What is lymphangiectasia
Lymphangiectasia also called lymphangiectases, refers to swollen lymphatic vessels caused by a wide range of scarring processes. Lymphangiectasia occurs as a consequence of lymphatic damage by an external cause, leading to obstruction of local lymphatic drainage. Lymphangiectasia are also termed acquired lymphangiomas. Acquired lymphangiectasia or acquired lymphangiomas arises after the lymphatic glands have been blocked or injured most commonly occur in adults as a late complication of mastectomy and radiation therapy. Patients usually present with numerous translucent vesicles in a chronic lymphedematous area several years after surgery with or without radiation therapy. Lymphangiectasia are often complicated by pain, copious fluid drainage, and recurrent attacks of cellulitis. Lymphangiectasia pose no potential for malignant transformation.
Nomenclature has not been consistent 1. Some authors apply the terms acquired lymphangioma and lymphangioma circumscriptum interchangeably. In both conditions, the typical cutaneous lesions are groups of small translucent vesicles, often compared with frog spawn. Although both share similar clinical and histologic features, the authors believe that they are 2 distinct entities. The term acquired lymphangioma (lymphangiectasia) is used when dilated lymphatic channels arise following damage to previously normal deep lymphatics, whereas lymphangioma circumscriptum is used when lymphatic channel dilation occurs because of congenital malformations of the lymphatic system involving the skin and the subcutaneous tissues. Acquired lymphangiectasia is more common in adults than in children. More generally, the condition occurs in patients between their fifth and seventh decades of life.
The pathogenesis of lymphangiectasia is not known 2; however, the vesicles associated with lymphangiectasia are suggested to represent saccular dilations of local superficial lymphatics. These vesicles develop secondary to increased intralymphatic pressure as a result of buildup of lymph in the superficial vessels caused by damage to previously normal deep lymphatics. This mechanism explains the accompanying lymphedema seen in most patients with lymphangiectasia. The lymphedema usually arises as a result of obstructed lymphatic drainage after mastectomy, radiation therapy, or tumor mass compression. Lymphangiectasia may also result from abnormal dermal structure and function, possibly related to photoaging and steroid-related atrophy 3.
Lymphangiectasia causes
Acquired lymphangiectasia can arise from a large number of external factors that cause structural damage to previously normal deep lymphatics.
Acquired lymphangiectasia may be associated with:
- Traumatic injury interrupting lymphatic drainage
- Lymph node dissection
- Removal of lymph glands for cancer
- Destruction of lymph glands by cancer.
Lymphangiectasia have been reported following radical mastectomy with or without radiation therapy 4; irradiation alone for various malignancies 5; metastatic lymph node obstruction; and various scarring processes, such as infections, keloids, scleroderma, and scrofuloderma 6. Acquired cutaneous lymphangiectasia has been described due to cervical cancer therapy 7.
Lymphangiectasia have been described on the penis and the scrotum after removal of a sacrococcygeal tumor; they may also arise on the vulva and the inner thigh after surgery for cervical or other pelvic cancers 8. Pelvic lymphatic obstruction may produce acquired vulvar lymphangiomas 9. Scrotal lymphangiectasia may be evident following scrofuloderma 10.
Acquired lymphangiectasia have been reported in the genital region of elderly patients without evidence of lymphatic obstruction.
Garcia-Doval et al 11 reported lymphangiectasis in a patient with cirrhotic ascites.
Lymphangiectasia symptoms
Acquired lymphangiectasia presents as clear or haemorrhagic papules often described like frogspawn. The small bumps tend to ooze a clear or milky fluid after minor injury.
Lymphangiectasia may affect the following sites:
- The armpits after a radical mastectomy
- The thighs after lymph node clearance for cancer
- The vulva after surgery or radiotherapy to cervical cancer
- The penis and scrotum after removal of a tumour at the base of the spine
Patients with lymphangiectasia typically present with numerous fluid-filled vesicles in a chronic lymphedematous area several years after surgery, more commonly due to a malignancy. The cutaneous lesions of lymphangiectasia can range from clear, fluid-filled blisters to smooth, flesh-colored nodules, often appearing along an incisional scar.
Coexisting lymphedema is present in most patients with acquired lymphangiectasia.
Patients can present with localized wetness or copious drainage of clear or milky fluid from ruptured vesicles. Pain and recurrent cellulitis are complications associated with lymphangiectasia.
Acquired lymphangiectasia may be associated upper limb lymphedema secondary to mastectomy, radiotherapy, keloids, chronic lymphedema, or scleroderma 12. Sometimes, they resolve in weeks without any treatment 13.
Rarely, lymphangiectasia may occur in pregnancy and spontaneously regress with childbirth 14. Acquired lymphangiectasia of the glans may also occur after circumcision 15.
Lymphangiectasia complications
Acquired lymphangiectasia can be painful. The poor lymphatic drainage may lead to bacterial infection (cellulitis).
Lymphangiectasia diagnosis
The diagnosis of lymphangiectasia is primarily based on clinical history and conventional light microscopy findings of skin biopsy confirming the presence of the thin-walled lymphatic vessels. Clinically, lymphangiectasia consists of several clusters of translucent, thick-walled, fluid-filled vesicles. The vesicles typically measure 2-10 mm in diameter. The affected area appears to be speckled by numerous translucent vesicles with normal-appearing skin among the lesions (see the image below). Some lymphangiectasia lesions may become pedunculated with a hyperkeratotic verrucous surface mimicking a wart 16.
Conjunctival lymphangiectasis may be evident as intermittent conjunctival swelling and dilated conjunctival vessels on ocular examination 17.
Although many patients without chronic lymphedema have been reported, it is a common physical finding in patients with acquired lymphangioma. Diffuse infiltration of subcutaneous tissue by lymphangiectasia may produce painless swelling at sites such as the subclavicular fossa 18.
Lymphangiectasia may also be evident on the penis. Benign transient lymphangiectasia of the penis may be evident 19. Acquired lymphangiectasia of the glans may occur after circumcision 15.
