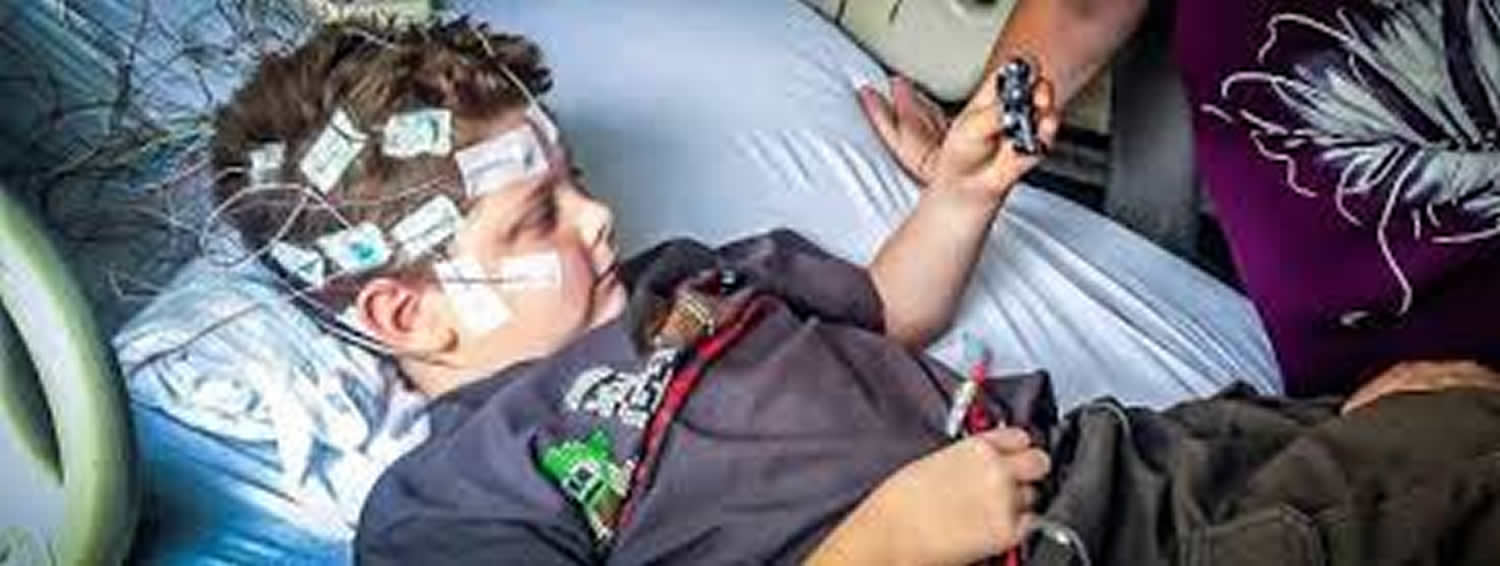
What is Lennox Gastaut syndrome
Lennox-Gastaut syndrome is a severe form of epilepsy characterized by recurrent seizures (epilepsy) that begin in infancy or early childhood, usually between ages 3 and 5 1. Affected individuals have multiple types of seizures that vary among individuals, a particular pattern of brain activity (called slow spike-and-wave) measured by a test called an electroencephalogram (EEG), and impaired mental abilities.
Lennox‐Gastaut syndrome is an age‐specific epileptic encephalopathy, characterized by epileptic seizures, slow spike‐waves in the waking electroencephalogram (EEG) and fast rhythmic bursts during sleep, psychomotor delay and personality disorders 2.
Lennox-Gastaut syndrome affects an estimated 1 to 2 per million people. Lennox-Gastaut syndrome accounts for less than 5 percent of all cases of childhood epilepsy. For unknown reasons, Lennox-Gastaut syndrome affects males slightly more often than females.
