Lymphoproliferative disorder
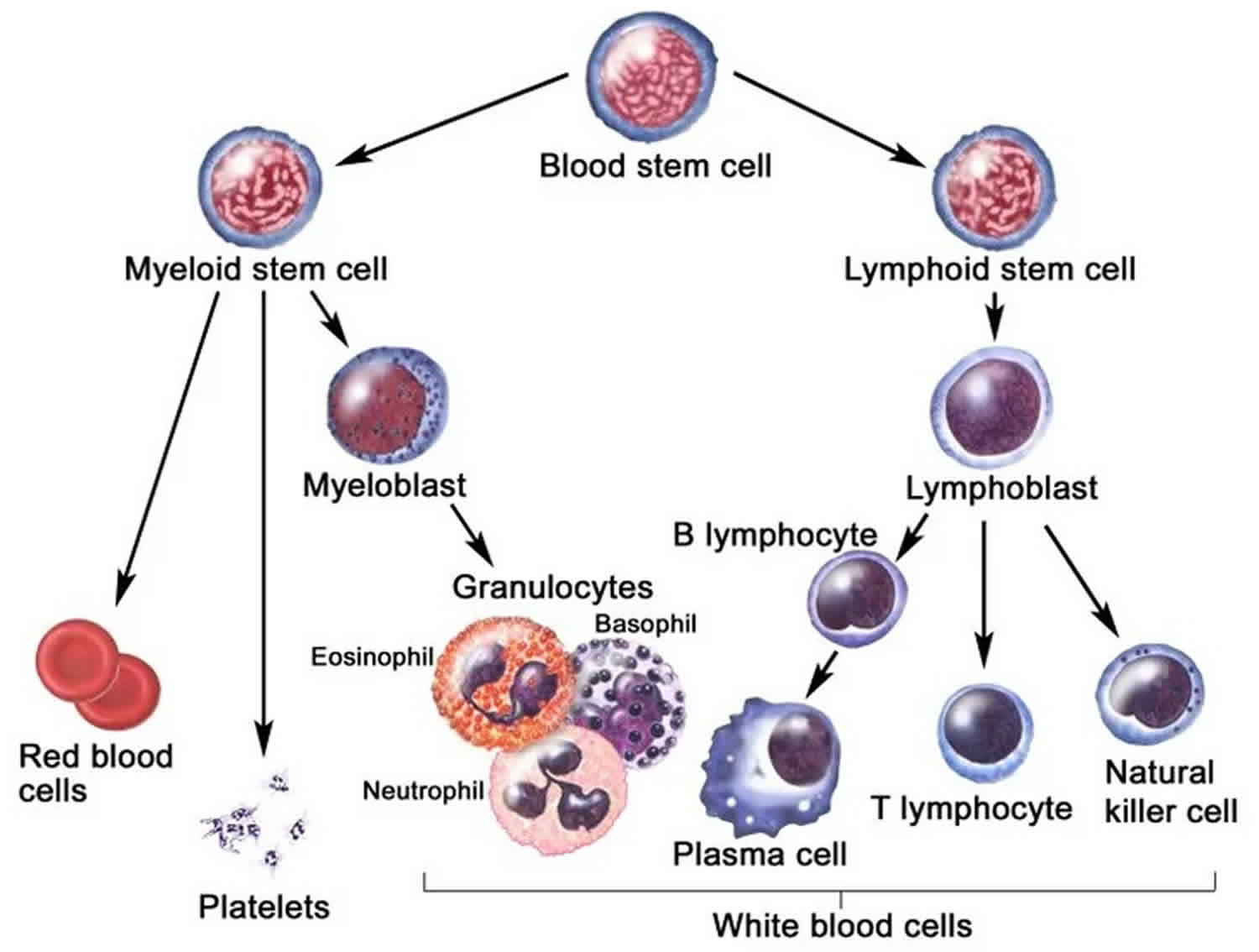
Lymphoproliferative disorder comprises a diverse group of diseases characterized by uncontrolled production of lymphocytes that cause monoclonal lymphocytosis, lymphadenopathy and bone marrow infiltration 1. Lymphoproliferative disorders are often treated like cancer. Lymphoproliferative disorder often occur in immunocompromised individuals. There are two subsets of lymphocytes: T and B cells that regenerate uncontrollably to produce immunoproliferative disorders, which are prone to immunodeficiency, a dysfunctional immune system, and lymphocyte dysregulation. Several gene mutations have been described as causes of lymphoproliferative disorder that can be iatrogenic or acquired. B-cell neoplasms are much more common than T-cell neoplasms in the United States and Europe.