Imaging and other tests may be necessary to establish whether there is a leak or blockage in the lymphatics.
Immunohistochemistry studies are important in differentiating lymphangiectasia from hemangiomas in difficult cases. Factor VIII–related antigen testing demonstrates positive results in the endothelial cells of hemangiomas but negative or weakly positive results in the endothelium of lymphangiomas. Ulex europaeus I testing results are positive in the endothelial cells of lymphangiectases and hemangiomas. Immunoperoxide staining with QBEnd10 (anti-CD34) antibodies shows positive reactivity results only in endothelial cells lining the blood vessels of the dermis.
Lymphangiectasia treatment
Acquired lymphangioma can be difficult to treat. No medical care has been proven to be effective for acquired lymphangiectasia because the responsible lymphatic vessels must be either excised or sealed to prevent recurrence. The affected area must be kept scrupulously clean daily with topical antibacterial agents and applying mupirocin ointment or silver sulfadiazine cream to reduce the risk of infection. Compression may be useful to reduce swelling caused by lymphoedema. Sometimes, electrosurgery, laser therapy 20, sclerotherapy 21, cryotherapy or surgical excision may be attempted.
Follow-up care is essential for early treatment of lymphangiectasia recurrences. Lymphangiosarcoma (Stewart-Treves syndrome) may occur in chronic edematous limbs, and early detection is critical. At times, severe recurrent cellulitis may warrant hospitalization in patients with lymphangiectasia, especially in patients who are immunocompromised. Intravenous antibiotic is required in patients with severe cellulitis.
Surgical care
Many surgical treatment modalities have been advocated in the care of lymphangiectasia; these modalities include electrodesiccation, laser therapy, sclerotherapy 21, cryotherapy, and surgical excision 16. In a small case series, liquid nitrogen cryotherapy was an effective treatment for localized conjunctival lymphangiectasia 22. Successful treatment of conjunctival lymphangiectasia has been described using a high-frequency radiowave electrosurgical device 23. Daily compression through bandaging or hosiery, in accessible areas, has yielded acceptable results. Although no consensus exists concerning therapy for lymphangiectasia, vulvar lymphangiectasia may be effectively handled with carbon dioxide laser therapy 24. Acquired balanic lymphangiectasia after circumcision was successfully treated with a 2940-nm erbium-doped yttrium aluminium garnet laser 25.
Lymphangiectasia prognosis
Lymphangiectasia has a good prognosis because most conditions respond well to treatment modalities 2. Lymphangiectasia is a nonfatal disease associated with a high tendency for local recurrence after treatment. Lymphangiectasia may be complicated by chronic copious drainage, pain, and recurrent bouts of cellulitis. In addition, lesions are often cosmetically undesirable.
Acquired lymphangiomas are not believed to have malignant potential, although associated chronic lymphedema places the patient at risk for lymphangiosarcoma (Stewart-Treves syndrome), which is an aggressive tumor with a dismal prognosis. Cutaneous angiosarcoma has also been reported in massive localized lymphedema in morbidly obese patients 26.
Conjunctival lymphangiectasia
Conjunctival lymphangiectasia is also called conjunctival lymphangiectasis, is a condition in which conjunctival swelling occurs as a result of dilated conjunctival lymphatic channels, most notably on the bulbar conjunctiva. The appearance at the slit lamp will show cystic appearing clear or yellowish, elevated conjunctival lymphatic channel(s) separated by translucent septate walls. Although the actual cause is unknown, the presumed underlying etiology is obstructed lymphatic channels. Conjunctival lymphangiectasia is thought of as benign, but can be associated with local inflammation, disruption of the tear film/dellen, or secondary hemorrhage (hemorrhagic lymphangiectasia of the conjunctiva) 27.
Intestinal lymphangiectasia
Intestinal lymphangiectasia is a rare, benign protein-losing gastroenteropathy characterized by dilatation of the intestinal lymphatics and loss of lymph fluid into the gastrointestinal tract, leading to the development of hypoproteinemia, edema, lymphocytopenia, hypogammaglobinemia, and immunologic anomalies 28. In addition to the loss of other serum components (eg, lipids), iron and certain trace metals may also be affected 29.
Intestinal lymphangiectasia can be primary (ie, congenital), in which case it affects children and young adults (mean age of onset, 11 y). The diagnosis in these cases often occurs during the first decade of life, with the first manifestations being persistent diarrhea and peripheral edema. This condition can also be secondary to other disease states, thereby affecting older adults 28. In a series from Japan, the average age at onset was 22.9 years.
Traditionally, protein-losing gastroenteropathies have been classified into three groups (depending on the mechanism of their cause) that include the following 30: (1) those causing mucosal damage leading to increased permeability to protein (usually not involving mucosal ulcerations), (2) those with mucosal erosions and/or ulcerations, and (3) those in which protein loss is secondary to mechanical lymphatic obstruction.
Intestinal lymphangiectasia causes
The following conditions can cause intestinal lymphangiectasia 31:
- Abdominal or retroperitoneal carcinoma
- Lymphoma
- Heart diseases (eg, constrictive pericarditis, congestive heart syndrome)
- Crohn disease
- Mesenteric tuberculosis
- Sarcoidosis
- Whipple disease
- Chronic pancreatitis
- Scleroderma
- Celiac disease
- Systemic lupus erythematosus (SLE)
- Retroperitoneal fibrosis
- Intestinal endometriosis
- Sclerosing mesenteritis
- Lymphenteric fistula
Intestinal lymphangiectasia symptoms
Patients usually present in childhood with edema and nonbloody diarrhea. Edema may be unilateral or bilateral, depending on the site of the lesion. Edema in primary intestinal lymphangiectasia is usually bilateral, whereas the secondary type often manifests as unilateral edema and is caused by various neoplastic, infiltrative, and inflammatory lesions affecting one side of the body.
If the onset of disease occurs during the early part of the first decade of life, growth retardation usually ensues.
Frequently, steatorrhea, malabsorption, lymphocytopenia, and hypogammaglobulinemia are present.