The following seizure types and EEG findings are associated with Lennox Gastaut syndrome 2:
- Tonic seizures (stiffening of the body, upward eye gaze, dilated pupils, and altered breathing patterns)
- Atypical absences (staring spells)
- Atonic seizures (brief loss of muscle tone, which could cause abrupt falls)
- Myoclonic seizures (sudden muscle jerks), and
- Generalized tonic-clonic seizures (muscle stiffness and rhythmic jerking).
1. Tonic axial seizures
These are the hallmark seizure type, and may be axial, appendicular or global, symmetrical, or unilateral. They consist of flexion of the neck and body, extension of the arms and legs, and contraction of the facial muscles. There may be associated apnoea, eye rolling and facial flushing. Consciousness is usually impaired. They are diurnal and nocturnal. They are usually brief, lasting seconds. The EEG shows discharges of fast bilateral bursts, particularly seen during sleep, predominately anteriorly or on the vertex.
2. Atypical absence seizures
These occur in the majority of cases and are frequently subtle. Loss of consciousness may be incomplete, allowing the individual to continue ongoing activities. However they are often accompanied by loss of muscle tone, myoclonic jerks and drooling 3. EEG shows irregular diffuse slow spike‐wave activity at 2 to 2.5 Hz.
3. Atonic seizures
These are less frequent than the tonic axial seizures. They are manifested by a rapid loss of tone that may involve a sudden head drop or fall to the ground and are associated with polyspikes and slow waves, or diffuse spike waves on the EEG 2.
4. Myoclonic seizures
These are considered rare and in children with prominent myoclonic jerks alternative diagnoses such as myoclonic astatic epilepsy or Dravet’s syndrome should be considered. Rarely they may precede atonic attacks as myoclonic‐atonic attacks. They can be associated with slow waves, polyspike waves, diffuse rapid spike waves or brief discharges predominantly in the anterior regions.
5. Tonic‐clonic and partial‐onset seizures
These are less commonly seen in Lennox Gastaut syndrome than other epilepsies, but may nonetheless be present in a minority of cases.
6. Status epilepticus and non‐convulsive status
Occurs in approximately two‐thirds of patients and usually consists of continuous absence seizures punctuated by recurring tonic seizures; may be difficult to recognize.
EEG (electroencephalogram)
This is abnormal in the vast majority of cases showing 2‐ to 2.5‐Hz slow spike‐wave discharges over both hemispheres with multifocal spikes and spike waves predominating in the frontal and temporal areas. In addition, the presence of fast (10 Hz) rhythms associated with tonic attacks or sometimes with minimal or no clinical manifestations, mainly during non‐REM sleep, is considered a necessary criteria by some authors. However, in some people the characteristic EEG abnormalities may be variable and even transient.
In Lennox-Gastaut syndrome, the most common seizure type is tonic seizures, which cause the muscles to stiffen (contract) uncontrollably. These seizures typically occur during sleep; they may also occur during wakefulness and cause sudden falls. Also common are atypical absence seizures, which cause a very brief partial or complete loss of consciousness. Additionally, many affected individuals have episodes called drop attacks, which cause sudden falls that can result in serious or life-threatening injuries. Drop attacks may be caused by sudden loss of muscle tone (described as atonic) or by abnormal muscle contraction (described as tonic). Other types of seizures have been reported less frequently in people with Lennox-Gastaut syndrome. Seizures associated with Lennox-Gastaut syndrome often do not respond well to therapy with anti-epileptic medications.
Although each seizure episode associated with Lennox-Gastaut syndrome is usually brief, more than two-thirds of affected individuals experience prolonged periods of seizure activity (known as status epilepticus) or episodes of many seizures that occur in a cluster.
Most children with Lennox-Gastaut syndrome have intellectual disability or learning problems even before seizures begin. These problems may worsen over time, particularly if seizures are very frequent or severe. Some affected children develop additional neurological abnormalities and behavioral problems. Many also have delayed development of motor skills such as sitting and crawling. As a result of their seizures and intellectual disability, most people with Lennox-Gastaut syndrome require help with the usual activities of daily living. However, a small percentage of affected adults live independently.
