Legius syndrome
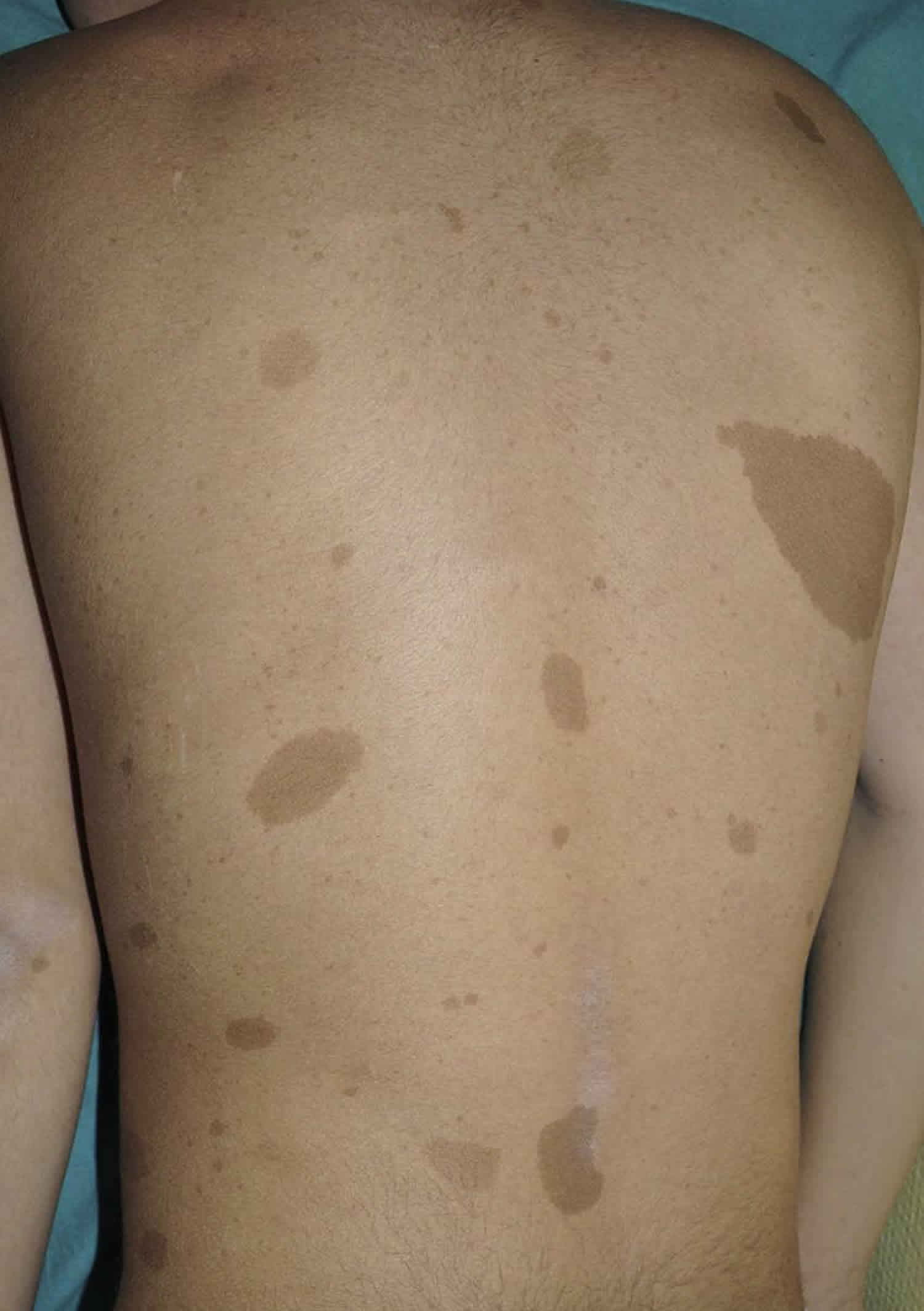
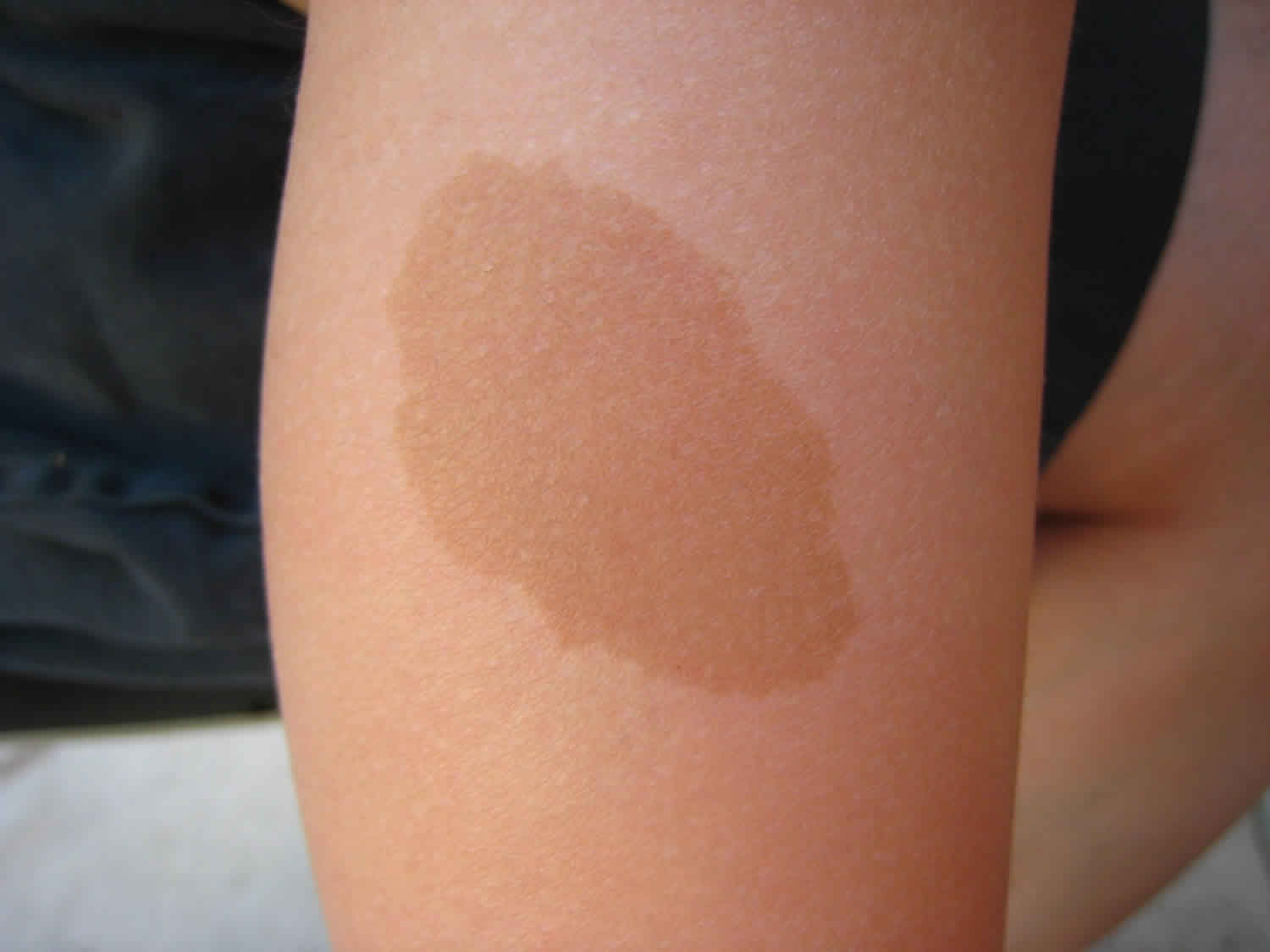
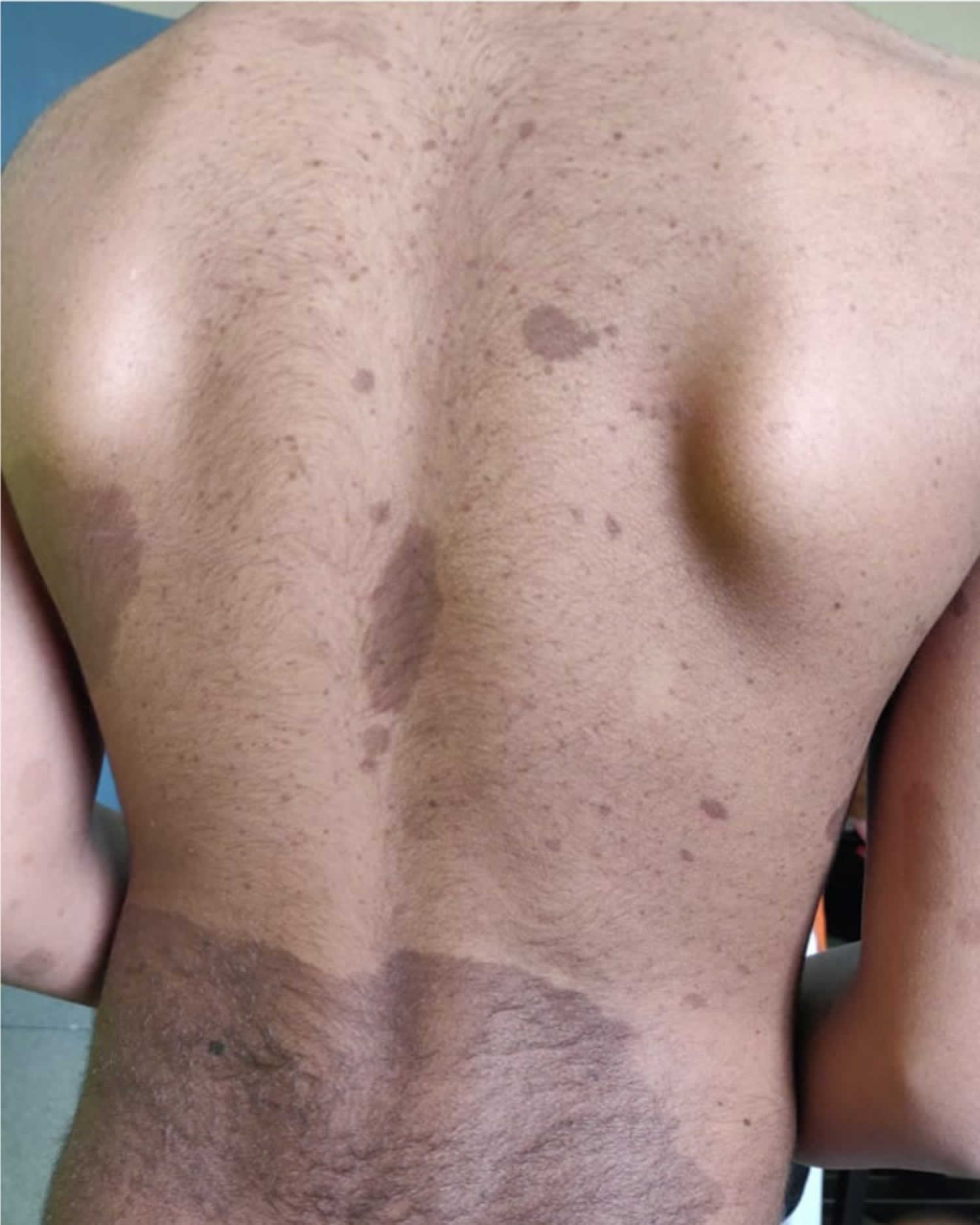
Legius syndrome also known as neurofibromatosis type 1-like syndrome, is a rare genetic skin pigmentation condition characterized by changes in skin coloring (pigmentation). Almost all affected individuals have multiple cafe-au-lait spots, which are flat light-brown patches on the skin that are darker than the surrounding area (see Figure 1 below). Another pigmentation change, freckles in the armpits and groin, may occur in some affected individuals. Legius syndrome may occur in anyone who has at least one biological parent with genetically confirmed Legius syndrome 1.
Other signs and symptoms of Legius syndrome may include an abnormally large head (macrocephaly) and unusual facial characteristics. Although most people with Legius syndrome have normal intelligence, some affected individuals have been diagnosed with learning disabilities, attention-deficit disorder (ADD), or attention-deficit-hyperactivity disorder (ADHD) 1.
