Triple A syndrome
Triple A syndrome also called “AAA syndrome”, Achalasia-Addisonianism-Alacrima syndrome or Allgrove syndrome, is an inherited condition characterized by three specific features: achalasia, Addison disease, and alacrima (a reduced or absent ability to secrete tears) 1. Achalasia is a disorder that affects the ability to move food through the esophagus, the tube that carries food from the throat to the stomach. Achalasia can lead to severe feeding difficulties and low blood sugar (hypoglycemia). Addison disease, also known as primary adrenal insufficiency, is caused by abnormal function of the small hormone-producing glands on top of each kidney (adrenal glands). The main features of Addison disease include fatigue, loss of appetite, weight loss, low blood pressure, and darkening of the skin. The third major feature of triple A syndrome is a reduced or absent ability to secrete tears (alacrima). Most people with triple A syndrome have all three of these features, although some have only two.
Several authors published descriptions of a more global autonomic disturbance associated with the original three characteristics, leading one author to suggest the name 4A syndrome (adrenal insufficiency, achalasia, alacrima, autonomic abnormalities). Specific autonomic disturbances described in this syndrome include abnormal pupillary reflexes, poor heart rate variability, and orthostatic hypotension. Affected individuals may also have developmental delay, intellectual disability, speech problems, a small head size, muscle weakness, movement problems, peripheral neuropathy, and optic atrophy. Many of the neurological symptoms of triple A syndrome worsen over time. Triple A syndrome is caused by mutations in the AAAS gene and is inherited in an autosomal recessive pattern 2. Alacrimia is treated with artificial tears while achalasia may need surgery with either pneumatic dilatation or Heller’s myotomy. Adrenal insufficiency is treated with glucocorticoid and if necessary mineralocorticoid replacement 2.
Many of the features of triple A syndrome are caused by dysfunction of the autonomic nervous system. This part of the nervous system controls involuntary body processes such as digestion, blood pressure, and body temperature. People with triple A syndrome often experience abnormal sweating, difficulty regulating blood pressure, unequal pupil size (anisocoria), and other signs and symptoms of autonomic nervous system dysfunction (dysautonomia).
People with this condition may have other neurological abnormalities, such as developmental delay, intellectual disability, speech problems (dysarthria), and a small head size (microcephaly). In addition, affected individuals commonly experience muscle weakness, movement problems, and nerve abnormalities in their extremities (peripheral neuropathy). Some develop optic atrophy, which is the degeneration (atrophy) of the nerves that carry information from the eyes to the brain. Many of the neurological symptoms of triple A syndrome worsen over time.
People with triple A syndrome frequently develop a thickening of the outer layer of skin (hyperkeratosis) on the palms of their hands and the soles of their feet. Other skin abnormalities may also be present in people with this condition.
Alacrima is usually the first noticeable sign of triple A syndrome, as it becomes apparent early in life that affected children produce little or no tears while crying. They develop Addison disease and achalasia during childhood or adolescence, and most of the neurologic features of triple A syndrome begin during adulthood. The signs and symptoms of this condition vary among affected individuals, even among members of the same family.
Triple A syndrome is a rare condition, although its exact prevalence is unknown. Only scattered family and case reports are noted in the literature. Review of multiple kindreds and analysis of a large, highly inbred kindred provide evidence that this is a rare syndrome with an autosomal recessive inheritance. The probable recurrence risk for future pregnancies from parents with a child affected with Triple A syndrome or Allgrove syndrome is 25%. The actual incidence is difficult to determine because of the variable presentation, including unexplained childhood death due to adrenal crisis and mild disease that is not apparent until adulthood.
My 15 year old daughter was recently diagnosed with triple A syndrome. Is there anything that can be done about her abnormal sweating?
Many of the features of triple A syndrome, including abnormally increased sweating (hyperhidrosis), are caused by dysfunction of the autonomic nervous system (dysautonomia), which controls involuntary body processes 1. In most cases, the goal of treating secondary hyperhidrosis (caused by a primary underlying condition) is to treat the underlying condition; unfortunately, there is no cure for dysautonomia so only symptomatic treatment may be possible 3.
Most of the treatments for hyperhidrosis discussed in the medical literature involve primary and/or focal hyperhidrosis, which is often idiopathic (of unknown cause). Treatment options for these have included medications, antiperspirants, iontophoresis, Botulinum toxin type A (Botox), endoscopic thoracic sympathectomy and surgical intervention 4. Several studies have mentioned that generalized hyperhidrosis may optionally be treated with systemic anticholinergic agents 5, which are used to prevent the stimulation of sweat glands. However, the associated adverse effects have somewhat limited their use 5.