People with Lennox-Gastaut syndrome have a higher risk of death than their peers of the same age. Although the increased risk is not fully understood, it is partly due to poorly controlled seizures and injuries from falls.
Lennox-Gastaut syndrome can be caused by a variety of conditions, including brain malformations, tuberous sclerosis, perinatal asphyxia, severe head injury, central nervous system infection, and inherited genetic and inherited degenerative or metabolic conditions. In 30-35 percent of individuals, no cause can be found.
Generally, three findings are necessary for the diagnosis: multiple generalized seizure types; a slow spike-and-wave pattern (less than 2.5 Hz) on electroencephalogram (EEG); and cognitive dysfunction 4. The International League Against Epilepsy Task Force most recently classified the disorder as an epileptic encephalopathy. Epileptic encephalopathies are a group of disorders in which seizure activity leads to progressive cognitive dysfunction.
Lennox-Gastaut syndrome can be very difficult to treat. A combination of seizure medications and other treatments may be used to improve seizure control and other associated conditions.
Other treatment options include dietary therapy with the ketogenic diet, vagus nerve stimulation, and epilepsy surgery (typically a corpus callostomy, which involves severing the band of nerve fibers that connect the two halves of the brain to prevent seizures from spreading). Medication may be combined with the other treatments to optimize seizure control. Children who improve initially may later show tolerance to a drug or have uncontrollable seizures.
How will the seizures in Lennox Gastaut syndrome keep changing?
Characteristics of Lennox Gastaut syndrome are likely to change in the adult years. As the patient reaches their 20s, only 30-50% maintain the characteristic EEG (slow spike-wave pattern) and clinical characteristics 5. In 33% of patients with cryptogenic Lennox Gastaut syndrome (no underlying cause) and 55% with symptomatic Lennox Gastaut syndrome (known cause), the characteristic Lennox Gastaut syndrome features disappeared with age 6. Seizure types often change or decrease in frequency, while behavior disturbances may persist or change with age.
Lennox Gastaut syndrome prognosis
The prognosis for individuals with Lennox-Gastaut syndrome varies. There is no cure for the disorder. Complete recovery, including freedom from seizures and normal development, is very rare.
Psychomotor delay and neuropsychiatric symptoms occur in 90% of people with Lennox Gastaut syndrome. Some children have delayed development prior to the onset of their seizures as part of a predisposing condition, for example West’s syndrome (infantile spasms). Nevertheless even in these individuals further regression of development is often seen after the onset of Lennox Gastaut syndrome. Language is frequently affected, with both slowness in ideation and expression in addition to difficulties of motor dysfunction. Severe behavioural disorders are nearly always present. There is also a tendency for psychosis to develop with time. The long‐term prognosis is poor; although the epilepsy often improves, complete seizure freedom is rare and conversely the mental and psychiatric disorders tend to worsen with time 2.
Lennox Gastaut syndrome life expectancy
The mortality rate associated with Lennox-Gastaut Syndrome ranges from 3 to 7%, with many deaths related to accidents 7. People with Lennox- Gastaut Syndrome have an increased risk of death compared to their peers of the same age. Although the increased risk is not fully understood, it is partly due to poorly controlled seizures and injuries from falls 8.
Lennox Gastaut syndrome symptoms
The symptoms of Lennox-Gastaut syndrome usually begin during infancy or childhood, most often between 3 to 5 years of age. Multiple types of seizures, which are basically electrical disturbances in the brain, affect children with Lennox-Gastaut syndrome. Most affected individuals experience multiple types of seizures, multiple times throughout the day. As affected individuals grow older, the types and frequency of seizure activity may change.