Many of the signs and symptoms of Legius syndrome also occur in a similar disorder called neurofibromatosis type 1 (NF1). It can be difficult to tell the two disorders apart in early childhood. However, the features of the two disorders differ later in life.
Legius syndrome was first described in 2007 by Brems and colleagues 2, who identified that a heterozygous mutation in SPRED1 gene was responsible of this mild neurofibromatosis phenotype. After 2007, more than 200 cases have been reported 3 and it has been demonstrated that up to 2% of patients fulfilling diagnostic criteria for neurofibromatosis type 1 (NF1), without NF1 gene mutations, have instead SPRED1 mutations 4. SPRED1 acts as a negative regulator of RAS pathway and interacts with neurofibromin, the product of the NF1 gene 5. Up to date, many different allelic variants have been found, but no genotype-phenotype correlation has been noted 6.
The prevalence of Legius syndrome is not known. Fewer than 300 cases have been reported to date. Many individuals with Legius syndrome are likely misdiagnosed because the signs and symptoms of Legius syndrome are similar to those of neurofibromatosis type 1 (NF1) 7. The incidence of neurofibromatosis type 1 is reported to be 1 per 3000, and about 2% of patients fulfilling diagnostic criteria for neurofibromatosis type 1 are found to have the genetic mutation underlying Legius syndrome (SPRED1).
Figure 1. Cafe-au-lait spots
Legius syndrome causes
Mutations in the SPRED1 gene on chromosome 15q14 cause Legius syndrome. Nearly 100 different mutations in this gene have been identified. The SPRED1 gene provides instructions for making the Spred-1 protein. This protein controls (regulates) an important cell signaling pathway called RAS-MAPK signal transduction pathway that is involved in the growth and division of cells (proliferation), the process by which cells mature to carry out specific functions (differentiation), cell movement, and the self-destruction of cells (apoptosis). Mutations in the SPRED1 gene lead to a nonfunctional protein that can no longer regulate the pathway, resulting in overactive signaling. It is unclear how mutations in the SPRED1 gene cause the signs and symptoms of Legius syndrome.
The proportion of cases related to de novo mutations (spontaneous mutations where no one in the family has it before and it appears to be a new thing in the family) is not yet known. No genotype-phenotype correlations have been found.
Legius syndrome inheritance pattern