The X-linked lymphoproliferative disorder is characterized by a mutation in the X chromosome that predisposes to natural killer cell lymphoproliferative disorder and T-cell lymphoproliferative disorder. Autoimmune lymphoproliferative syndrome is a type of lymphoproliferative disorder caused by a mutation in the gene that encodes for a Fas protein which is located in the long arm of chromosome 10. Males with X-linked immunodeficiency syndrome are susceptible to lymphoproliferative disorder and at risk for acquiring Epstein Barr Virus (EBV) and further development of lymphoma.
Individuals with common variable immunodeficiency, severe combined immunodeficiency, Wiskott-Aldrich syndrome, ataxia-telangiectasia, Chediak–Higashi syndrome and viral infections including HIV, are also prone to lymphoproliferative disorder. Others at risk include patients undergoing tissue transplantation and the use of immunosuppressive drugs such as cyclosporin, sirolimus, and tacrolimus. Invasive fungal infections have also been linked to this pathology 2.
The chronic lymphoproliferative disorders are immuno-morphologically and clinically diverse. Common features of these processes include various immunophenotypes (T, B, and NK cells) and terminal deoxynucleotidyl transferase negativity. The B-cell lymphocytic disorders include B-cell chronic lymphocytic leukemia, B-cell prolymphocytic leukemia, non-Hodgkin lymphoma (including mantle cell lymphoma) in leukemic phase, hairy cell leukemia and splenic lymphoma with villous lymphocytes. The T-cell chronic lymphoproliferative disorders include Sezary syndrome, T-cell prolymphocytic leukemia, adult T-cell leukemia-lymphoma, and large granulated lymphocyte leukemia 3.
Lymphoproliferative disorder causes
Epstein-Barr virus (EBV) is an etiological factor for most lymphoproliferative disorders. EBV infects 90% of people during their lives 1. Epstein Barr Virus (EBV) presumably spreads by saliva or droplets. It has an incubation period of 4 to 5 weeks. In early childhood, it causes few symptoms, but in adolescents and young adults, it may cause infectious mononucleosis.
Lymphoproliferative disorders originate when physiological mechanisms of control of proliferation of both T and B cells break down, resulting in the uncontrolled and autonomous increase of immune cells leading to lymphocytosis and lymphadenopathy, and often involvement of extranodal sites, e.g., bone marrow.
In immunocompromised patients, EBV can cause mild disease. However, in immune-suppressed transplant patients, immunosurveillance may be compromised by the lack of T cells, leading to a proliferation of EBV-infected B-lymphocytes and post-transplant lymphoproliferative disorder. Polyclonal post-transplant lymphoproliferative disorder can form tumor masses and presents with symptoms of a mass effect. Monoclonal forms of post-transplant lymphoproliferative disorder can manifest as a disseminated malignant lymphoma.
Generation of T-cell depletion by use of anti-T lymphocyte antibodies in the prevention/treatment of graft rejection can further increase the risk of developing the post-transplant lymphoproliferative disorder. Such antibodies include anti-lymphocyte globulin, muromonab-CD3 (OKT3), and anti-thymocyte globulin (ATG).
Inherited causes of lymphoproliferative disorders
X-linked lymphoproliferative disorder
Mutations in the SH2D1A and XIAP genes cause X-linked lymphoproliferative disorder. SH2D1A gene mutations cause X-linked lymphoproliferative disorder type 1, and XIAP gene mutations cause X-linked lymphoproliferative disorder type 2. The SH2D1A gene provides instructions for making a protein called signaling lymphocyte activation molecule (SLAM) associated protein (SAP). This protein is involved in the functioning of lymphocytes that destroy other cells (cytotoxic lymphocytes) and is necessary for the development of specialized T cells called natural killer T cells. The SAP protein also helps control immune reactions by triggering self-destruction (apoptosis) of cytotoxic lymphocytes when they are no longer needed.