The most common types of seizures associated with Lennox-Gastaut syndrome are tonic and atonic seizures. Tonic seizures cause increased muscle tone and muscle stiffness. They are characterized by sustained muscle contractions that can cause mild abnormalities such as a slight bend of the body and brief interruption of breathing or more significant problems such as muscle spasms of the face and flexion or extension of the arms and legs. Affected children may extend their arms over their heads similar to a ballet dancer. Tonic seizures are usually brief (lasting between a few seconds and a minute) and are especially prevalent at night during sleep, but can also occur during the day. There is usually a brief loss of consciousness during a tonic seizure. Tonic seizures that occur when awake can cause affected individuals to fall.
Atonic seizures cause a sudden loss of muscle tone and limpness. They can cause the head to drop or nod, problems with posture or sudden falls. Atonic seizures are also known as drop attacks. Atonic seizures can lead to injuries of the head and face because of sudden, unexpected falls. When sitting, affected individuals may collapsed forward or backward at the waist. Atonic seizures may only partially affect consciousness and usually last only a few seconds.
A third type of seizure commonly associated with Lennox-Gastaut syndrome is atypical absence seizures. This type of seizure is associated with a period of unconsciousness usually marked by unresponsive staring. Absence seizures usually begin and end abruptly and the affected individual usually resumes activity with no memory of the episode. Absence seizures do not cause convulsions and may be so mild that they go unnoticed. They usually last only a couple to several seconds. If the child is developmentally delayed, the parents may only notice a subtle change in function or responsiveness.
Additional types of seizures can affect individuals with Lennox-Gastaut syndrome less often. These include myoclonic seizures, which are characterized by abnormal, jerky movements and may occur alone or in conjunction with atypical absence seizures; tonic-clonic seizures (once known as grand mal seizures), which last a couple of minutes and are characterized by stiffening of the limbs and then jerking of the limbs and face; and partial seizures, which involve electrical abnormalities in a limited area of the brain and come in a variety of forms. Some individuals with Lennox-Gastaut syndrome experience prolonged, uninterrupted seizure activity that lasts for more than 30 minutes (nonconvulsive status epilepticus). Nonconvulsive status epilepticus may be associated with a child being unaware or inattentive and, in some cases, may be so subtle that is goes unnoticed. Nonconvulsive status epilepticus requires medical intervention.
Intelligence is usually, but not always, affected in children with Lennox-Gastaut syndrome. Affected children may experience varying degrees of cognitive dysfunction and delays in reaching developmental milestones such as sitting, crawling or walking. Children with Lennox-Gastaut syndrome may develop normally before the onset of seizures, and then lose previously acquired skills (psychomotor regression).Because the seizures associated with Lennox-Gastaut syndrome are usually resistant to treatment, intellectual impairment and learning problems may worsen over time. Children with Lennox-Gastaut syndrome may also develop behavioral problems ranging from hyperactivity and irritability to autistic symptoms and psychosis.
In some cases, individuals with Lennox-Gastaut syndrome may have been initially affected by infantile spasms. Infantile spasms, which are also known as West syndrome, are characterized by sudden, involuntary contractions of the head, neck, and trunk and/or uncontrolled extension of the legs and/or arms.
Lennox Gastaut syndrome causes
Lennox-Gastaut syndrome can have many different causes. Lennox-Gastaut syndrome likely has a genetic component, although the specific genetic factors are not well understood.
In approximately 70-80 percent of patients, Lennox-Gastaut syndrome has an identifiable cause. Most cases of Lennox-Gastaut syndrome are caused by an existing neurological abnormality. These cases may be referred to as symptomatic Lennox-Gastaut syndrome, can be associated with brain injuries that occur before or during birth, reduced oxygen supply that occurs before birth (perinatal hypoxia), problems with blood flow in the developing brain, stroke, brain infections such as encephalitis or meningitis, or other disorders affecting the nervous system. Lennox-Gastaut syndrome can also result from brain malformations such as forms of cortical dysplasia, which are abnormalities in the outer surface of the brain (cerebral cortex). Many people with Lennox-Gastaut syndrome have a history of epilepsy beginning in infancy (infantile spasms) or a related condition called West syndrome before developing the features of Lennox-Gastaut syndrome.