Despite hypogammaglobulinemia, opportunistic infections rarely occur, although lymphocytopenia predisposes patients to abnormal cellular immunities, including homograft rejection and cutaneous anergy.
Ascites (often chylous ascites) and chylous pleural effusions are also reported in patients with long-standing lymphangiectasia.
Intestinal lymphangiectasia complications
Primary intestinal lymphangiectasia is associated with an increased risk of lymphoma.
Fibrotic entrapment of the small bowel is reported in patients with congenital intestinal lymphangiectasia.
Oral manifestations include gingivitis caused by poor lymphocytic function and enamel defects caused by poor calcium absorption.
Intestinal lymphangiectasia diagnosis
Primary intestinal lymphangiectasia
Peripheral edema is noted on physical examination in patients with primary intestinal lymphangiectasia 32.
Macular edema on funduscopic examination has been reported and is a cause of reversible blindness 33.
Pachydermoperiostosis has been associated with protein-losing enteropathy due to intestinal lymphangiectasia 34. Pachydermoperiostosis is a rare hereditary disease characterized by clubbing of the fingers, periostosis, and skin changes.
Secondary intestinal lymphangiectasia
Secondary intestinal lymphangiectasia may involve multiple physical findings, depending on the cause.
Laboratory Studies
Serum protein levels
The most common laboratory finding in intestinal lymphangiectasia is hypoproteinemia. Hypoalbuminemia is most prominent, and lymphocytopenia and hypogammaglobulinemia (eg, immunoglobulin A [IgA], immunoglobulin G [IgG], immunoglobulin M [IgM]) are also prominent. Cholesterol levels are not usually elevated.
Alpha1-antitrypsin levels
In random dry stools, levels of alpha1-antitrypsin has been used to indirectly measure protein leakage in the gastrointestinal (GI) tract. Alpha1-antitrypsin is negligibly broken down by intestinal proteases and, thus, is excreted intact in the stool. However, although measurement of stool alpha1-antitrypsin may serve as a good screening examination for protein loss, several studies have shown poor correlation between the value of alpha1-antitrypsin in the stool and its clearance measurement. In part, this is because of increased degradation of alpha1-antitrypsin in different milieus. For example, the breakdown of alpha1-antitrypsin is higher in environments where the pH level is less than 3, as in the stomach or small bowel in hyperacidity states.
The most specific test for protein loss in the alimentary tract is direct measurement of alpha1-antitrypsin clearance from plasma. Values greater than 24 mL/day in patients without diarrhea (diarrhea increases alpha1-antitrypsin clearance) and over 56 mL/day in those with diarrhea indicate protein loss in the gastrointestinal tract. GI bleeding has also been shown to increase alpha1-antitrypsin clearance as a result of whole blood loss.
Imaging studies
Double-contrast radiographs of the small bowel may be helpful, because they may show thickened folds due to intestinal edema from hypoproteinemia, nodular protrusions, and absence of mucosal ulcerations.
Ultrasonography and computed tomography (CT) scanning are also useful in identifying dilated intestinal loops, regular and diffuse thickening of the intestinal walls, plical hypertrophy, and mesenteric edema. CT scans may help show circumferential thickening of the small bowel wall with low attenuation (<30 H).
Multiple detector computed tomography (MDCT) scanning after direct lympangiography appears to provide an accurate evaluation and diagnosis of primary intestinal lymphangiectasia. In a retrospective study of 55 affected patients, all of whom underwent multidetector CT after direct lymphangiography, investigators noted that multiple detector computed tomography identified intra-intestinal, extra-intestinal, and lymphatic vessel abnormalities, including different degrees of intestinal dilatation, small bowel wall thickening, ascites, mesenteric edema, mesenteric nodules, lumbar trunk and intestinal trunk reflux 35.
Procedures
Endoscopy
Repeatedly, the role of endoscopy has been proven useful. Small bowel enteroscopy not only helps detect mucosal changes suggestive of the disease but also allows acquisition of histologic samples to establish a diagnosis 36.
White villi and/or spots (dilated lacteals), white nodules, and submucosal elevations are observed. Xanthomatous plaques are often visualized.
Capsule endoscopy
Capsule endoscopy has also been used to help identify the characteristic changes of intestinal lymphangiectasia not reachable with standard endoscopy.
Jejunal biopsy
Jejunal biopsy establishes a definitive diagnosis and shows dilation of mucosal and submucosal lymphatic channels. To increase the diagnostic yield, large biopsy forceps should be used when available. In addition, because of the patchy involvement of the small bowel, obtaining multiple biopsy samples from different areas is recommended.
Intestinal biopsy results reveal the characteristic dilatation of the lymph vessels of the mucosa and submucosa without any evidence for inflammation.
Intestinal lymphangiectasia treatment
Treatment of patients with primary intestinal lymphangiectasia involves control of symptoms with the use of dietary, pharmaceutical, and behavioral modifications, such as the following:
- Dietary modifications include a low-fat diet and substitution of long-chain fatty acids with medium-chain fatty acids 37. A logical step might be to reduce the amount of salt intake, although this has not been proven to decrease edema. In a literature review, Desai et al 37 investigated the efficacy of a medium-chain fatty acid diet in the treatment of primary intestinal lymphangiectasia in 27 patients compared to results of 28 control patients. In the fatty acid group, complete symptom resolution occurred in 17 patients (63%), compared to 10 patients (35.7%) in the non-fatty acid group. In addition, there was 1 death (3.7%) in the fatty acid group, whereas the second group experienced 5 (17.8%) deaths. The authors concluded that a medium-chain fatty acid diet is a valid option for the treatment of pediatric patients 37.
- Medications that may be used include over-the-counter remedies (eg, bulking agents, drugs to control diarrhea). Treatment of secondary causes of lymphangiectasia target the underlying disease. In several reports, octreotide has demonstrated efficacy in refractory cases 38. In a case report of a 42-year-old man with primary intestinal lymphangiectasia, Troskot et al found that only octreotide provided therapeutic resolution 39. Following the use of a slow-release form of octreotide, the patient had a partial remission. A case of intestinal lymphangiectasia refractory to octreotide and nutritional manipulations was successfully treated with tranexamic acid. This patient presented with refractory anemia due to continued gastrointestinal [GI] blood loss.
