (severe congenital neutropenia) lead to the production of neutrophils that die off quickly or do not function properly. Some gene mutations result in unstable proteins that build up in neutrophils, leading to cell death. Other gene mutations result in proteins that impair the maturation or function of neutrophils, preventing these cells from responding appropriately to immune signals.
The unfolded protein response (UPR) has been recently proposed as a potential explanation for increased apoptosis seen in severe congenital neutropenia. Increased endoplasmic reticulum stress leads to the activation of the UPR. When an accumulation of unfolded or misfolded proteins occurs in the lumen of the endoplasmic reticulum, the UPR works to protect the cell against the damage caused by these improperly folded proteins. The goals of the UPR are to maintain homeostasis in the cell by arresting protein translation and promoting signaling pathways that lead to increased production of molecular chaperones to help with protein folding. If this does not happen, the UPR initiates apoptosis 4.
About half of all cases of Kostmann syndrome (severe congenital neutropenia) are caused by mutations in the ELANE gene. Another 10 percent are caused by mutations in the HAX1 gene. The other genes each account for only a small percentage of all cases of this condition. In about one-third of people with Kostmann syndrome (severe congenital neutropenia), the cause of the disorder is unknown.
Kostmann syndrome inheritance pattern
Most cases of Kostmann syndrome (severe congenital neutropenia) are classified as sporadic and occur in people with no apparent history of the disorder in their family. Some of these cases are associated with changes in specific genes; however in some cases the cause of the disorder is unknown.
When Kostmann syndrome (severe congenital neutropenia) is caused by mutations in the ELANE gene, it is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. Mutations in a few other genes that cause this condition are also inherited in an autosomal dominant pattern.
When Kostmann syndrome (severe congenital neutropenia) is caused by mutations in the HAX1 gene, it is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. Many cases of this condition are caused by genetic mutations that are inherited in an autosomal recessive pattern.
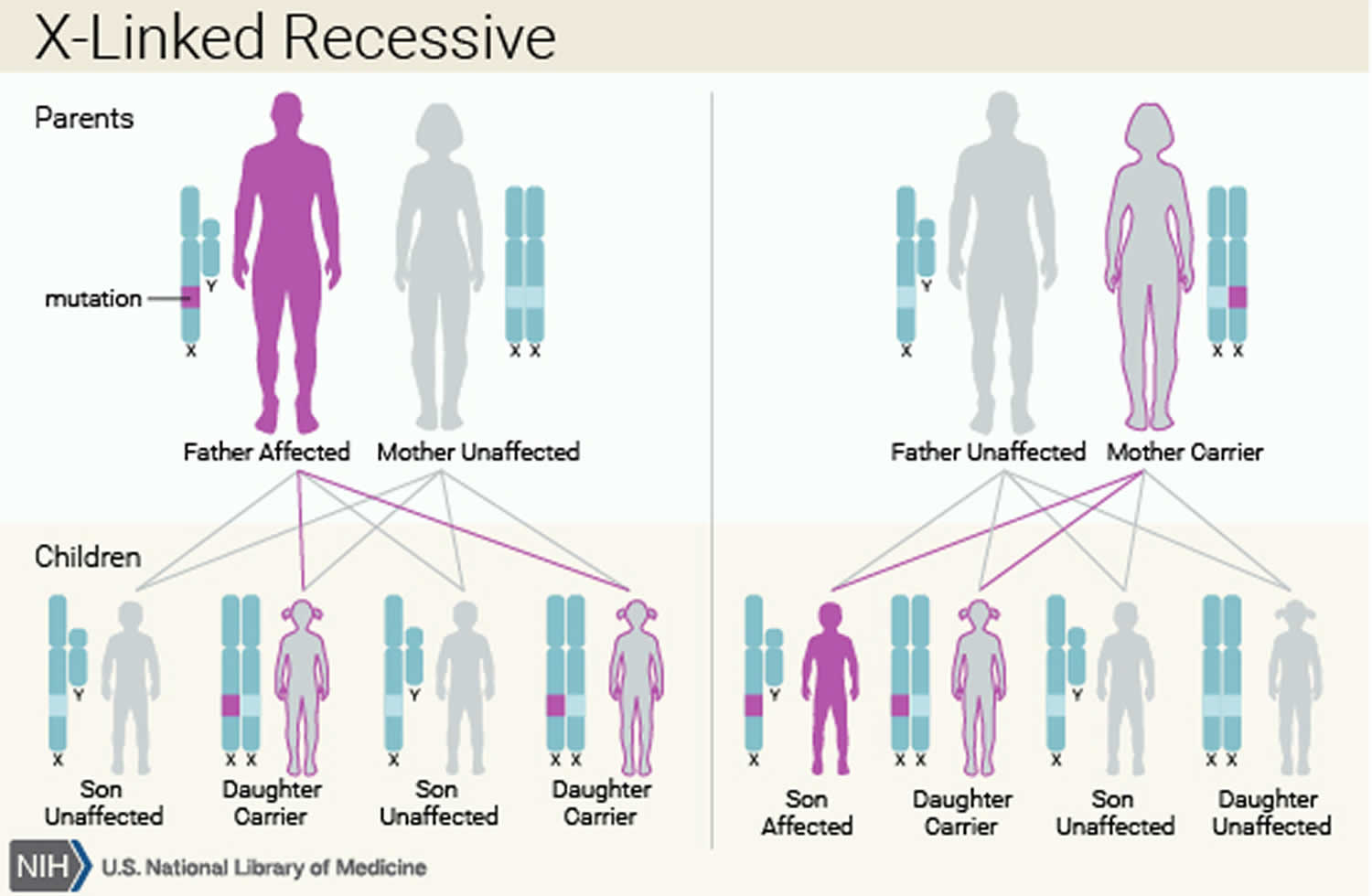
In rare cases, Kostmann syndrome (severe congenital neutropenia) is inherited in an X-linked recessive pattern. In these cases, the gene that causes the condition is located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation would have to occur in both copies of the gene to cause the disorder. Because it is unlikely that females will have two altered copies of this gene, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Figure 1. Autosomal dominant inheritance pattern
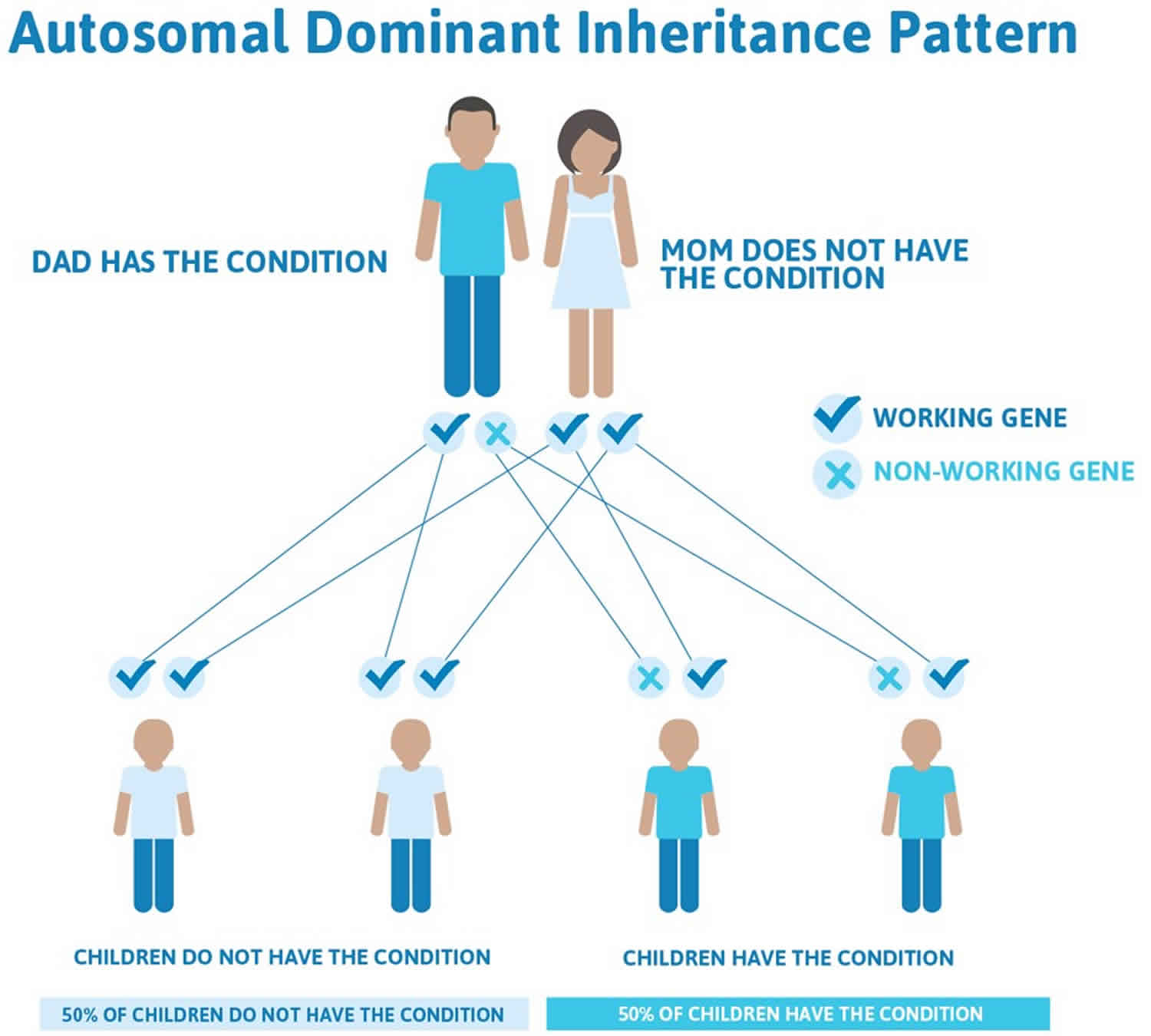
Figure 2. Autosomal recessive inheritance pattern
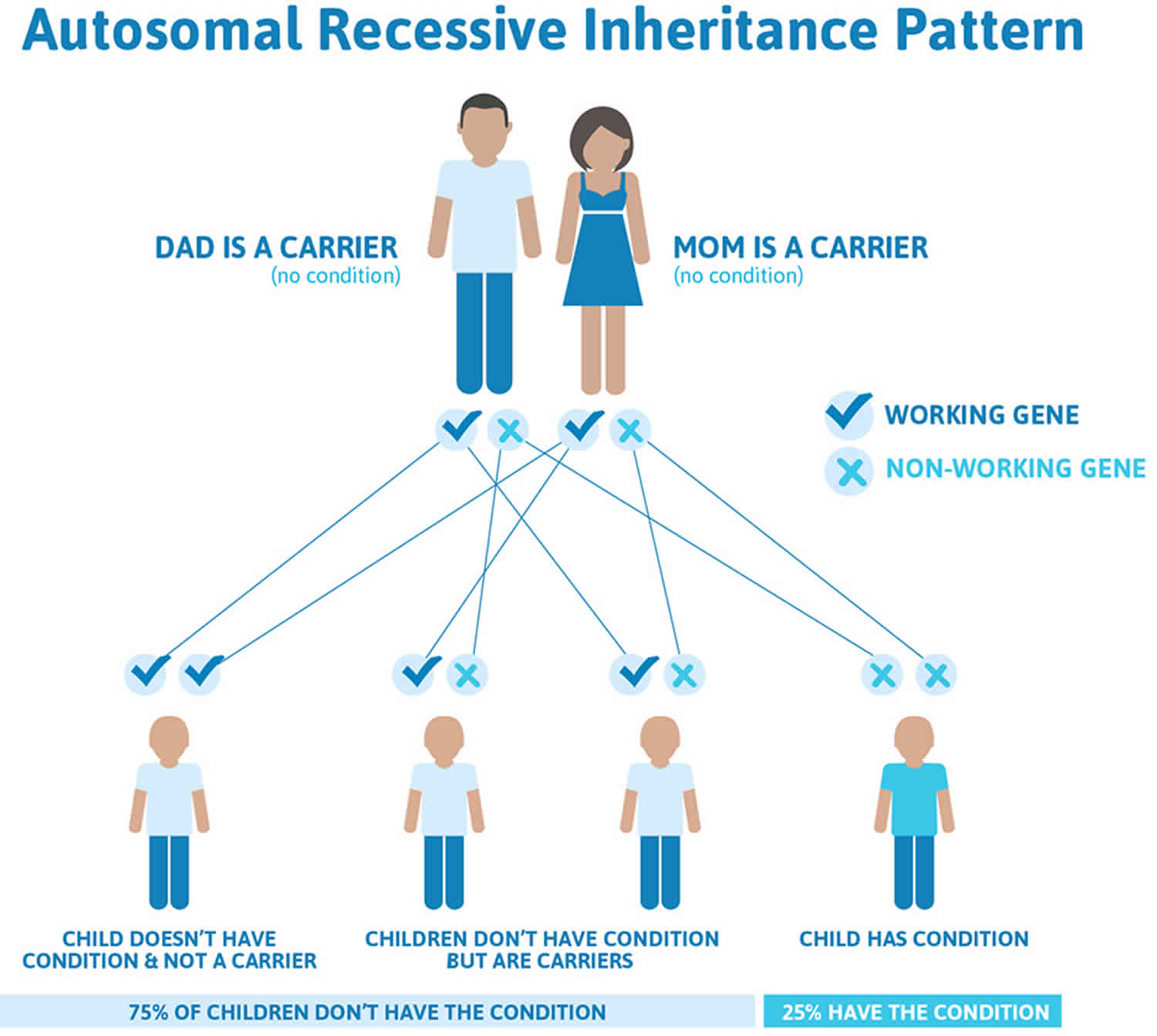
Figure 3. X-linked recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Kostmann’s syndrome symptoms
Signs and symptoms of Kostmann disease include the following:
- Oral ulcers
- Gingivitis, which may lead to early loss of permanent teeth
- Pharyngitis
- Sinusitis, otitis media
- Lymphadenopathy, lymphadenitis