In addition, mutations in several genes have been associated with Lennox-Gastaut syndrome, each in a small number of affected individuals. These genes are involved in the function of nerve cells in the brain, but it is unclear how changes in them contribute to the development of Lennox-Gastaut syndrome. The condition can also occur as part of a genetic disorder such as tuberous sclerosis complex.
Approximately 17-30 percent of individuals with Lennox-Gastaut syndrome have a previous history of West syndrome. In general, these cases tend to be more severe.
In about 10 percent of affected individuals, Lennox-Gastaut syndrome may also be classified as cryptogenic, in which the cause is unknown or cannot be determined after evaluation. These individuals have no history of seizures, neurological problems, or delayed development. Cryptogenic cases are presumed to result from an unidentified condition (secondary Lennox-Gastaut syndrome). Individuals with cryptogenic Lennox-Gastaut syndrome do not have a previous history of seizure activity, prior neurological problems or cognitive impairment before the development of the disorder. Cryptogenic cases generally have a later onset than symptomatic cases.
In some cases of Lennox-Gastaut syndrome no associated condition is present or presumed and the cause of the disorder is unknown.
Although the cause of Lennox-Gastaut is known in most cases, the exact underlying mechanisms that ultimately bring about the various seizures that characterize the disorder are unknown. Researchers have not discovered any genes that are associated with Lennox-Gastaut syndrome, although the disorder may have a genetic component that contributes to its development. More research is necessary to determine the specific factors, including any potential genetic factors that are involved in the development of Lennox-Gastaut syndrome.
Lennox Gastaut syndrome inheritance pattern
Most cases of Lennox-Gastaut syndrome are sporadic, which means they occur in people with no history of the disorder in their family. When Lennox-Gastaut syndrome is associated with a genetic change, the mutation is usually not inherited but occurs as a random (de novo) event during the formation of reproductive cells (eggs or sperm) in an affected person’s parent or in early embryonic development. However, 3 to 30 percent of people with this condition have a family history of some type of epilepsy, indicating that inherited genetic factors may play a role in some cases of Lennox-Gastaut syndrome.
Lennox Gastaut syndrome diagnosis
Lennox-Gastaut syndrome is defined as having a clinical triad that must be identified for a diagnosis. This triad consists of multiple seizures of different types, a distinctive EEG brain wave pattern (slow [1.5- to 2.5-Hz] spike-and-wave pattern) and some degree of cognitive impairment and behavioral abnormalities. However, these symptoms may not all be present at the onset of the disorder, making an accurate diagnosis of Lennox-Gastaut syndrome difficult. The wide variety of potential causes of Lennox-Gastaut syndrome also complicates the diagnosis.
A diagnosis of Lennox-Gastaut syndrome is usually made based upon a thorough clinical evaluation, a detailed patient history and a complete physical and neurological evaluation including advanced imaging techniques, such as electroencephalography (EEG) and magnetic resonance imaging (MRI). During an EEG, the brain’s electrical impulses are recorded. In individuals with Lennox-Gastaut syndrome, such EEG testing typically reveals the distinctive brain wave pattern (slow [1.5- to 2.5-Hz] spike-and-wave pattern). During a MRI scan, three-dimensional images are produced that reflect the brain’s anatomy; such scanning helps physicians examine brain structure and potentially locate the cause of the seizure activity.
Lennox Gastaut syndrome treatment
Lennox-Gastaut syndrome can be very difficult to treat. A combination of seizure medications and other treatments may be used to improve seizure control and other associated conditions. However, no specific therapy for Lennox-Gastaut syndrome is effective in all cases and the disorder has proven particularly resistant to most therapeutic options. The three main forms of treatment of Lennox-Gastaut syndrome are anti-epileptic drugs, dietary therapy (typically the ketogenic diet) or device/surgery (vagus nerve stimulation therapy or corpus callosotomy). Rarely, resective surgery is an option.