Antidepressant drugs such as amitriptyline and paroxetine, as well as antihypertensive drugs (blood pressure medications) such as beta blockers, calcium channel antagonists, alpha antagonists, and alpha-2 agonists have been described in single case reports as only slightly to moderately effective 6.
Do all individuals with triple A syndrome develop achalasia?
Most individuals with triple A syndrome develop achalasia. It usually develops during childhood or adolescence, although in some cases it does not occur until early adulthood 1.
What are the neurological features of triple A syndrome?
Many of the neurological features of triple A syndrome are caused by dysfunction of the autonomic nervous system. This part of the nervous system controls involuntary body processes such as digestion, blood pressure, and body temperature. People with triple A syndrome often experience abnormal sweating, difficulty regulating blood pressure, unequal pupil size (anisocoria), and other signs and symptoms of autonomic nervous system dysfunction (dysautonomia) 1.
People with this condition may have other neurological abnormalities, such as developmental delay, intellectual disability, speech problems (dysarthria), and a small head size (microcephaly). In addition, affected individuals commonly experience muscle weakness, movement problems, and nerve abnormalities in their extremities (peripheral neuropathy). Some develop optic atrophy, which is the degeneration (atrophy) of the nerves that carry information from the eyes to the brain 1.
Most of the neurologic features of triple A syndrome begin during adulthood, and they often worsen over time 1.
How might neuropathy associated with triple A syndrome be treated?
There is limited information available regarding the treatment of neuropathy specific to triple A syndrome. However, the following is general information about how the symptoms of peripheral and autonomic neuropathies are sometimes managed. Individuals that are interested in learning about how to treat their own neuropathy should speak with their health care provider or a neurologist for individual treatment options.
For peripheral neuropathy, a major goal of treatment is to first manage the condition causing the neuropathy. Another goal of treatment is to relieve the painful symptoms. Medications may be used to relieve the pain of peripheral neuropathy. Mild symptoms may be relieved by over-the-counter pain medications. For more severe symptoms, a doctor may recommend prescription painkillers. Anti-seizure medications were originally developed to treat epilepsy; however, some doctors also prescribe them for nerve pain. A cream containing capsaicin, a substance found naturally in hot peppers can cause modest improvements in peripheral neuropathy symptoms. Tricyclic antidepressant medications were originally developed to treat depression; however, they have been found to help relieve pain as well. Another type of therapy called transcutaneous electrical nerve stimulation (TENS) uses electrodes that are placed on the skin, sending a gentle electric current through the electrodes; some people report this therapy improves symptoms. Other potential remedies include exercise, which may reduce neuropathy pain and can help control blood sugar levels; and massaging of hands and feet, which may improve circulation, stimulate nerves and temporarily relieve pain 7.
Treatment of autonomic neuropathy, like that of peripheral neuropathy, also includes treating the underlying disease and managing the specific symptoms. Gastrointestinal symptoms may be managed by modifying the diet or using dietary supplements. Medications to ease constipation such as laxatives may help ease constipation; increasing the amount of fiber in the diet may also ease constipation. Some doctors may prescribe tricyclic antidepressants to treat diarrhea and abdominal pain. Autonomic neuropathy can cause a number of heart rate and blood pressure problems, which may be managed with medication. For individuals that experience excessive sweating, a doctor may prescribe a medication that decreases perspiration. There is no medication to increase sweating if an individual has lost the ability to sweat 8.
Triple A syndrome causes
Mutations in the AAAS (ADRACALIN) gene cause triple A syndrome. The AAAS gene provides instructions for making a protein called ALADIN whose function is not well understood 9. Within cells, ALADIN is found in the nuclear envelope, the structure that surrounds the nucleus and separates it from the rest of the cell. Based on its location, ALADIN is thought to be involved in the movement of molecules into and out of the nucleus.
Mutations in the AAAS gene change the structure of ALADIN in different ways; however, almost all mutations prevent this protein from reaching its proper location in the nuclear envelope. The absence of ALADIN in the nuclear envelope likely disrupts the movement of molecules across this membrane. Researchers suspect that DNA repair proteins may be unable to enter the nucleus if ALADIN is missing from the nuclear envelope. DNA damage that is not repaired can cause the cell to become unstable and lead to cell death. Although the nervous system is particularly vulnerable to DNA damage, it remains unknown exactly how mutations in the AAAS gene lead to the signs and symptoms of triple A syndrome.