Some SH2D1A gene mutations impair SAP function. Others result in an abnormally short protein that is unstable or nonfunctional, or prevent any SAP from being produced. The loss of functional SAP disrupts proper signaling in the immune system and may prevent the body from controlling the immune reaction to EBV infection. In addition, lymphomas may develop when defective lymphocytes are not properly destroyed by apoptosis.
The XIAP gene provides instructions for making a protein that helps protect cells from undergoing apoptosis in response to certain signals. XIAP gene mutations can lead to an absence of XIAP protein or decrease the amount of XIAP protein that is produced. It is unknown how a lack of XIAP protein results in the signs and symptoms of X-linked lymphoproliferative disorder, or why features of this disorder differ somewhat between people with XIAP and SH2D1A gene mutations.
X-linked agammaglobulinemia
Mutations in the BTK gene cause X-linked agammaglobulinemia. This gene provides instructions for making the BTK protein, which is important for the development of B cells and normal functioning of the immune system. Most mutations in the BTK gene prevent the production of any BTK protein. The absence of functional BTK protein blocks B cell development and leads to a lack of antibodies. Without antibodies, the immune system cannot properly respond to foreign invaders and prevent infection.
Autoimmune lymphoproliferative syndrome
Mutations in the FAS gene cause autoimmune lymphoproliferative syndrome in approximately 75 percent of affected individuals; these mutations are associated with the classic form of the disorder. The FAS gene provides instructions for making a protein involved in cell signaling that results in the self-destruction of cells (apoptosis).
When the immune system is turned on (activated) to fight an infection, large numbers of lymphocytes are produced. Normally, these lymphocytes undergo apoptosis when they are no longer required. FAS gene mutations lead to an abnormal protein that interferes with apoptosis. As a result, excess lymphocytes accumulate in the body’s tissues and organs and often begin attacking them, leading to autoimmune disorders. Interference with apoptosis allows cells to multiply without control, leading to the lymphomas that often occur in people with this disorder.
Non-classic forms of autoimmune lymphoproliferative syndrome may be caused by mutations in additional genes, some of which have not been identified.
Chediak-Higashi syndrome
Chediak-Higashi syndrome is caused by mutations in the LYST gene. This gene provides instructions for making a protein known as the lysosomal trafficking regulator. Researchers believe that this protein plays a role in the transport (trafficking) of materials into structures called lysosomes and similar cell structures. Lysosomes act as recycling centers within cells. They use digestive enzymes to break down toxic substances, digest bacteria that invade the cell, and recycle worn-out cell components.
Mutations in the LYST gene impair the normal function of the lysosomal trafficking regulator protein, which disrupts the size, structure, and function of lysosomes and related structures in cells throughout the body. In many cells, the lysosomes are abnormally large and interfere with normal cell functions. For example, enlarged lysosomes in certain immune system cells prevent these cells from responding appropriately to bacteria and other foreign invaders. As a result, the malfunctioning immune system cannot protect the body from infections.
In pigment cells called melanocytes, cellular structures called melanosomes (which are related to lysosomes) are abnormally large. Melanosomes produce and distribute a pigment called melanin, which is the substance that gives skin, hair, and eyes their color. People with Chediak-Higashi syndrome have oculocutaneous albinism because melanin is trapped within the giant melanosomes and is unable to contribute to skin, hair, and eye pigmentation.
Researchers believe that abnormal lysosome-like structures inside blood cell fragments called platelets underlie the abnormal bruising and bleeding seen in people with Chediak-Higashi syndrome. Similarly, abnormal lysosomes in nerve cells probably cause the neurological problems associated with this disease.