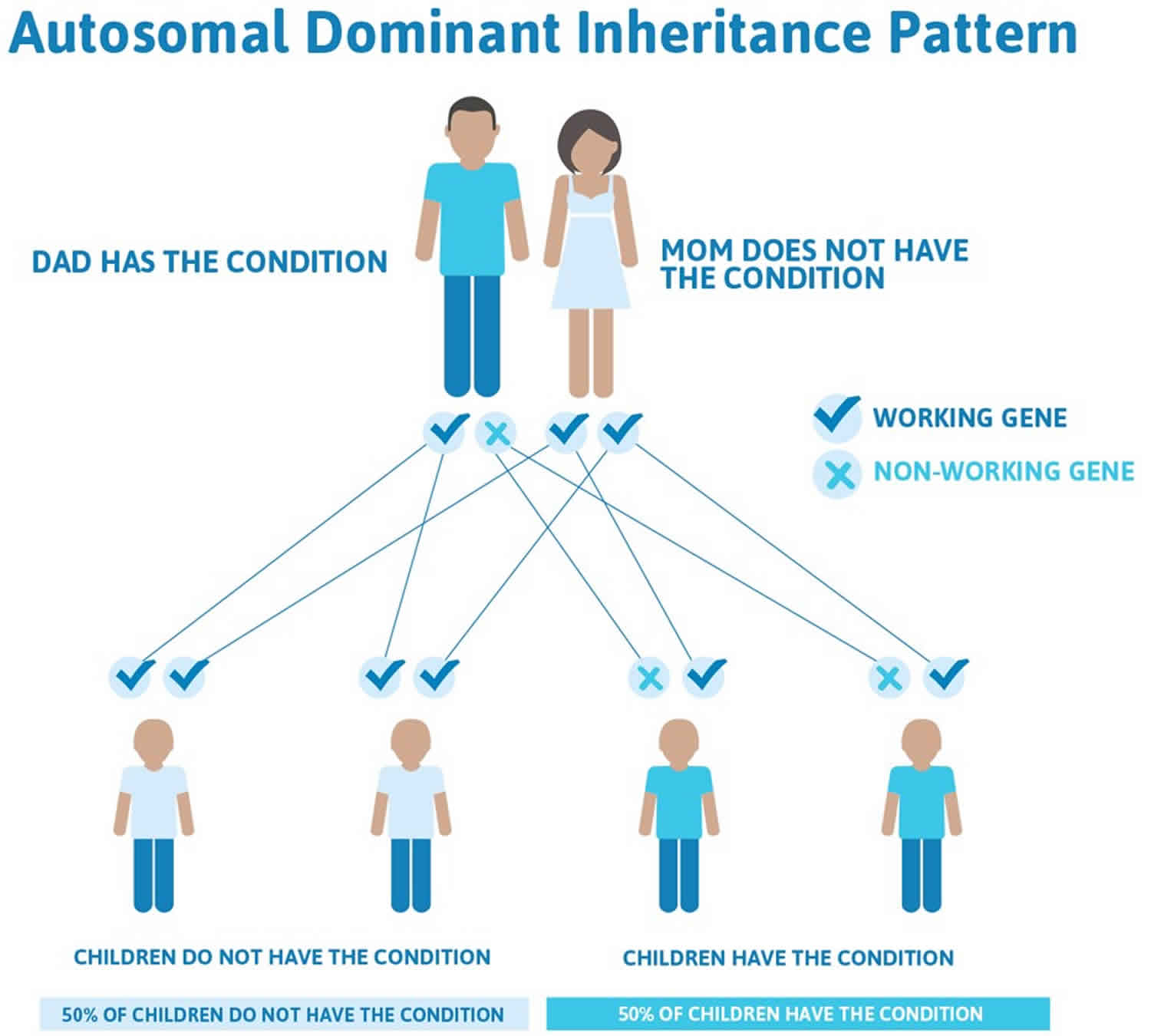
Legius syndrome is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
Often autosomal dominant conditions can be seen in multiple generations within the family. If one looks back through their family history they notice their mother, grandfather, aunt/uncle, etc., all had the same condition. In cases where the autosomal dominant condition does run in the family, the chance for an affected person to have a child with the same condition is 50% regardless of whether it is a boy or a girl. These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
- When one parent has the abnormal gene, they will pass on either their normal gene or their abnormal gene to their child. Each of their children therefore has a 50% (1 in 2) chance of inheriting the changed gene and being affected by the condition.
- There is also a 50% (1 in 2) chance that a child will inherit the normal copy of the gene. If this happens the child will not be affected by the disorder and cannot pass it on to any of his or her children.
Figure 2 illustrates autosomal dominant inheritance. The example below shows what happens when dad has the condition, but the chances of having a child with the condition would be the same if mom had the condition.
Figure 2. Legius syndrome autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Legius syndrome symptoms
The clinical features of Legius syndrome vary significantly in nature and severity from one person to another. The clinical presentation of Legius syndrome is very similar to that of neurofibromatosis type 1 (NF1). Almost all patients with Legius syndrome present with multiple café-au-lait spots sometimes associated with intertriginous freckling, but lack Lisch nodules, optic pathway gliomas, bone abnormalities, neurofibromas or other tumor manifestations 1. The number of these café-au-lait spots tends to increase with age during childhood 8. Other less common manifestations include short stature, macrocephaly, Noonan-like facies, pectus excavatum/carinatum, lipomas, hypopigmented macules, vascular lesions, learning disabilities, attention deficit/hyperactivity disorder (ADHD), and developmental delay.
Other common skin features may include:
- Freckles in the armpit and groin regions
- Lipomas 1.
Non-skin features may include:
- Macrocephaly
- Unusual facial characteristics, similar to Noonan syndrome
- Short stature
- Pectus excavatum (sunken breastbone) or pectus carinatum (protruding breastbone) 1.