Some individuals with triple A syndrome do not have an identified mutation in the AAAS gene. The genetic cause of the disorder is unknown in these individuals.
Triple A syndrome inheritance pattern
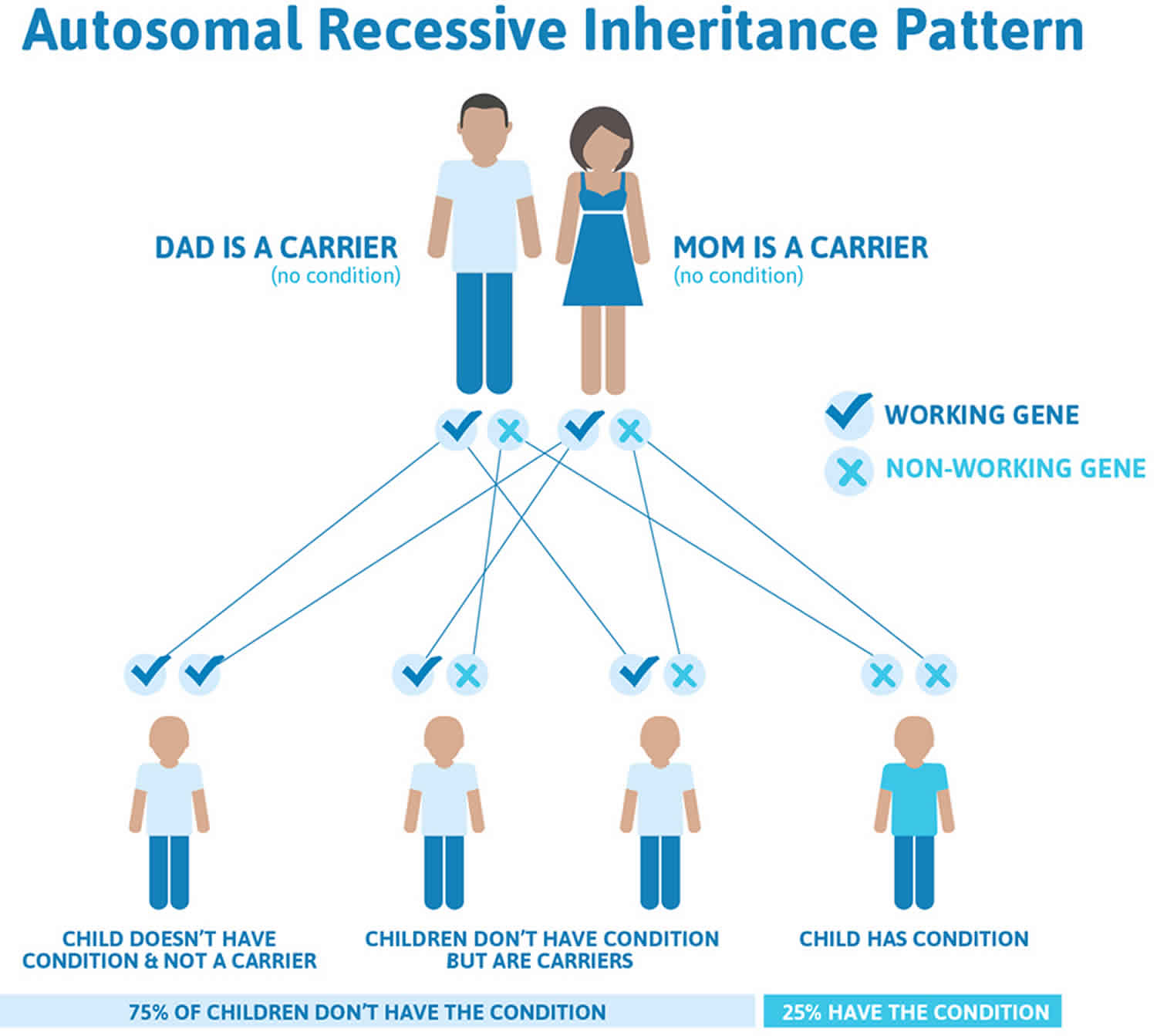
Triple A syndrome is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 1 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 1. Triple A syndrome autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Triple A syndrome symptoms
Many cases of Triple A syndrome present with classic symptoms of primary adrenal insufficiency, including hypoglycemic seizures and shock. Less frequently, a child may be evaluated initially for recurrent vomiting, dysphagia, and failure to thrive (achalasia) or for ocular symptoms associated with alacrima.