- Bronchitis, pneumonia
- Cellulitis
- Cutaneous abscess, boils
- Omphalitis
- Perianal abscess
- Lung abscess
- Liver abscess
- Peritonitis
- Enteritis with chronic diarrhea and vomiting
- Bacteremia and/or septicemia, most commonly caused by streptococci or staphylococci. Other commonly encountered organisms include Pseudomonas, fungi, and, in rare cases, Clostridium species.
- Urinary tract infection (UTI)
- Fractures or bone pain
- Splenomegaly
- Neurological symptoms, including epilepsy and neuropsychological deficits 5
Neutropenic patients are usually infected by organisms of their endogenous flora, the resident bacteria of the mouth and oropharynx, gastrointestinal tract, and skin. A cardinal feature of these infections is a lack of purulence. By age 2 years, oral ulcers and tooth decay are almost always present. The stomatologic disorders are distinguished by erosive, hemorrhagic, and painful gingivitis associated with papules on the tongue and the cheek mucosa, as well as perirectal inflammation or cellulitis. Abscesses, pneumonia, and septicemia may also occur.
The most common microbes seen are Staphylococcus aureus and gram-negative organisms. Patients with severe congenital neutropenia also are at risk for significant bacterial infections such as otitis media, bronchitis, pneumonia, osteomyelitis, or cellulitis. In addition, patients can also experience diffuse gastrointestinal lesions leading to abdominal pain and diarrhea that can mimic Crohn disease. They are not initially predisposed to other fungal, parasitic, or viral infections, but have a higher risk of fungal infection with prolonged neutropenia or extended antibiotic use 1.