- Pollack and colleagues 40 reported a case of primary intestinal lymphangiectasia in a female patient with tuberous sclerosis complex (TSC) and a tuberous sclerosis complex-2 mutation in whom a trial of the mTOR inhibitor rapamycin was very effective. There was improvement in her clinical symptoms of PIL as well as in abnormal laboratory values. The investigators concluded that these findings suggest that PIL is a rare manifestation of TSC, thereby justifying the use of mTOR inhibitors in future studies 40. Tan et al 41 also reported a patient in whom primary intestinal lymphangiectasia was the first manifestation of tuberous sclerosis complex.
Treatment of patients with secondary causes of intestinal lymphangiectasia involves management of the underlying disease.
Intestinal lymphangiectasia prognosis
The clinical course of intestinal lymphangiectasia is highly variable with about 23% of patients showing improvement and 64% remaining unchanged; the mortality rate is 13%.
For patients with primary intestinal lymphangiectasia with an onset early in life (usually during the first decade), growth retardation usually occurs. The prognosis of patients with secondary intestinal lymphangiectasia depends on the extent and severity of the underlying disease.
Morbidity/mortality
Morbidity is related to the pathophysiology of this disease. Edema and diarrhea are predominant clinical features; however, the following negative sequelae are also observed:
- Lymphocytopenia, hypogammaglobulinemia
- Hypoalbuminemia, hypocalcemia, trace metal deficiency
- Chylous pleural effusions, ascites (Chylous ascites and transudative ascites are reported.)
Primary intestinal lymphangiectasia
Primary intestinal lymphangiectasia also called Waldmann’s disease, is a rare digestive disorder in which the lymph vessels supplying the lining of the small intestine are enlarged. Primary intestinal lymphangiectasia is characterized by dilated intestinal lacteals resulting in lymph leakage into the small bowel lumen and responsible for protein-losing enteropathy leading to lymphopenia, hypoalbuminemia and hypogammaglobulinemia 42. The cause of primary intestinal lymphangiectasia is still unknown. Primary intestinal lymphangiectasia is usually diagnosed before three years of age but is sometimes diagnosed later in life. Primary intestinal lymphangiectasia signs and symptoms include swelling of the legs and abdominal discomfort, loss of lymphatic fluid into the gastrointestinal tract, protein-losing enteropathy, too little albumin in the blood, reduced levels of antibodies, and immunodeficiency. Edema may be moderate to severe with anasarca and includes pleural effusion, pericarditis or chylous ascites. Fatigue, abdominal pain, weight loss, inability to gain weight, moderate diarrhea or fat-soluble vitamin deficiencies due to malabsorption may also be present 42. In some patients, limb lymphedema is associated with primary intestinal lymphangiectasia and is difficult to distinguish lymphedema from edema. Exsudative enteropathy is confirmed by the elevated 24-hour stool α1-antitrypsin clearance. Primary intestinal lymphangiectasia treatment involves a special long-term diet 43.
Primary intestinal lymphangiectasia causes
The cause of primary intestinal lymphangiectasia is unknown. Multiple affected family members have been reported rarely.
Primary intestinal lymphangiectasia signs and symptoms
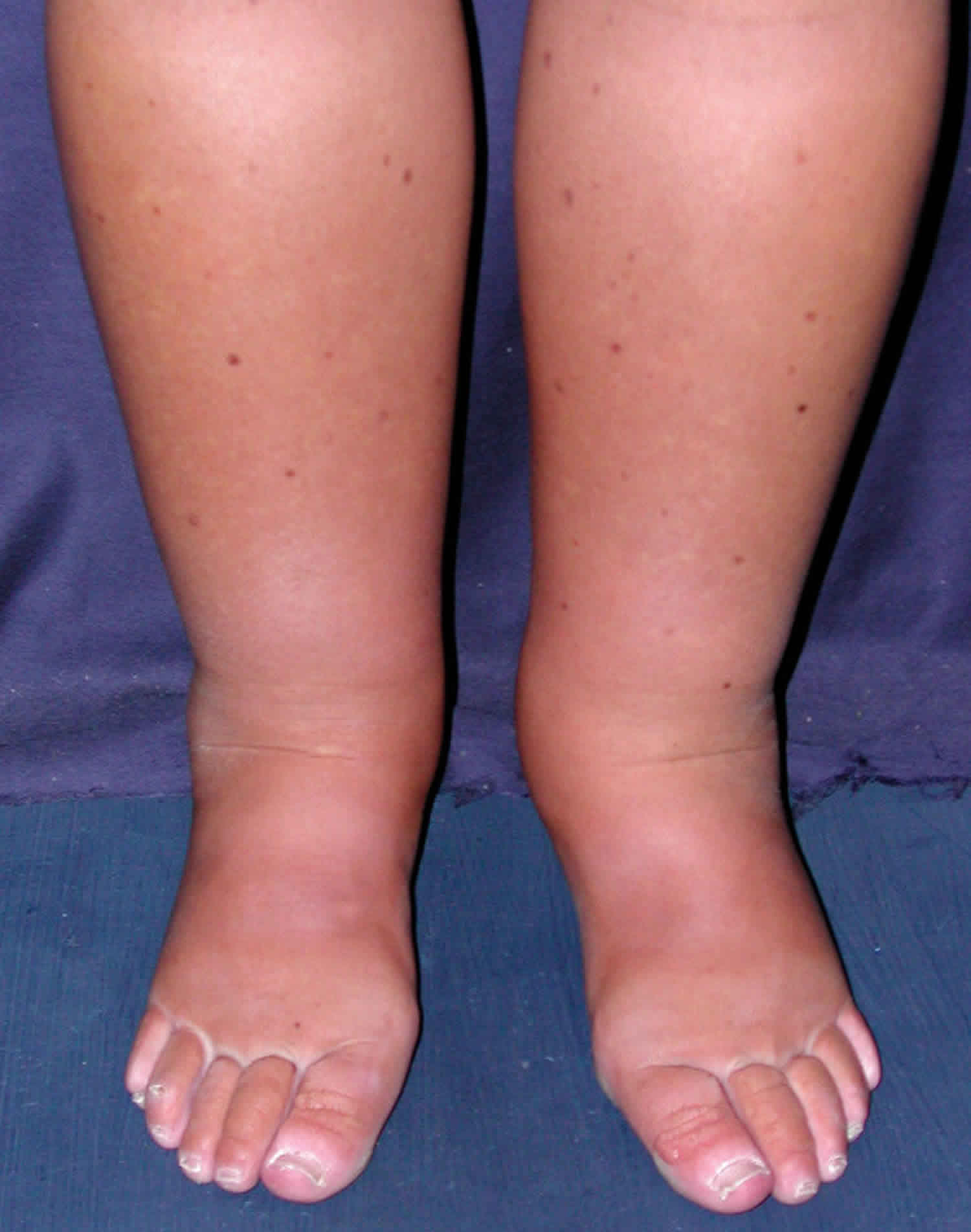
The most obvious sign of the disorder is moderate to severe swelling in the lower limbs, eventually face, abdomen and external genitalia due to fluid retention (edema). Fluid is retained because the blood protein (albumin) levels are low. Lymphedema may also be associated and not easy to differentiate from edema.