At presentation, review of systems may be positive for crying without tears, hyperpigmentation, developmental delay, seizures, dysphagia, hypernasal speech, and symptoms related to orthostatic hypotension.
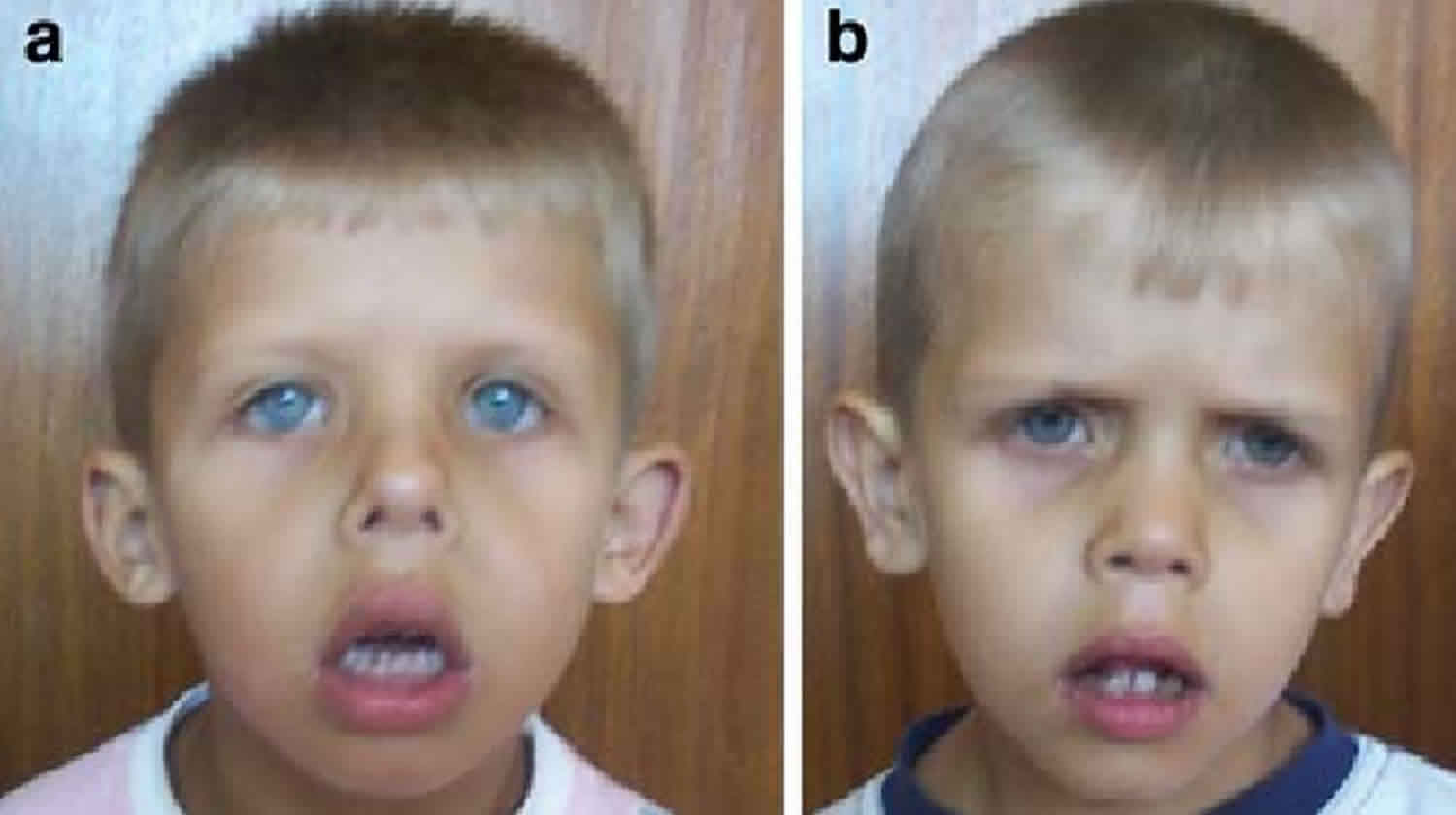
A distinct facial appearance associated with triple A syndrome consists of a long thin face with a long philtrum, narrow upper lip, and a down-turned mouth. These features are not seen in unaffected siblings 10.