Wiskott-Aldrich syndrome
Mutations in the WAS gene cause Wiskott-Aldrich syndrome. The WAS gene provides instructions for making a protein called WASP. This protein is found in all blood cells. WASP is involved in relaying signals from the surface of blood cells to the actin cytoskeleton, which is a network of fibers that make up the cell’s structural framework. WASP signaling triggers the cell to move and attach to other cells and tissues (adhesion). In white blood cells, this signaling allows the actin cytoskeleton to establish interactions between cells and the foreign invaders that they target (immune synapses).
WAS gene mutations that cause Wiskott-Aldrich syndrome lead to a lack of any functional WASP. Loss of WASP signaling disrupts the function of the actin cytoskeleton in developing blood cells. White blood cells that lack WASP have a decreased ability to respond to their environment and form immune synapses. As a result, white blood cells are less able to respond to foreign invaders, causing many of the immune problems related to Wiskott-Aldrich syndrome. Similarly, a lack of functional WASP in platelets impairs their development, leading to reduced size and early cell death.
Because they all have the same genetic cause, Wiskott-Aldrich syndrome, X-linked thrombocytopenia, and severe congenital neutropenia are sometimes collectively referred to as WAS-related disorders.
Ataxia telangiectasia
Mutations in the ATM gene cause ataxia-telangiectasia. The ATM gene provides instructions for making a protein that helps control cell division and is involved in DNA repair. This protein plays an important role in the normal development and activity of several body systems, including the nervous system and immune system. The ATM protein assists cells in recognizing damaged or broken DNA strands and coordinates DNA repair by activating enzymes that fix the broken strands. Efficient repair of damaged DNA strands helps maintain the stability of the cell’s genetic information.
Mutations in the ATM gene reduce or eliminate the function of the ATM protein. Without this protein, cells become unstable and die. Cells in the part of the brain involved in coordinating movements (the cerebellum) are particularly affected by loss of the ATM protein. The loss of these brain cells causes some of the movement problems characteristic of ataxia-telangiectasia. Mutations in the ATM gene also prevent cells from responding correctly to DNA damage, which allows breaks in DNA strands to accumulate and can lead to the formation of cancerous tumors.
Post-transplant lymphoproliferative disorder
Lymphoproliferative disorders associated with transplantation and concomitant immunosuppressive therapy are increasingly common. Post-transplant lymphoproliferative disorders are varied and somewhat depend on the nature of the allograft and on the immunosuppressive agents used to prevent graft (or host) rejection. In most cases, the lymphoproliferative disorder is of B-cell origin; however, in rare cases, T-cell lymphoproliferative disorders are described 4.
Most post-transplant lymphoproliferative disorders occur in the setting of a solid organ transplantation. The primary risk factor appears to be Epstein Barr Virus (EBV) seronegativity at time of transplant. The type of organ transplanted has also been identified as a risk factor. Lung, small bowel, and multiple organ grafts are identified as high risk compared with kidney, heart 5 and liver. The more T-cell specific the immunosuppression used, the higher the incidence of posttransplant lymphoproliferative disorder.
The incidence of posttransplant lymphoproliferative disorder following bone marrow transplantation is lower than post-transplant lymphoproliferative disorder following solid organ transplantation. Essentially all posttransplant lymphoproliferative disorder following bone marrow transplantation is associated with EBV. Any factors that either stimulate B-cell proliferation and/or decrease or delay T-cell immunity increase the risk of posttransplant lymphoproliferative disorder. For allogeneic recipients, the risk of posttransplant lymphoproliferative disorder has consistently been found to be strongly associated with human leukocyte antigen (HLA) disparity.
Weintraub et al 6 conducted a retrospective chart review of pediatric solid-organ transplant recipients (aged 0 to 21 years) at a single institution between 2001 and 2009 to identify risk factors for the development of posttransplant lymphoproliferative disease (posttransplant lymphoproliferative disorder). A total of 350 pediatric patients received a solid organ transplant during the study period. Of those patients, 90 (25.7%) developed Epstein-Barr virus (EBV) viremia. Of those, 28 (31%) developed posttransplant lymphoproliferative disorder. The median age at time of transplant was 11.5 months in the posttransplant lymphoproliferative disorder group and 21.5 months in the EBV viremia–only group. All patients who developed posttransplant lymphoproliferative disorder had 1 or more clinical symptoms. Younger age at transplant, increased immunosuppression before the development of EBV viremia, higher peak EBV level, and the presence of clinical symptoms were found to be predictive of the development of posttransplant lymphoproliferative disorder in solid-organ transplant recipients who had EBV viremia 6.