Treatment may require the coordinated efforts of a team of specialists. Pediatricians, neurologists, pediatric neurologists, surgeons, and/or other healthcare professionals may need to systematically and comprehensively plan an affected child’s treatment. Families need to work with healthcare professionals to develop a treatment plan that covers various potential situations such as seizure emergencies, routine medical illnesses, or what to do if an affected individual misses a dosage of medication. Families should also keep a list of which medications can possibly worsen Lennox-Gastaut syndrome. An affected individual’s treatment regimen will need repeated revisions throughout a person’s life as the types and frequency of seizures may change and the effectiveness of a particular therapy can lessen. Other healthcare providers are frequently consulted, including social workers, neuropsychologists, psychiatrists and rehabilitation services (occupational, physical and speech therapy).
Anti-epileptic drugs are usually given to individuals with Lennox-Gastaut syndrome, but the individual response is highly variable. In some cases, it is possible that treatment with anti-epileptic drugs may help reduce or control various types of seizure activity associated with Lennox Gastaut syndrome. The medication valproate is generally considered a first-line therapy for various seizure types. However, because individuals with Lennox-Gastaut syndrome have different types of seizures, they often require therapy with multiple types of anti-epileptic drugs. Such medications may include clobazam, clonazepam, felbamate (closely monitored), lamotrigine, rufinamide, topiramate, and cannabidiol. However, the optimum treatment for Lennox Gastaut syndrome remains uncertain and no study to date has shown any one drug to be highly efficacious; rufinamide, lamotrigine, topiramate and felbamate may be helpful as add‐on therapy, clobazam may be helpful for drop seizures. Until further research has been undertaken, clinicians will need to continue to consider each patient individually, taking into account the potential benefit of each therapy weighed against the risk of adverse effects 9. In addition, anti-epileptic drugs may be associated with significant side effects, especially in individuals who receive multidrug, high-dose regimens. anti-epileptic drugs can also become less effective over time. Being on multiple medications, which may cause sedation, can sometimes worsen seizure control.
Valproate (valproic acid) is generally considered the first-line therapy for Lennox-Gastaut syndrome because it is effective against a wide spectrum of seizures. Valproate is usually first given alone (monotherapy) and if ineffective another drug such as lamotrigine, topiramate, rufinamide or clobazam may be added.
A variety of specific drugs have been approved by the Food and Drug Administration (FDA) for the treatment of Lennox-Gastaut syndrome including topiramate (Topamax). Topiramate has been approved as an add-on (adjunctive) therapy for children and adults. The drug is manufactured by Ortho-McNeil Neurologics.
The FDA has also approved the anticonvulsant drug felbamate (Felbatol) for the treatment of seizures in children with Lennox-Gastaut syndrome. Due to the occurrence of rare but serious side effects from the drug, physicians should become familiar with the medication and know how to monitor for side-effects before prescribing the medication. This drug, while effective, is not typically first or second line because of the side-effect concerns.
In addition, the FDA has approved the lamotrigine (Lamictal) as an add-on (adjunctive) therapy (i.e., as a medication to be used in association with other appropriate anticonvulsant medications) for the treatment of generalized seizures associated with Lennox-Gastaut syndrome.
In 2008, the FDA approved rufinamide (Banzel) for the use as an adjunctive (add-on) treatment for seizures associated with Lennox-Gastaut syndrome. Rufinamide decreases seizure frequency in some individuals and seems to be particularly effective for atonic or drop attack seizures.
Clobazam (Onfi) was approved by the FDA in 2011 to treat the seizures associated with Lennox-Gastaut syndrome.
In June 2018 the U.S. Food and Drug Administration approved cannabidiol (Epidolex, derived from marijuana) for the treatment of seizures associated with Lennox-Gastaut syndrome in individuals ages 2 and older. The drug contains only small amount of the psychoactive element in marijuana and does not induce euphoria associated with the drug.