Microcephaly is associated frequently with this disorder, but whether this is a primary manifestation or simply a reflection of recurrent hypoglycemia and/or malnutrition is unclear.
Conjunctival injection and irritation may be the only obvious signs of alacrima. Slit lamp examination may reveal punctate keratopathy or corneal ulceration 11.
Definitive diagnosis of alacrima can be made at bedside with the Schirmer test. This test evaluates the wetting of a special strip placed in the conjunctival sac for 5 minutes. Less than 10 mm of wetting is abnormal.
Cardiac examination findings may be abnormal due to a number of autonomic nervous system defects that may accompany Triple A syndrome. Orthostatic hypotension and diminished heart rate variations during deep breathing and Valsalva maneuver are well documented. Abnormal findings on respiratory examination may be secondary to recurrent aspiration accompanying achalasia.
Skin examination of patients may reveal abnormal findings that assist in confirming diagnosis. Hyperpigmentation is common but may be observed less frequently than in other forms of primary adrenal failure. Hyperkeratosis and fine fissuring of the palms of the hands and soles of the feet represent a unique feature of this syndrome.
Neurologic features are varied and have been the subject of several case reports and reviews 12. The most commonly described abnormal features of the neurologic examination are hyperreflexia, dysarthria, hypernasal speech with palatopharyngeal incompetence, and ataxia.
Adults may exhibit progressive neural degeneration, develop parkinsonian features, and show mental deterioration.
A family history of early unexplained infant deaths and familial consanguinity provides important clues. Evaluate siblings for early signs, particularly alacrima because this defect is frequently present from birth.
Although mental retardation and hyperpigmentation in the parents or grandparents of patients have been reported, these are not common or consistent findings and are not expected with autosomal recessive inheritance.
Triple A syndrome diagnosis
Assess adrenal function in patients with triple A syndrome. Patients who present with the combination of achalasia and alacrima should undergo a complete evaluation of their pituitary-adrenal axis to exclude adrenal insufficiency. Incidence of glucocorticoid deficiency in patients with isolated achalasia is low, and endocrine evaluation is not warranted unless symptoms consistent with glucocorticoid deficiency are present. Because no such data are available for patients with isolated alacrima, other clinical features must guide testing in this population. In patients with symptoms of cortisol deficiency or combined alacrima and achalasia, draw baseline adrenocorticotropic hormone (ACTH) and cortisol values and perform an ACTH stimulation test to assess adrenal function.
Esophageal motility tests are pertinent in patients presenting with dysphagia, food regurgitation, or both.
Determine serum sodium, potassium, aldosterone, and renin levels. Although aldosterone levels are usually normal in 4A syndrome, several cases of mineralocorticoid deficiency have been reported.
The presence of plasma antiadrenal antibodies should direct the investigation to the possibility of Addison disease.
Look for normal plasma very long chain fatty acids (hexa-eicosanoate) to exclude adrenoleukodystrophy.
If malnutrition is present, a comprehensive metabolic panel and complete blood count (CBC) count are warranted.
For patients presenting with a seizure, obtain a baseline serum glucose concentration and perform a lumbar puncture.
Although none of the above tests are specific for triple A syndrome, they may provide clues for making this diagnosis.
Imaging studies
MRI or CT scanning of the head (if neurologic problems are observed)
Patients frequently reveal atrophic lacrimal glands on computed tomography (CT) of the orbits.
If the patient presents with a seizure, magnetic resonance imaging of the brain is useful to exclude other causes of new-onset seizures.
Abdominal CT scanning may reveal cortical atrophy of the adrenal glands, similar to that observed with primary adrenal insufficiency. However, this is typically not necessary to make the diagnosis.
Barium esophagography, esophageal manometry, and endoscopy
Various methods are used to demonstrate achalasia of the esophagus.
Perhaps the most readily available and commonly used test is barium esophagography, although esophageal manometry 10 and endoscopy are also used.
Barium esophagography typically demonstrates a dilated esophagus with minimal, if any, peristaltic movement. The meal frequently passes slowly through a tight lower esophageal sphincter.