Recipients of solid organs or allogeneic hematopoietic stem cells are at increased risk of developing lymphoma, which can be secondary to immunosuppression caused by Epstein-Barr virus (EBV) 7.
Other causes of lymphoproliferative disorder
Common variable immune deficiency
The cause in common variable immune deficiency is unknown in approximately 90 percent of cases. It is likely that this condition is caused by both environmental and genetic factors. While the specific environmental factors are unclear, the genetic influences in common variable immune deficiency are believed to be mutations in genes that are involved in the development and function of immune system cells called B cells. B cells are specialized white blood cells that help protect the body against infection. When B cells mature, they produce special proteins called antibodies (also known as immunoglobulins). These proteins attach to foreign particles, marking them for destruction. Mutations in the genes associated with common variable immune deficiency result in dysfunctional B cells that cannot make sufficient amounts of antibodies.
In about 10 percent of cases, a genetic cause for common variable immune deficiency is known. Mutations in at least 13 genes have been associated with common variable immune deficiency. The most frequent mutations occur in the TNFRSF13B gene. The protein produced from this gene plays a role in the survival and maturation of B cells and in the production of antibodies. TNFRSF13B gene mutations disrupt B cell function and antibody production, leading to immune dysfunction. Other genes associated with common variable immune deficiency are also involved in the function and maturation of immune system cells, particularly of B cells; mutations in these genes account for only a small percentage of cases.
All individuals with common variable immune deficiency have a shortage (deficiency) of two or three specific antibodies. Some have a deficiency of the antibodies called immunoglobulin G (IgG) and immunoglobulin A (IgA), while others, in addition to lacking IgG and IgA, are also deficient in immunoglobulin M (IgM). A shortage of these antibodies makes it difficult for people with this disorder to fight off infections. Abnormal and deficient immune responses over time likely contribute to the increased cancer risk. In addition, vaccines for diseases such as measles and influenza do not provide protection for people with common variable immune deficiency because they cannot produce an antibody response.
HIV infection
HIV infection is the most common cause for acquired immunodeficiency. HIV is a retrovirus that occurs as two types: HIV-1 and HIV-2. Both types are spread through direct contact with HIV-infected body fluids, such as blood, semen, and vaginal fluids, or from a mother who has HIV to her child during pregnancy, labor and delivery, or breastfeeding (through breast milk).
HIV infection weakens your immune system, making you much more likely to develop numerous infections and certain types of cancers. Cancers common to HIV/AIDS:
- Kaposi’s sarcoma. A tumor of the blood vessel walls, this cancer is rare in people not infected with HIV, but common in HIV-positive people. It usually appears as pink, red or purple lesions on the skin and mouth. In people with darker skin, the lesions may look dark brown or black. Kaposi’s sarcoma can also affect the internal organs, including the digestive tract and lungs.
- Lymphoma. This cancer starts in the white blood cells. The most common early sign is painless swelling of the lymph nodes in your neck, armpit or groin.
Lymphoproliferative disorder symptoms
X-linked lymphoproliferative disease
X-linked lymphoproliferative disease is a disorder of the immune system and blood-forming cells that is found almost exclusively in males. More than half of individuals with X-linked lymphoproliferative disease experience an exaggerated immune response to the Epstein-Barr virus (EBV). EBV is a very common virus that eventually infects most humans. In some people it causes infectious mononucleosis (commonly known as “mono”). Normally, after initial infection, EBV remains in certain immune system cells (lymphocytes) called B cells. However, the virus is generally inactive (latent) because it is controlled by other lymphocytes called T cells that specifically target EBV-infected B cells.