Additional therapies that have been used to treat individuals with Lennox-Gastaut syndrome include the ketogenic diet, Vagus nerve stimulation Therapy and various surgical techniques. These options are generally reserved for individuals who do not respond to or no longer respond to drug therapy, and are typically combined with drug therapy (The exception is dietary therapy, which is typically added to drug therapy, but rarely maybe successful by itself in this population.).
The ketogenic diet may reduce seizure activity in some individuals with Lennox-Gastaut syndrome. The ketogenic diet is a high fat, low carbohydrate diet that makes the body burn fat for energy instead of sugar (glucose). It is a strict diet that requires rigid compliance and commitment. The ketogenic diet can have side effects and individuals following the diet should be routinely monitored by their physicians and a trained nutritionist. The effectiveness of the ketogenic diet varies from one individual to another. Researchers do not understand why the diet is effective in treating seizures or why it is effective for some people, but not others.
Some individuals with Lennox-Gastaut syndrome, especially those who have not responded to other forms of therapy, may be treated with surgical therapies including complete corpus callosotomy or vagus nerve stimulation.
A corpus callosotomy is a surgical procedure in which the cerebral hemispheres are disconnected by cutting the corpus callosum, which is a large bundle of nerves that connects the two halves (hemispheres) of the brain and allows them to share information. This procedure does not include the cutting of brain tissue. This procedure is generally reserved for individuals who suffer from intractable seizures that lead to injuries (e.g., drop seizures or frequent generalized tonic-clonic seizures) or are potentially life-threatening. It is most effective for atonic, tonic and tonic-clonic seizures.
Vagus nerve stimulation is a procedure in which a device called a pulse generator is inserted into the chest and a wire is run underneath the skin to the vagus nerve in the neck. The pulse generator is similar to a pacemaker and transmits mild, electrical impulses to the brain via the vagus nerve. These impulses prevent seizures from occurring. The intensity and timing of the nerve impulses are determined based upon each individual’s needs. This is combined with drug therapy and most effective for drop seizures and generalized tonic-clonic seizures.
References- Lennox-Gastaut Syndrome Information Page. https://www.ninds.nih.gov/Disorders/All-Disorders/Lennox-Gastaut-Syndrome-Information-Page
- Beaumanoir A, Blume W. The Lennox‐Gastaut syndrome. In: Roger J, Bureau M, Dravet C, Tassinari C, Wolf P editor(s). Epileptic Syndromes in Infancy, Childhood and Adolescence. 4th Edition. London: John Libby Eurotext, 2005:125‐48.
- Aicardi J. Lennox‐Gastaut syndrome. In: Wallace editor(s). Epilepsy in Children. First Edition. London: Chapman & Hall Medical, 1996:249‐62.
- Lennox-Gastaut Syndrome. https://rarediseases.org/rare-diseases/lennox-gastaut-syndrome
- Lennox-Gastaut syndrome in adulthood: clinical and EEG features. Epilepsy Res. 2010 May;89(2-3):271-7. doi: 10.1016/j.eplepsyres.2010.01.012. Epub 2010 Feb 10. https://www.ncbi.nlm.nih.gov/pubmed/20149600
- Long-term prognosis of Lennox-Gastaut syndrome. Epilepsia. 1996;37 Suppl 3:44-7. https://www.ncbi.nlm.nih.gov/pubmed/8681912
- Lennox gastaut syndrome, review of the literature and a case report. Head & Face Medicine20084:9 https://doi.org/10.1186/1746-160X-4-9
- Increased risk of death among children with Lennox-Gastaut syndrome and infantile spasms. J Child Neurol. 2010 Apr;25(4):441-7. doi: 10.1177/0883073809348355. Epub 2009 Dec 18. https://www.ncbi.nlm.nih.gov/pubmed/20023065
- Hancock EC, Cross JH. Treatment of Lennox‐Gastaut syndrome. Cochrane Database of Systematic Reviews 2013, Issue 2. Art. No.: CD003277. DOI: 10.1002/14651858.CD003277.pub3 https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD003277.pub3/full





{kind=link}