Abdominal pain and/or nausea, vomiting and diarrhea may also be present. Affected individuals may experience fatigue, weight loss, and an inability to gain weight in childhood. The blood lymphocyte count is usually low as are blood protein (albumin, globulins because protein in the lymph leaks into the intestine and the feces called exudative enteropathy) and blood cholesterol levels (because cholesterol from food is not properly absorbed).
Swelling of the membrane surrounding the heart (pericarditis) and fluid in the chest (pleural effusion) or ascites (abdominal effusion) can occur. Extreme generalized swelling of the body (anasarca) can be a rare life threatening complication in children.
Primary intestinal lymphangiectasia diagnosis
The diagnosis of primary intestinal lymphangiectasia is made by viewing the intestine with a flexible scope (endoscope), removing tissue samples from several areas (biopsy) and examining these tissues for signs of abnormal dilation. This exam is rarely normal and videocapsule endoscopy may be useful when endoscopic findings are not contributive. Intestinal lymph oozing may be confirmed by the increased clearance of alpha-1 antitrypsin in the stools.
Primary intestinal lymphangiectasia may be suspected on a prenatal ultrasound if edema of lower limbs or generalized edema is noted.
Primary intestinal lymphangiectasia treatment
Treatment of primary intestinal lymphangiectasia may include a strictly low-fat long-term diet supplemented by medium-chain triglycerides to supply essential fatty acids and nutrients associated with fat-soluble vitamin such as vitamin D. The need for dietary control appears to be permanent, because clinical and biochemical findings reappear after low-fat diet interruption. The administration of water pills (diuretics) may sometimes be helpful. Albumin infusion is sometimes proposed in patients with important serous effusion or uncomfortable lower limb edema. Very occasionally surgical removal of the diseased portion of the intestine may be beneficial if the damage is limited to a local area. In some cases, octreotide is proposed in association with the diet. Compression stocking is used to stabilize in associated lower limb lymphedema.
Congenital pulmonary lymphangiectasia
Congenital pulmonary lymphangiectasia is a rare developmental disorder that affects the lungs. Congenital pulmonary lymphangiectasia is present from birth and usually becomes apparent in the first few days of life with respiratory failure 44. Congenital pulmonary lymphangiectasia sometimes is apparent before birth with non-immune hydrops fetalis and pleural effusion (fluid in the lung) 44. Infants with congenital pulmonary lymphangiectasia often develop severe, potentially life-threatening, respiratory distress shortly after birth. They may also develop cyanosis caused by low oxygen levels in the blood, which causes the skin to have a bluish tint. Symptoms are due to abnormally wide (dilated) lymphatic vessels within the lungs 44. These vessels drain a fluid called lymph from different areas of the body. They are an important part of the lymphatic system, which helps the immune system protect the body against infection and disease 45.
Infants with congenital pulmonary lymphangiectasia often develop severe, potentially life-threatening, respiratory distress shortly after birth. Affected infants may also develop cyanosis, a condition marked by abnormal bluish discoloration of the skin that occurs because of low levels of circulating oxygen in the blood.
The exact cause of congenital pulmonary lymphangiectasia is unknown. It can occur as a primary or secondary disorder (due to another underlying condition). Primary congenital pulmonary lymphangiectasia occurs as an isolated defect or as part of a generalized form of lymphatic disease affecting the whole body. Secondary congenital pulmonary lymphangiectasia can occur due to a variety of heart abnormalities or lymphatic obstruction 45. Some cases of congenital pulmonary lymphangiectasia have been associated with genetic disorders 44.
Treatment aims to relieve the symptoms of the disorder and may include CPAP, intubation, and/or fluid drainage. While much of the older literature suggests a very high mortality rate, recent studies suggest that congenital pulmonary lymphangiectasia does not have as poor an outlook 45.
Are there dietary recommendations for infants with congenital pulmonary lymphangiectasia?
We are not aware of published dietary guidelines for infants with congenital pulmonary lymphangiectasia. However, as nutrition can play a role in limiting lymphatic production, considerations that have been suggested in the literature include total parenteral nutrition (TPN) and/or supplementation with medium-chain triglycerides (MCTs) 46. Enteral nutrition with medium-chain triglycerides and TPN have been successfully used 47. If chylothorax occurs, a number of components are lost, including fats (mainly phospholipids, cholesterol, and triglycerides); proteins (mainly albumin, immunoglobulins, and fibrinogen); electrolytes; and fat-soluble vitamins in concentrations similar to those found in plasma 47.