Neuropsychiatric features may include:
- Learning difficulties
- Developmental delay
- Attention-deficit hyperactivity disorder (ADHD) 1.
The intellectual disabilities are generally less severe in patients with Legius syndrome compared to patients with neurofibromatosis type 1 9.
Importantly, Legius syndrome does not cause neurofibromas and Lisch nodules, which are typically seen in neurofibromatosis type 1 1.
Legius syndrome diagnosis
Legius syndrome is difficult to diagnose on a clinical basis alone given its similar cutaneous clinical presentation to other disorders with multiple café-du-lait spots.
Where Legius syndrome is suspected, genetic testing can be used to confirm the diagnosis. The diagnosis of Legius syndrome is established with suggestive findings and a heterozygous pathogenic variant in SPRED1 identified by molecular genetic testing. This involves:
- Sequence analysis of SPRED1
- Deletion/duplication analysis (if sequence analysis is unremarkable)
- A multi-gene panel for SPRED1 and other genes of interest 1.
Suspicious findings that may warrant genetic testing for Legius syndrome include:
- Café-au-lait macules without other clinical features, suggesting neurofibromatosis type 1
- A parent with café-au-lait macules without clinical features, again suggesting neurofibromatosis type 1 1.
Legius syndrome treatment
Children with Legius syndrome should be under regular screening and evaluation for developmental delay, cognitive impairment, and behavioral issues 1.
The treatment of Legius syndrome is primarily supportive and should be focused on the specific problems of the affected individual.
Appropriate management, if indicated, may include:
- Physical, speech, and/or occupational therapy
- Behavioral-modification therapy and pharmacological therapy (ie, for ADHD) 1.
Legius syndrome prognosis
Patients with Legius syndrome have a generally good prognosis with appropriate problem-based management.
An affected individual must consider the 50% risk of genetic transmission to each child.
References- Legius E, Stevenson D. Legius Syndrome. 2010 Oct 14 [Updated 2020 Aug 6]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK47312
- Brems H, Chmara M, Sahbatou M, Denayer E, Taniguchi K, Kato R, Somers R, Messiaen L, De Schepper S, Fryns JP, Cools J, Marynen P, Thomas G, Yoshimura A, Legius E. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nat Genet. 2007;39:1120–1126. doi: 10.1038/ng2113
- Brems H, Legius E. Legius syndrome, an Update. Molecular pathology of mutations in SPRED1. Keio J Med. 2013;62(4):107–112. doi: 10.2302/kjm.2013-0002-RE
- Messiaen L, Yao S, Brems H, Callens T, Sathienkijkanchai A, Denayer E, Spencer E, Arn P, Babovic-Vuksanovic D, Bay C, Bobele G, Cohen BH, Escobar L, Eunpu D, Grebe T, Greenstein R, Hachen R, Irons M, Kronn D, Lemire E, Leppig K, Lim C, McDonald M, Narayanan V, Pearn A, Pedersen R, Powell B, Shapiro LR, Skidmore D, Tegay D, et al. Clinical and mutational spectrum of Neurofibromatosis type 1-like syndrome. JAMA. 2009;302(19):2111–8. doi: 10.1001/jama.2009.1663
- Stowe IB, Mercado EL, Stowe TR, Bell EL, Oses-Prieto JA, Hernández H, Burlingame AL, McCormick F. A shared molecular mechanism underlies the human rasopathies Legius syndrome and Neurofibromatosis-1. Genes Dev. 2012;26(13):1421–1426. doi: 10.1101/gad.190876.112
- Benelli E, Bruno I, Belcaro C, Ventura A, Berti I. Legius syndrome: case report and review of literature. Ital J Pediatr. 2015;41:8. Published 2015 Feb 8. doi:10.1186/s13052-015-0115-9 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4323213
- Legius syndrome. https://ghr.nlm.nih.gov/condition/legius-syndrome
- Legius syndrome. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=137605
- Benelli E, Bruno I, Belcaro C, Ventura A, Berti I. Legius syndrome: case report and review of literature. Ital J Pediatr. 2015;41:8. Published 2015 Feb 8. doi:10.1186/s13052-015-0115-9





{kind=link}