Other tests
Brainstem auditory evoked response (BAER)
Numerous investigators have demonstrated hearing deficits associated with triple A syndrome. Brainstem auditory evoked response (BAER) testing is useful in determining which patients have hearing deficits. Both normal and abnormal responses compatible with bilateral sensorineural hearing loss are found.
Autonomic testing
Investigation of the autonomic nervous system, including tilt-table and heart rate variability testing, is useful in demonstrating and following autonomic dysfunction 13. Many patients have diminished heart rate variability and exaggerated orthostatic responses on tilt-table. Formal pupillometry, when available, may demonstrate anisocoria and slowed constriction velocity.
Ophthalmologic evaluation for lacrimal dysfunction
Ophthalmologic testing is warranted in children with triple A syndrome.
A Schirmer test provides a semiquantitative measure of tearing. It consists of placing a standardized test strip in the conjunctival sac and measuring the wetting of this strip over a 5-minute interval. Less than 10 mm of wetting during this time is defined as alacrima.
Other ophthalmologic testing, including slit lamp examination and fluorescein staining, is helpful to identify patients with corneal pathology secondary to poor lacrimation.
Neurologic evaluation
A complete neurologic evaluation and developmental study may highlight the impaired neurologic and developmental function associated with this syndrome. Palatopharyngeal incompetence, sensory impairment, ataxia, and muscle weakness are among the documented findings.
Triple A syndrome treatment
There is no cure for triple A syndrome at this time; treatment typically focuses on managing individual signs and symptoms of the condition.
Glucocorticoid deficiency in individuals with known adrenal insufficiency (present with Addison disease) is typically treated by replacement of glucocorticoids. This may be important for avoiding an adrenal crisis and allowing for normal growth in children. In adult individuals, as well as those who have difficulty with compliance, replacing hydrocortisone with prednisone or dexamethasone is sometimes recommended. It is usually recommended that affected individuals wear a medical alert bracelet or necklace and carry the emergency medical information card supplied with it.
Achalasia is typically managed with surgical correction. Individuals may be monitored for pulmonary complications (due to reflux and aspiration). Gastric acid reduction therapy in individuals with reflux after surgical intervention is usually recommended. The symptoms in individuals with achalasia may be improved partially with pneumatic dilatation (also called balloon dilation). For those who remain symptomatic after this, other surgeries may be recommended.
Alacrima is typically managed by applying topical lubricants (such as artificial tears or ointments), and with punctal occlusion (a procedure used to close the tear ducts that drain tears from the eye). The symptoms of alacrima typically improve with punctal occlusion. However, this procedure is usually only done when therapy with topical lubricants is unsuccessful 2.
Glucocorticoid deficiency
Careful replacement of glucocorticoids in patients with known adrenal insufficiency is critical to avoid an adrenal crisis and to allow for normal growth in children. Growth must be closely monitored because overtreatment with glucocorticoids impairs linear growth. Providing stress doses of corticosteroids during illness or injury is also important.
Every patient should always wear a medical alert bracelet or necklace and carry the emergency medical information card supplied with it.
In adult patients, as well as those who have difficulty with compliance, replacing cortisone with an equipotent dose of prednisone or dexamethasone is appropriate.
Prednisone and dexamethasone are less preferred for maintenance than hydrocortisone which has balanced 1:1 effects of mineralocorticoid vs. glucocorticoid. Also, these corticosteroids are not preferred in children due to potential for growth-suppressive effects with greater potency and longer duration of action compared to hydrocortisone.
Patients must be instructed on the appropriate management of stress dosing of glucocorticoids.
A medical alert bracelet or necklace that states “adrenal insufficiency” or similar language should be worn at all times.
Because of the possibility of severe stress or trauma in a situation where medical assistance is not immediately available, the patient and his or her family members should be instructed to inject hydrocortisone or dexamethasone intramuscularly in a dose appropriate for the size of the patient, typically 100 mg hydrocortisone or 2 mg dexamethasone for adolescents and adults. The injection should be given whenever the patient cannot tolerate enteral stress dosing (eg, vomiting, loss of consciousness, severe diarrhea), followed promptly by medical attention at the closest facility.
Achalasia
Achalasia is best managed with surgical correction. Monitoring patients for pulmonary complications (due to reflux and aspiration) and providing gastric acid reduction therapy in patients with symptomatic reflux after surgical intervention is important.
Alacrima
Alacrima is managed with regular application of topical lubricants and with punctal occlusion. Children may need to be frequently reminded to use artificial tears. Children must have an annual ophthalmologic evaluation.