Congenital pulmonary lymphangiectasia causes
The exact cause of congenital pulmonary lymphangiectasia is unknown. Most cases occur randomly, for no apparent reason (sporadically). Congenital pulmonary lymphangiectasia may be caused by a congenital defect in the development of the lung or result from obstruction of the lymph vessels in the lungs (pulmonary lymphatics). Some cases have been associated with genetic multisystem disorders including Noonan syndrome, Turner syndrome, Hennekam syndrome or Fryns syndrome. congenital pulmonary lymphangiectasia may be inherited as dominant, recessive, or X-linked inheritance pattern.
Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
All individuals carry 4-5 abnormal genes. Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
Dominant genetic disorders occur when only a single copy of an abnormal gene is necessary to cause a particular disease. The abnormal gene can be inherited from either parent or can be the result of a new mutation (gene change) in the affected individual. The risk of passing the abnormal gene from affected parent to offspring is 50% for each pregnancy. The risk is the same for males and females.
X-linked genetic disorders are conditions caused by an abnormal gene on the X chromosome and manifest mostly in males. Females that have a defective gene present on one of their X chromosomes are carriers for that disorder. Carrier females usually do not display symptoms because females have two X chromosomes and only one carries the defective gene. Males have one X chromosome that is inherited from their mother and if a male inherits an X chromosome that contains a defective gene he will develop the disease.
Female carriers of an X-linked disorder have a 25% chance with each pregnancy to have a carrier daughter like themselves, a 25% chance to have a non-carrier daughter, a 25% chance to have a son affected with the disease and a 25% chance to have an unaffected son.
If a male with X-linked disorders is able to reproduce, he will pass the defective gene to all of his daughters who will be carriers. A male cannot pass an X-linked gene to his sons because males always pass their Y chromosome instead of their X chromosome to male offspring.
Pulmonary lymphangiectasia may occur as a secondary condition to a variety of heart (cardiac) disorders including hypoplastic left heart syndrome, cor triatum, and congenital mitral valve stenosis. Infectious agents have also been suggested as a possible cause of this disorder.
Congenital pulmonary lymphangiectasia symptoms
Much of the older medical literature suggests that congenital pulmonary lymphangiectasia has an extremely high mortality rate. However, recent studies suggest that the disorder does not have as poor a prognosis as described and that symptoms may improve with age in some cases.
Symptoms associated with congenital pulmonary lymphangiectasia often develop during the newborn (neonatal) period shortly after birth. During intrauterine period, the occurrence of non-immune hydrops fetalis associated with pleural effusion may be linked to congenital pulmonary lymphangiectasia. In some cases, symptoms develop later during infancy. The symptoms of congenital pulmonary lymphangiectasia vary in severity from case to case; in most cases, the earlier the presentation the more severe the symptoms.
Affected infants often develop severe respiratory failure and abnormal bluish discoloration of the skin that occurs because of low levels of circulating oxygen in the blood (cyanosis). Infants with congenital pulmonary lymphangiectasia may also exhibit coughing or wheezing, progressive difficulty breathing (dyspnea), coughing up blood (hemoptysis), and swelling due to accumulation of lymphatic fluid (lymphedema). Affected infants may exhibit growth failure during infancy.
Additional symptoms eventually develop including an abnormally rapid rate of breathing (tachypnea), chylous pleural effusion, known as chylothorax and recurrent respiratory infections. Chyle is a fat-laden cloudy fluid that is absorbed during digestion by the lymphatic vessels located around the intestine. Chylothorax is the accumulation of chyle or lymph fluid in the pleural cavity. Chyle is composed by fats (mainly phospholipids), proteins (albumin in particular), and a significant amount of lymphocytes. Chyle normally flows through lymphatic vessels into the upper chest (thoracic duct) and is then deposited into veins, where it mixes with blood. In some cases, chyle may accumulate in the abdomen causing chylous ascites.
Some infants with congenital pulmonary lymphangiectasia may develop heart abnormalities including a limited ability to circulate blood to the lungs and the rest of the body resulting in fluid buildup in the heart, lung and various body tissues (congestive heart failure).
Some infants with congenital pulmonary lymphangiectasia develop gastroesophageal reflux, a digestive disorder characterized by the passage or flowing back (reflux) of the contents of the stomach or small intestines (duodenum) into the esophagus. The esophagus is the tube that carries food from the mouth to the stomach (esophagus). Symptoms of gastroesophageal reflux may include a sensation of warmth or burning rising up to the neck area (heartburn or pyrosis), swallowing difficulties (dysphagia), and chest pain. This problem is a possible complication of congenital pulmonary lymphangiectasia, but not a direct consequence or typical symptom.
Congenital pulmonary lymphangiectasia diagnosis
A diagnosis of congenital pulmonary lymphangiectasia may be made based upon a thorough clinical evaluation, identification of characteristic symptoms and a variety of specialized imaging tests. These may include high resolution helical chest computed tomography (CT) scans. During CT scanning, a computer and x-rays are used to create a film showing cross-sectional images of an organ’s tissue structure. In individuals with congenital pulmonary lymphangiectasia, CT scans may reveal fluid build up in the chest cavity or lung tissue, showing the characteristic diffuse thickening of the interstitium. An imaging procedure called lymphoscintigraphy may be used to provide pictures of the lymphatic system, and it is very useful to detect diffuse aspects of congenital lymphatic dysplasia, such as lymphedema.
If pleural effusion is present, bronchoscopy and lung biopsy may be considered. During bronchoscopy, a thin, flexible tube (bronchoscope) is inserted through the nose or mouth, allowing a physician to examine the throat, larynx, trachea and lower airways. Lung biopsy involves the surgical removal and microscopic evaluation of affected lung tissue.
Congenital pulmonary lymphangiectasia treatment
The treatment of congenital pulmonary lymphangiectasia is symptomatic and supportive. Newborns with serious complications may require a variety of procedures shortly after birth including the use of a machine that creates a controlled flow of air (continuous positive airway pressure, CPAP) to an affected individual’s airways to support spontaneous breathing or the placement of a tube into the windpipe (trachea) to assist breathing performing mechanical ventilation (tracheal intubation). In some cases, at birth the immediate drainage of excess fluid from the chest cavity (pleural effusion) with assisted ventilation may improve respiratory distress. A chest tube may be inserted to drain fluid in some cases.