Severe congenital neutropenia patients also may experience a variety of extrahematopoietic manifestations depending on their genetic abnormalities, such as neurologic involvement in HAX1 mutations, dermatologic manifestations in G6PC3 mutations, or monocytopenia in WASp mutations.
Kostmann syndrome diagnosis
An absolute neutrophil count (ANC) of less than 200 cells/mm³ is seen in classic cases of severe congenital neutropenia.
Monocytosis and eosinophilia may be evident. Total leukocyte counts are frequently normal because of the monocytosis. Mild anemia may be present from chronic inflammation, and thrombocytosis may be present.
Quantitative immunoglobulins may show hypergammaglobulinemia. Patients have a normal response to vaccinations.
Complement levels typically are normal.
Antineutrophil antibodies are absent but should be checked to exclude an autoimmune etiology when the diagnosis is entertained in the first few months of life.
Electrolyte levels and renal and liver function test results are within the normal range.
Imaging studies
Imaging studies are performed only as clinically indicated to evaluate for infection. Bone density (duel-energy x-ray absorptiometry [DEXA] scanning) is performed to evaluate for osteopenia or osteoporosis.
Genetic testing
Genetic testing can be considered to differentiate between the variants of severe congenital neutropenia depending on the clinical findings.
Bone marrow biopsy
Bone marrow biopsy can be very helpful in the diagnosis of severe congenital neutropenia. Bone marrow is also necessary to rule out malignant conditions, evaluate myeloid maturation, determine cellularity, and elucidate an etiology 6.