People with X-linked lymphoproliferative disease may respond to EBV infection by producing abnormally large numbers of T cells, B cells, and other lymphocytes called macrophages. This proliferation of immune cells often causes a life-threatening reaction called hemophagocytic lymphohistiocytosis. Hemophagocytic lymphohistiocytosis causes fever, destroys blood-producing cells in the bone marrow, and damages the liver. The spleen, heart, kidneys, and other organs and tissues may also be affected. In some individuals with X-linked lymphoproliferative disease, hemophagocytic lymphohistiocytosis or related symptoms may occur without EBV infection.
About one-third of people with X-linked lymphoproliferative disease experience dysgammaglobulinemia, which means they have abnormal levels of some types of antibodies. Antibodies (also known as immunoglobulins) are proteins that attach to specific foreign particles and germs, marking them for destruction. Individuals with dysgammaglobulinemia are prone to recurrent infections.
Cancers of immune system cells (lymphomas) occur in about one-third of people with X-linked lymphoproliferative disease.
X-linked lymphoproliferative disease can be divided into two types based on its genetic cause and pattern of signs and symptoms: X-linked lymphoproliferative disease type 1 also known as classic X-linked lymphoproliferative disease and X-linked lymphoproliferative disease type 2. People with X-linked lymphoproliferative disease type 2 have not been known to develop lymphoma, are more likely to develop hemophagocytic lymphohistiocytosis without EBV infection, usually have an enlarged spleen (splenomegaly), and may also have inflammation of the large intestine (colitis). Some researchers believe that these individuals should actually be considered to have a similar but separate disorder rather than a type of X-linked lymphoproliferative disease.
Without treatment, most people with X-linked lymphoproliferative disease survive only into childhood. Death usually results from hemophagocytic lymphohistiocytosis.
Autoimmune lymphoproliferative syndrome
Autoimmune lymphoproliferative syndrome presents with an increased size of lymphoid organs including lymph nodes and spleen in up to 90% of patients. The liver may be enlarged in up to 40% of patients. Autoimmune cytopenia is common including autoimmune hemolytic anemia, neutropenia, and thrombocytopenia. Symptoms that resemble systemic lupus erythematosus may be present. Autoimmune lymphoproliferative syndrome is associated with other autoimmune disorders like autoimmune cerebellar ataxia, transverse myelitis, Guillain–Barre syndrome, and autoimmune glomerulonephritis.
Clinical features of chronic hematological malignancies (chronic leukemias and lymphomas) include:
- Males are more affected than women
- Asthenia
- Anemia
- Thrombocytopenia
- Granulocytopenia
- Loss of weight
- Lymphadenopathy (for example, peripheral, mesenteric, and retroperitoneal).
- Splenomegaly
- Hepatomegaly
- Metastatic disease affecting several organs including jaw, liver, ovaries, central nervous system (CNS), and gastrointestinal (GI) tract
- Recurrent infections
- Skin rash
Lymphoproliferative disorder diagnosis
General tests in lymphoproliferative disorders
- Clinical findings indicate local or distant adenopathy and hepatosplenomegaly.
- In certain conditions, the gastrointestinal (GI) tract or lung tissue may also be affected.
Biochemical panel
- Perform serologic tests for cytomegalovirus and Epstein-Barr virus (EBV).
- Measure the erythrocyte sedimentation rate.
- Evaluate electrolyte, blood urea nitrogen (BUN), creatinine, phosphate, calcium, and uric acid levels to rule out tumor lysis syndrome.
- Assess lactate dehydrogenase levels to assess the neoplastic burden.
Imaging studies
CT scans obtained with intravenous or oral contrast material can help in determining the true extent of abdominal adenopathy, infiltration of the bowel wall, and the accurate sizes of tumorous masses. This information is important for staging and assessing therapeutic response.
MRI studies of soft-tissue infiltrative processes can refine the clinician’s understanding of the tumor burden and the potential that vital structures might be compromised.
Chest radiography is performed in patients with pulmonary lesions to follow the progression or regression of disease. In some patients, pulmonary functional tests can provide further objective evidence of disease progression or a therapeutic response.
Ultrasonography: Ultrasonography is sometimes helpful.
Small bowel follow-through study: In children with GI lesions, this test helps in diagnosing ileal disease.