As affected infants age supplemental oxygen may be necessary. Symptomatic treatment for associated conditions such as coughing, wheezing and recurrent infections may also be necessary. Affected children should be monitored for the development of bronchitis. Since nutritional considerations can play a role in limiting lymphatic production, nutritional supplementation may also be recommended.
Since nutritional considerations can play a role in limiting lymphatic production, nutritional supplementation with Medium Chain Triglycerides (MCT) may also be recommended.
Congenital pulmonary lymphangiectasia prognosis
Conflicting data have been reported regarding the outcome for children with congenital pulmonary lymphangiectasia 48. The prognosis has previously been reported to be very poor, with mortality at about 100% before the 1990s 49. However, more recent reports suggest that congenital pulmonary lymphangiectasia is not a uniformly fatal condition, and that in survivors, the condition improves. This might be due to advances in modern intensive care therapy and also to the fact that severity can vary among affected children 49. It has been suggested that children with generalized lymphangiectasia who have pulmonary involvement may have less severe congenital pulmonary lymphangiectasia and a better prognosis; having a primary developmental defect of the pulmonary lymphatic vessels tends to be associated with a higher mortality 50. Those with congenital pulmonary lymphangiectasia who do survive infancy often continue to have medical problems that are characteristic of chronic lung disease. Gastroesophageal reflux and poor growth are also not uncommon during the first year of life, especially between six and twelve months of age, and are closely related to chronic lung disease 48.
References- Verma SB. Lymphangiectasias of the skin: victims of confusing nomenclature. Clin Exp Dermatol. 2009 Jul. 34(5):566-9.
- Lymphangiectasia. https://emedicine.medscape.com/article/1086917-overview
- Back SJ, Kim YJ, Choi DK, et al. Cutaneous lymphangiectasia associated with photoageing and topical corticosteroid application. Clin Exp Dermatol. 2009 Apr. 34(3):352-4.
- Sener SF, Milos S, Feldman JL, et al. The spectrum of vascular lesions in the mammary skin, including angiosarcoma, after breast conservation treatment for breast cancer. J Am Coll Surg. 2001 Jul. 193(1):22-8.
- Schwab RA, McCollough ML. Acquired vulvar lymphangiomas: a sequela of radiation therapy. Cutis. 2001 Mar. 67(3):239-40.
- Di Leonardo M, Jacoby RA. Acquired cutaneous lymphangiectasias secondary to scarring from scrofuloderma. J Am Acad Dermatol. 1986 Apr. 14(4):688-90.
- Hong JY, Jung GJ, Li K. Acquired Cutaneous Lymphangiectasia Secondary to Cervical Cancer Treatment. Am J Dermatopathol. 2019 May. 41 (5):396-397.
- Mendiratta V, Harjai B, Sardana K. Tubercular lymphadenitis with lymphangiectases of the vulva. J Eur Acad Dermatol Venereol. 2005 Mar. 19(2):264-5.
- Chang MB, Newman CC, Davis MD, Lehman JS. Acquired lymphangiectasia (lymphangioma circumscriptum) of the vulva: Clinicopathologic study of 11 patients from a single institution and 67 from the literature. Int J Dermatol. 2016 Mar 9.
- Bandyopadhyay D. Scrotal lymphangiectasia following scrofuloderma. Indian J Dermatol Venereol Leprol. 2017 May-Jun. 83 (3):397-398.
- Garcia-Doval I, de la Torre C, Losada A, Ocampo C, Rodriguez T, Cruces MJ. Acquired cutaneous lymphangiectasia in a patient with cirrhotic ascites. J Eur Acad Dermatol Venereol. 1999 Sep. 13(2):109-12.
- Rao AG. Acquired lymphangiectasis following surgery and radiotherapy of breast cancer. Indian J Dermatol. 2015 Jan-Feb. 60 (1):106.
- Valdes F, Peteiro C, Toribio J. [Acquired lymphangiectases and breast cancer]. Actas Dermosifiliogr. 2007 Jun. 98(5):347-50.
- Verma S. Pregnancy-induced lymphangiectasias of the vulva. Int J STD AIDS. 2008 Mar. 19(3):211-2.
- Zhang RZ, Yang YH, Zhu WY. Acquired lymphangiectasia of the glans following circumcision. J Dtsch Dermatol Ges. 2014 Jul. 12(7):623-4.
- Lee DJ, Jung SE, Kim YC. What is your diagnosis? acquired lymphangiectasia. Cutis. 2015 Feb. 95 (2):67, 89-90.
- Kiliç A, Gül A, Cinal A. Conjunctival Lymphangiectasis. Ophthalmic Surg Lasers Imaging. 2010 Apr 2. 1-2.
- Varron L, Vignes S, Green L, Morelec I, Broussolle C, Seve P. Recurrent lymphangiectasia of the left supraclavicular fossa: an unusual cause of paroxystic swelling. Arch Dermatol. 2011 Nov. 147(11):1337-8.
- Misson A, Deswysen AC, Tennstedt D, Muschart X. A case of transient lymphangiectasis of the penis. Acta Clin Belg. 2014 Aug. 69(4):294-5.
- Landthaler M, Hohenleutner U, Braun-Falco O. Acquired lymphangioma of the vulva: palliative treatment by means of laser vaporization carbon dioxide. Arch Dermatol. 1990 Jul. 126(7):967-8.
- Ahmed DD, Waldorf JC, Randle HW. Cutaneous lymphangiectasis: treatment with sclerotherapy. Plast Reconstr Surg. 1998 Feb. 101(2):434-6.
- Fraunfelder FW. Liquid nitrogen cryotherapy for conjunctival lymphangiectasia: a case series. Trans Am Ophthalmol Soc. 2009 Dec. 107:229-32.
- Choi SM, Jin KH, Kim TG. Successful treatment of conjunctival lymphangiectasia accompanied by corneal dellen using a high-frequency radiowave electrosurgical device. Indian J Ophthalmol. 2019 Mar. 67 (3):409-411.