When leukemic or myelodysplastic transformation occurs, bone marrow cytogenetics exhibit monosomy 7 in 50% of cases. Granulocyte colony-stimulating factor (G-CSF) receptor mutations occur within an intracellular part of the receptor and can be identified from either blood or bone marrow samples. As G-CSF receptor mutations (CSF3R) are not present at birth, they do not represent the underlying defect for the disease.
The bone marrow findings of severe congenital neutropenia typically demonstrate normal or decreased cellularity with an arrest of neutrophil precursor maturation at the promyelocyte or myelocyte level. This is typically seen in the severe congenital neutropenia due to ELANE, HAX1, WASp, G6PC3, and CSF3R mutations. Maturation arrest in the promyelocyte phase is often associated with bone marrow hypereosinophilia and monocytosis 6.
Kostmann syndrome treatment
Fevers higher than 39°C and low monocyte count (< 100/μL) are associated with more serious infections. Oral antibiotic therapy can be used for superficial or ear/nose/throat infection in moderate neutropenia with close monitoring if inflammatory markers are low (C-reactive protein, < 15 mg/L).
Patients with severe congenital neutropenia and sepsis require hospitalization. Blood, urine, and infectious-site cultures, as well as chest radiography, should be performed as clinically indicated. Aggressive intravenous parenteral antibiosis with a combination of a third-generation cephalosporin and an aminoglycoside should be considered. If fevers persist beyond 48 hours, the addition of antifungal agents is recommended.
In patients with severe infections that are worrisome, G-CSF should be started at the known dose that patient responds to, or standard dose of 5 μg/kg/d. There is an induction phase with G-CSF to evaluate the response of individual, with an increase in absolute neutrophil count (ANC) (>1500/μL) and clinical improvement after 10-15 days. The initial daily dose is 5 μg/kg subcutaneously. If there is no response after 15 days, the daily dose is increased by 5 μg/kg. If the response is rapid or excessive (ANC >5000/μL), the dose is halved.
Antibiotic prophylaxis
Antimicrobial prophylaxis may be useful in preventing recurrent infections. Oral sulfamethoxazole/trimethoprim sulfate (Bactrim) as a once daily, 50 mg/kg/day dose has been used. This only partially prevents the gingivostomatitis associated with severe congenital neutropenia. Concurrent therapy with metronidazole, which covers oral saprophytic flora, especially anaerobes, also may be added.
Hematopoietic growth factors
Hematopoietic growth factors (granulocyte colony-stimulating factor [G-CSF] and granulocyte macrophage colony-stimulating factor [GM-CSF]) are used to correct neutropenia. G-CSF has been used since the late 1980s and has shown a greater than 90% response rate. G-CSF is more efficacious and tolerable than GM-CSF, with less flu-like syndrome and less marked eosinophilia. There are 2 forms of G-CSF available: filgrastim (Neupogen in 480- or 330-µg vials) and lenograstim (Granocyte in 340- or 130-µg vials). Lenograstim is the glycosylated form of G-CSF.
Pegfilgrastim (Neulasta)
Pegfilgrastim is the pegylated, covalent conjugate of G-CSF, a combination of filgrastim and polyethylene glycol, with a half-life of 15-80 hours, which decreases the number of injections needed from daily to once weekly. Pegfilgrastim has been shown to be clinically efficacious and improve compliance and quality of life in various case reports 7. However, in an observational study of 17 patients from the French Severe Chronic Neutropenia Registry who received pegfilgrastim, only half the patients prescribed were able to continue this medication long term because of adverse events and lack of efficacy 8. Further studies evaluating long-term outcomes are required.
Filgrastim or lenograstim (G-CSF)
There is an induction phase with G-CSF to evaluate the response of individuals, with an increase in absolute neutrophil count (ANC) (>1500/μL) and clinical improvement after 10-15 days. The initial daily dose is 5 μg/kg subcutaneously. If there is no response after 15 days, the daily dose is increased by 5 μg/kg. If the response is rapid or excessive (ANC >5000 /μL), the dose is halved. Once the minimal daily dose is determined, the maintenance phase can begin, with monitoring of neutrophil counts every 3-6 months. Almost two thirds of patients respond to a daily dose of 2-10 µg/kg, while 20% respond to 10-20 μg/kg. A small percentage of patients require higher doses, up to 100 μg/kg. Around 10% of patients are unresponsive to G-CSF therapy 9.