Surgical care
The symptoms of alacrima improve with punctal occlusion. This procedure is only necessary when therapy with topical lubricants is unsuccessful because of poor compliance.
The symptoms of lower esophageal sphincter spasm in patients with achalasia can be ameliorated partially with pneumatic dilatation 14. In patients who remain symptomatic after pneumatic dilatation, an anterior cardiomyotomy (modified Heller operation) may be performed. This surgical procedure involves directly cutting the muscles of the spastic sphincter. Both procedures have a risk of esophageal perforation and a high rate of postsurgical reflux.
Patients with triple A syndrome who undergo surgery must be treated with stress doses of glucocorticoids in the perioperative period.
Triple A syndrome prognosis
Provided the patient is effectively managed, a normal lifespan and childbirth are possible. Cases of parkinsonism, peripheral neuropathy, and seizures developing in patients have been reported. Whether these also occur in patients who received an early diagnosis and long-term effective medical and surgical management is unclear.
The primary cause of mortality is unrecognized adrenal crisis. The most frequent initial presentation is a hypoglycemic seizure secondary to glucocorticoid deficiency. Most patients have previously unrecognized alacrima at the time of presentation. This leads to severe keratopathy and corneal melting (dehydration-induced ulceration). Achalasia leading to frequent vomiting or regurgitation also commonly occurs and may lead to growth failure. Most children who are diagnosed with achalasia in the general population have isolated esophageal dysfunction and do not have any other features of triple A syndrome.
Although the 3 main features produce the primary morbidities associated with triple A syndrome, a slow neurologic deterioration occurs in many patients. This most frequently includes mild mental retardation and autonomic neuropathy but may include ataxia and muscle weakness as well.
In the pediatric population, developmental delay is common. Determining if this impairment is a primary feature of the syndrome or simply a reflection of the episodic hypoglycemia that occurs in association with glucocorticoid deficiency is difficult.
References- Triple A syndrome. https://ghr.nlm.nih.gov/condition/triple-a-syndrome
- Allgrove (AAA) Syndrome. https://emedicine.medscape.com/article/919360-overview
- Dysautonomia Information Page. https://www.ninds.nih.gov/Disorders/All-Disorders/Dysautonomia-Information-Page
- Hyperhidrosis. https://dermnetnz.org/topics/hyperhidrosis
- M.A. Callejas, R. Grimalt and E. Cladellasa. Hyperhidrosis Update. Actas Dermo-Sifiliográficas. 2010; 101(2):110-118.
- Schlereth T, Dieterich M, Birklein F. Hyperhidrosis–causes and treatment of enhanced sweating. Dtsch Arztebl Int. 2009;106(3):32-37. doi:10.3238/arztebl.2009.0032 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2695293
- Peripheral neuropathy. https://www.mayoclinic.org/diseases-conditions/peripheral-neuropathy/symptoms-causes/syc-20352061
- Autonomic neuropathy. https://www.mayoclinic.org/diseases-conditions/autonomic-neuropathy/symptoms-causes/syc-20369829
- Li W, Gong C, Qi Z, Wu DI, Cao B. Identification of AAAS gene mutation in Allgrove syndrome: A report of three cases. Exp Ther Med. 2015 Oct. 10 (4):1277-1282.
- Alhussaini B, Gottrand F, Goutet JM, Scaillon M, Michaud L, Spyckerelle C, et al. Clinical and manometric characteristics of Allgrove syndrome. J Pediatr Gastroenterol Nutr. 2011 Sep. 53(3):271-4.
- Moschos MM, Margetis I, Koehler K, Gatzioufas Z, Huebner A. New ophthalmic features in a family with triple A syndrome. Int Ophthalmol. 2011 Jun. 31(3):239-43.
- Vallet AE, Verschueren A, Petiot P, Vandenberghe N, Nicolino M, Roman S, et al. Neurological features in adult Triple-A (Allgrove) syndrome. J Neurol. 2012 Jan. 259(1):39-46.
- Chu ML, Berlin D, Axelrod FB. Allgrove syndrome: documenting cholinergic dysfunction by autonomic tests. J Pediatr. 1996 Jul. 129(1):156-9.
- Alakeel A, Raynaud C, Rossi M, Reix P, Jullien D, Souillet AL. [Allgrove syndrome]. Ann Dermatol Venereol. 2015 Feb. 142 (2):121-4.





{kind=link}