- Hamida MB, Baccouche D, El Fekih N, Fazaa B, Kamoun R. Lymphangiectasia of the vulva, treatment with CO 2 laser. Indian J Dermatol Venereol Leprol. 2012 Jan. 78(1):122.
- Shi G, He XZ, Fan YM. Successful Treatment of Acquired Balanic Lymphangiectasia Following Circumcision Using 2,940-nm Erbium-Doped Yttrium Aluminium Garnet Laser. Dermatol Surg. 2016 May 26.
- Shon W, Ida CM, Boland-Froemming JM, Rose PS, Folpe A. Cutaneous angiosarcoma arising in massive localized lymphedema of the morbidly obese: a report of five cases and review of the literature. J Cutan Pathol. 2011 Apr 26.
- Conjunctival lymphangiectasis. https://webeye.ophth.uiowa.edu/eyeforum/atlas/pages/conjunctival-lymphangiectasis.htm
- Alshikho MJ, Talas JM, Noureldine SI, et al. Intestinal lymphangiectasia: Insights on management and literature review. Am J Case Rep. 2016 Jul 21. 17:512-22.
- Isa HM, Al-Arayedh GG, Mohamed AM. Intestinal lymphangiectasia in children. A favorable response to dietary modifications. Saudi Med J. 2016 Feb. 37(2):199-204.
- Levitt DG, Levitt MD. Protein losing enteropathy: comprehensive review of the mechanistic association with clinical and subclinical disease states. Clin Exp Gastroenterol. 2017. 10:147-68.
- Intestinal lymphangiectasia. https://emedicine.medscape.com/article/179571-overview
- Wang X, Jin H, Wu W. Primary intestinal lymphangiectasia manifested as unusual edemas and effusions: A case report. Medicine (Baltimore). 2016 Mar. 95(10):e2849.
- Lai Y, Yu T, Qiao XY, Zhao LN, Chen QK. Primary intestinal lymphangiectasia diagnosed by double-balloon enteroscopy and treated by medium-chain triglycerides: a case report. J Med Case Rep. 2013 Jan 14. 7(1):19.
- Sethuraman G, Malhotra AK, Khaitan BK, et al. Familial pachydermoperiostosis in association with protein-losing enteropathy. Clin Exp Dermatol. 2006 Jul. 31(4):531-4.
- Sun X, Shen W, Chen X, Wen T, Duan Y, Wang R. Primary intestinal lymphangiectasia: Multiple detector computed tomography findings after direct lymphangiography. J Med Imaging Radiat Oncol. 2017 Oct. 61(5):607-13.
- Takenaka H, Ohmiya N, Hirooka Y, et al. Endoscopic and imaging findings in protein-losing enteropathy. J Clin Gastroenterol. 2012 Aug. 46(7):575-80.
- Desai AP, Guvenc BH, Carachi R. Evidence for medium chain triglycerides in the treatment of primary intestinal lymphangiectasia. Eur J Pediatr Surg. 2009 Aug. 19(4):241-5.
- Al Sinani S, Rawahi YA, Abdoon H. Octreotide in Hennekam syndrome-associated intestinal lymphangiectasia. World J Gastroenterol. 2012 Nov 21. 18(43):6333-7.
- Troskot R, Jurcic D, Bilic A, Gomercic Palcic M, Tezak S, Brajkovic I. How to treat an extensive form of primary intestinal lymphangiectasia?. World J Gastroenterol. 2015 Jun 21. 21(23):7320-5.
- Pollack SF, Geffrey AL, Thiele EA, Shah U. Primary intestinal lymphangiectasia treated with rapamycin in a child with tuberous sclerosis complex (TSC). Am J Med Genet A. 2015 Sep. 167A (9):2209-12.
- Tan NBL, Tamblyn S, Hinds R. Primary intestinal lymphangiectasia as a first manifestation of tuberous sclerosis complex. J Pediatr Gastroenterol Nutr. 2017 Oct. 65(4):e96.
- Vignes S, Bellanger J. Primary intestinal lymphangiectasia (Waldmann’s disease). Orphanet J Rare Dis. 2008;3:5. Published 2008 Feb 22. doi:10.1186/1750-1172-3-5 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2288596
- Primary Intestinal Lymphangiectasia. https://rarediseases.org/rare-diseases/primary-intestinal-lymphangiectasia
- Toru HS, Sanhal CY, Yilmaz GT, Ozbudak IH, Mendilcioglu I, Ozbilim G. Rare congenital pulmonary malformation with diagnostic challenging: congenital pulmonary lymphangiectasia, report of four autopsy cases and review of literature. J Matern Fetal Neonatal Med. August, 2015; 28(12):1457-1460.
- Congenital Pulmonary Lymphangiectasia. https://rarediseases.org/rare-diseases/congenital-pulmonary-lymphangiectasia
- Reiterer F, Grossauer K, Morris N, Uhrig S, Resch B. Congenital pulmonary lymphangiectasis. Paediatr Respir Rev. September, 2014; 15(3):275-280.
- Carlo Bellini, Francesco Boccardo, Corradino Campisi, and Eugenio Bonioli. Congenital pulmonary lymphangiectasia. Orphanet J Rare Dis. 2006; 1:43:
- Carlo Bellini, Francesco Boccardo, Corradino Campisi, and Eugenio Bonioli. Congenital pulmonary lymphangiectasia. Orphanet J Rare Dis. 2006; 1:43:
- Reiterer F, Grossauer K, Pfleger A, Häusler M, Resch B, Eber E, Popper H, Urlesberger B. Severe primary pulmonary lymphangiectasis in a premature infant: management and follow up to early childhood. Pediatr Int. 2015; 57(1):166-169.
- Hwang JH, Kim JH, Hwang JJ, Kim KS, Kim SY. Pneumonectomy case in a newborn with congenital pulmonary lymphangiectasia. J Korean Med Sci. April, 2014; 29(4):609-613.





{kind=link}