Short-term tolerability
G-CSF has good short-term tolerability with occasional immediate and local reactions (< 1 per 100). Flu-like reactions are another infrequent adverse effect. Bone pain occurs in 2-5% of patients, which resolves within 24 hours and does not recur with lower doses 1.
Long-term tolerability
G-CSF acts principally on granulocytes, but various blood cell abnormalities can be seen transiently during treatment. Monocytosis (above 1500/μL) is frequent and eosinophilia can be amplified by G-CSF, but lymphocytosis and hemoglobin levels are usually not affected. Occasionally, reticulocytosis and elevated hemoglobin levels can be seen if there is an initial inflammatory anemia at the start of treatment.
Thrombocytopenia is the most common hematologic adverse effect and is usually moderate and resolves when the G-CSF dose is reduced. Thrombocytopenia can also be from hypersplenism. The spleen is usually enlarged at the start of treatment. Spleen rupture requiring splenectomy can sometimes occur.
Uricemia can arise from long-term treatment but has no clinical consequences. Gout exacerbations have been seen in short-course therapy, however.
The first cases of leukocytoclastic vasculitis were seen after short-term G-CSF treatment and seem to be due to the increase in neutrophil adhesion molecule expression.
Two cases of mesangioproliferative glomerulonephritis have been seen with long-term treatment, but they resolved after dose reduction or stoppage of treatment.
Osteoporosis occurs in up to one fourth of patients with severe congenital neutropenia who receive G-CSF therapy, and 2 cases of pathological fractures have been reported. However, severe congenital neutropenia alone seems to be associated with osteopenia, which is usually present before treatment starts.
Surgical care
Surgical drainage of abscesses may be required for infections refractory to intravenous antibiotics.
Hematopoietic stem cell transplantation (HSCT) can be curative for severe congenital neutropenia and is the only option when patients continue to have severe infections despite G-CSF therapy. Hematopoietic stem cell transplantation (HSCT) is indicated when there is G-CSF resistance (>50 μg/kg/day) and myelodysplasia/leukemic transformation.
Patients chronically dependent on high doses of G-CSF (at least 20 μg/kg per injection at least 3 months a year) should be considered for bone marrow grafting on a case-by-case basis since there is a high risk of leukemic transformation, taking into account whether related donors are available. Survival with hematopoietic stem cell transplantation (HSCT) is now is over 70%, even with malignant transformation in most cases.
Diet
No dietary restrictions are necessary in patients with severe congenital neutropenia, so long as they are on adequate doses of G-CSF and their absolute neutrophil counts are maintained above 1000/μL.
Further outpatient care
Obtain a complete blood count (CBC) count with differential twice per week during the first 4 weeks after the initiation of granulocyte-colony stimulating factor (G-CSF) or for 2 weeks following any dosage adjustments. Thereafter, obtain a CBC count with differential monthly for 6 months. When the minimum daily dose is found, the maintenance phase can be started, with monitoring of absolute neutrophil counts every 3-6 months.
Routine clinical follow-up every 3 months for patients who are stable is recommended.
Kostmann syndrome prognosis
The mortality rate for Kostmann syndrome (severe congenital neutropenia) prior to the antibiotic era (1950s) was as high as 90% 10 and even with antibiotics, >80% of patients would die from severe bacterial infections 11. A breakthrough in the treatment of severe congenital neutropenias occurred with the availability of recombinant granulocyte-colony-stimulating factor (G-CSF) for clinical use 12.
Before the introduction of granulocyte colony-stimulating factor (G-CSF), patients would often die during the first year of life. The mortality rate is 70% within the first year of life in the absence of medical intervention with granulocyte colony-stimulating factor (G-CSF), bone marrow transplantation, or peripheral blood stem cell transplantation 13. After the introduction of G-CSF, patients survive into adulthood but are at significant risk of leukemia or myelodysplasia 6. On the basis of national averages of premature deliveries in the general population, an estimated 8% of patients with Kostmann disease are delivered prematurely and admitted to neonatal intensive care units. Although neutropenia is common in ill preterm infants and severe congenital neutropenia is relatively rare, severe congenital neutropenia should be considered if other etiologies for the neutropenia are not found. Therapy with G-CSF can save lives in this clinical setting because of the innate risks of infection in a preterm infant.