Lymphoproliferative disorder treatment
- Epstein–Barr virus (EBV)-associated T-cell and/or NK-cell (EBV T/NK-cell) lymphoproliferative disorders can be cured in most cases with allogeneic hematopoietic stem cell transplantation (HSCT). Primary-EBV infection-associated hemophagocytic lymphohistiocytosis that is an EBV T/NK-cell lymphoproliferation may be managed with the use of steroids, cyclosporine A, and etoposide. Remission is known to occur in some patients but may require multi-drug block chemotherapy 8.
- In autoimmune lymphoproliferative syndrome, one should treat the underlying autoimmune disease. The first-line therapy includes the use of corticosteroids and intravenous immunoglobulins. Second-line treatment includes mycophenolate mofetil. Sirolimus, which is a mammalian target of rapamycin (mTOR) can lead to near-complete resolution of the autoimmune disease 9.
- Post-transplant lymphoproliferative disorder can spontaneously regress with cessation or reduction of immunosuppressant therapy and can additionally be treated with antiviral therapy.
- One can also treat hematological malignancies with multiple-agent chemotherapy, including cyclophosphamide and prednisone in combination with vincristine and doxorubicin.
- Rituximab has been used to treat CD20-positive hematological malignancies since 1997 and now can be replaced by anti-CD20 biosimilar that is more effective 10.
Lymphoproliferative disorder survival rate
Overall, lymphoproliferative disorder has a poor outcome, but myeloablative hematopoietic stem cell transplantation may be beneficial in some patients 11.
References- Justiz Vaillant AA, Stang CM. Lymphoproliferative Disorders. [Updated 2019 Jun 18]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK537162
- Calabrò ML, Sarid R. Human Herpesvirus 8 and Lymphoproliferative Disorders. Mediterr J Hematol Infect Dis. 2018;10(1):e2018061
- Litz CE, Brunning RD. Chronic lymphoproliferative disorders: classification and diagnosis. Baillieres Clin. Haematol. 1993 Dec;6(4):767-83.
- Absalon MJ, Khoury RA, Phillips CL. Post-transplant lymphoproliferative disorder after solid-organ transplant in children. Semin Pediatr Surg. 2017 Aug. 26 (4):257-266.
- Ohta H, Fukushima N, Ozono K. Pediatric post-transplant lymphoproliferative disorder after cardiac transplantation. Int J Hematol. 2009 Sep. 90(2):127-36.
- Weintraub L, Weiner C, Miloh T, Tomaino J, Joashi U, Benchimol C, et al. Identifying predictive factors for posttransplant lymphoproliferative disease in pediatric solid organ transplant recipients with epstein-barr virus viremia. J Pediatr Hematol Oncol. 2014 Nov. 36(8):e481-6.
- Dierickx D, Habermann TM. Post-Transplantation Lymphoproliferative Disorders in Adults. N. Engl. J. Med. 2018 Feb 08;378(6):549-562.
- Sawada A, Inoue M. Hematopoietic Stem Cell Transplantation for the Treatment of Epstein-Barr Virus-Associated T- or NK-Cell Lymphoproliferative Diseases and Associated Disorders. Front Pediatr. 2018;6:334.
- Teachey DT, Obzut DA, Axsom K, Choi JK, Goldsmith KC, Hall J, Hulitt J, Manno CS, Maris JM, Rhodin N, Sullivan KE, Brown VI, Grupp SA. Rapamycin improves lymphoproliferative disease in murine autoimmune lymphoproliferative syndrome (ALPS). Blood. 2006 Sep 15;108(6):1965-71.
- Freeman CL, Sehn L. Anti-CD20 Directed Therapy of B Cell Lymphomas: Are New Agents Really Better? Curr Oncol Rep. 2018 Nov 27;20(12):103.
- Cohen JM, Sebire NJ, Harvey J, Gaspar HB, Cathy C, Jones A, Rao K, Cubitt D, Amrolia PJ, Davies EG, Veys P. Successful treatment of lymphoproliferative disease complicating primary immunodeficiency/immunodysregulatory disorders with reduced-intensity allogeneic stem-cell transplantation. Blood. 2007 Sep 15;110(6):2209-14.





{kind=link}