Patients with severe congenital neutropenia are at an increased risk of bacterial and fungal infections, with most frequent infections involving the skin, mucosa, ears, nose, throat, and lungs. Stomatitis starts after age 2 years with erosive hemorrhagic gingivitis and painful aphthouslike papules on the tongue and cheeks, contributing significantly to morbidity. Chronic periodontal disease has been attributed to deficiency in a defensin, the antimicrobial peptide LL-37 14. Diffuse gastrointestinal lesions may cause abdominal pain and diarrhea, resembling bacterial enteritis. Bacterial infections involve Staphylococcus aureus and Staphylococcus epidermidis, streptococci, enterococci, pneumococci, Pseudomonas aeruginosa, gram-negative bacilli, and fungal infections with Candida or Aspergillus species.
About 1 in 5 patients with severe congenital neutropenia develop secondary malignancies. The incidence of acute myelogenous leukemia (AML) or myelodysplastic syndrome (MDS) in severe congenital neutropenia after 10 years of G-CSF treatment is 21%. Acquired mutations in G-CSF receptor CSF3R are a highly predictive marker for the progression of severe congenital neutropenia to leukemia 15. Of patients with severe congenital neutropenia, 20-30% have acquired mutations in the CSF3R gene, which produce C-terminally truncated hyperresponsive forms of the G-CSFRhyper and a strong predisposition for myelodysplastic syndrome and AML.
References- Donadieu J, Fenneteau O, Beaupain B, Mahlaoui N, Chantelot CB. Congenital neutropenia: diagnosis, molecular bases and patient management. Orphanet J Rare Dis. 2011. 6:26.
- Kostmann syndrome. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=99749
- https://ghr.nlm.nih.gov/condition/severe-congenital-neutropenia
- Nanua S, Murakami M, Xia J, Grenda DS, Woloszynek J, Strand M. Activation of the unfolded protein response is associated with impaired granulopoiesis in transgenic mice expressing mutant Elane. Blood. 2011 Mar 31. 117(13):3539-47.
- Carlsson G, van’t Hooft I, Melin M, et al. Central nervous system involvement in severe congenital neutropenia: neurological and neuropsychological abnormalities associated with specific HAX1 mutations. J Intern Med. 2008 Oct. 264(4):388-400.
- Zeidler C, Germeshausen M, Klein C, Welte K. Clinical implications of ELA2-, HAX1-, and G-CSF-receptor (CSF3R) mutations in severe congenital neutropenia. Br J Haematol. 2009 Feb. 144(4):459-67.
- Fioredda F, Calvillo M, Lanciotti M, Lanza T, Giunti L, Castagnola E. Pegfilgrastim in children with severe congenital neutropenia. Pediatr Blood Cancer. 2010 Mar. 54(3):465-7.
- Beaupain B, Leblanc T, Reman O, Hermine O, Vannier JP, Suarez F. Is pegfilgrastim safe and effective in congenital neutropenia? An analysis of the French Severe Chronic Neutropenia registry. Pediatr Blood Cancer. 2009 Dec. 53(6):1068-73.
- Ward AC, Dale DC. Genetic and molecular diagnosis of severe congenital neutropenia. Curr Opin Hematol. 2009 Jan. 16(1):9-13.
- Chown G, Gelfand AS. Agranulocytosis. Can Med Assoc J. 1933;29:128–134.
- Gilman PA, Jackson DP, Guild HG. Congenital agranulocytosis: prolonged survival and terminal acute leukemia. Blood. 1970;36:576–585.
- Souza LM, et al. Recombinant human granulocyte colony-stimulating factor: effects on normal and leukemic myeloid cells. Science. 1986;232:61–65.
- Connelly JA, Choi SW, Levine JE. Hematopoietic stem cell transplantation for severe congenital neutropenia. Curr Opin Hematol. 2012 Jan. 19(1):44-51.
- Putsep K, Carlsson G, Boman HG, Andersson M. Deficiency of antibacterial peptides in patients with morbus Kostmann: an observation study. Lancet. 2002 Oct 12. 360(9340):1144-9.
- Germeshausen M, Welte K, Ballmaier M. In vivo expansion of cells expressing acquired CSF3R mutations in patients with severe congenital neutropenia. Blood. 2009 Jan 15. 113(3):668-70.





{kind=link}