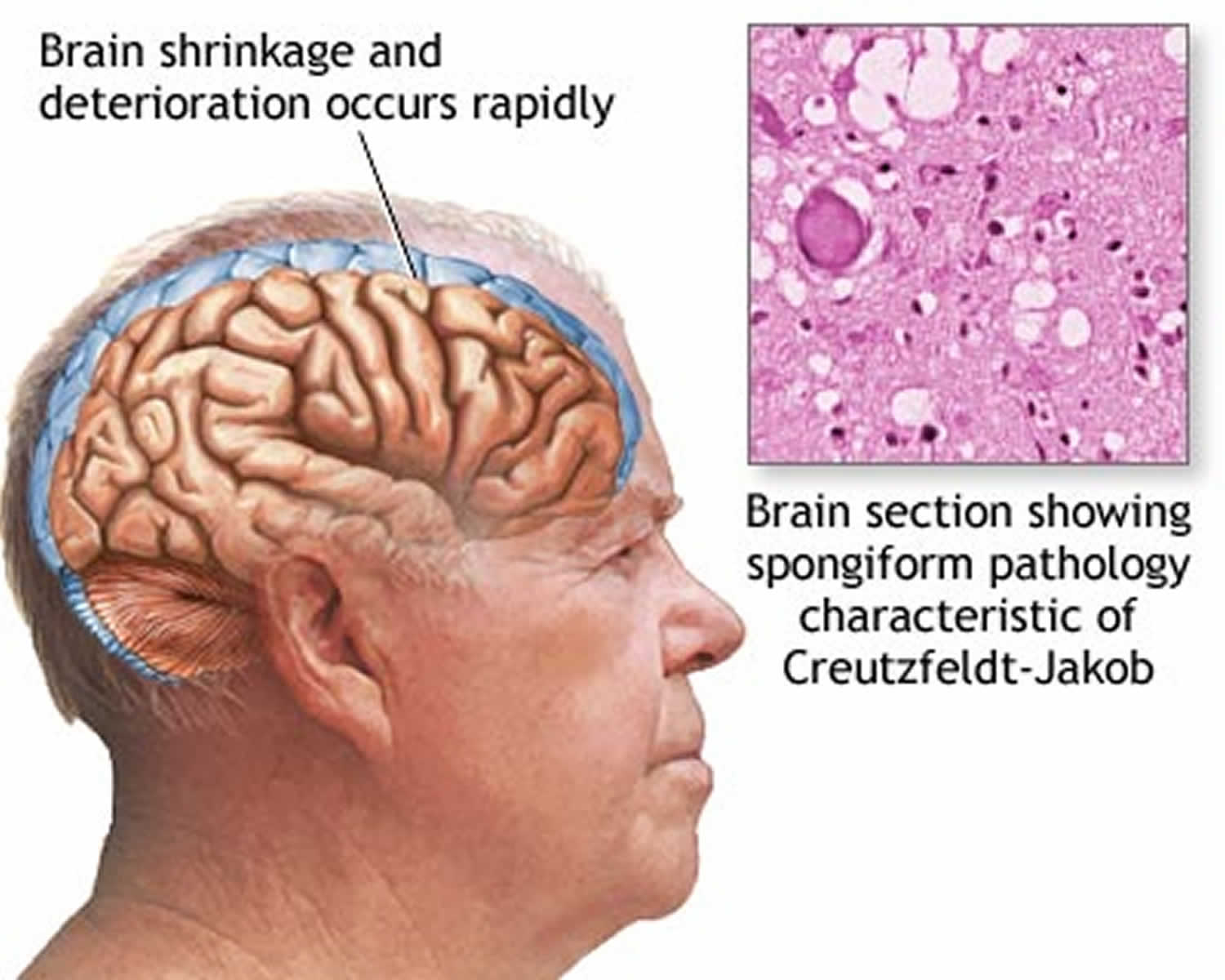
What is mad cow disease
Mad Cow Disease is also known as bovine spongiform encephalopathy (BSE), is a progressive neurological disorder of cattle that results from infection by an unusual transmissible agent called a prion 1. “Bovine” means that the disease affects cows, “spongiform” refers to the way the brain from a sick cow looks spongy under a microscope, and “encephalopathy” indicates that it is a disease of the brain. Most scientists think that mad cow disease is caused by a protein called a prion. For reasons that are not yet understood, the normal prion protein changes into an abnormal prion protein that is harmful that then damages the central nervous system of cattle. The body of a sick cow does not even know the abnormal prion is there. Without knowing it is there, the cow’s body cannot fight off the disease. Mad Cow Disease (bovine spongiform encephalopathy) damages a cow’s central nervous system (brain and spinal cord).
Normal (harmless) prion proteins are found in almost all body tissues, but are at the highest levels in brain and nerve cells. The exact role of normal prion proteins is unknown, but it’s thought they may play a role in transporting messages between certain brain cells. Mistakes sometimes occur during protein folding and the prion protein can’t be used by the body. Normally, these misfolded prion (abnormal prion) proteins are recycled by the body, but they can build up in the brain if they aren’t recycled. This causes the brain cells to die, releasing more prions to infect other brain cells. Eventually, clusters of brain cells are killed and deposits of misfolded prion protein called plaques may appear in the brain. Prion infections also cause small holes to develop in the brain, so it becomes sponge-like. The damage to the brain causes the mental and physical impairment associated with Creutzfeldt-Jakob disease, and eventually leads to death. Prions can survive in nerve tissue, such as the brain or spinal cord, for a very long time, even after death.
It is worth noting that there are two types of bovine spongiform encephalopathy, classical and atypical. Classical bovine spongiform encephalopathy is caused by contaminated feed fed to cows. Atypical bovine spongiform encephalopathy is rarer and happens spontaneously, usually in cows 8-years-old or older.
Research indicates that the first probable infections of bovine spongiform encephalopathy in cows occurred during the 1970’s with two cases of mad cow disease being identified in 1986. Mad cow disease possibly originated as a result of feeding cattle meat-and-bone meal that contained bovine spongiform encephalopathy-infected products from a spontaneously occurring case of mad cow disease or scrapie-infected sheep products. Scrapie is a prion disease of sheep. There is strong evidence and general agreement that the outbreak was then amplified and spread throughout the United Kingdom cattle industry by feeding rendered, prion-infected, bovine meat-and-bone meal to young calves.
The mad cow disease epizootic in the United Kingdom peaked in January 1993 at almost 1,000 new cases per week. Since then, the annual numbers of mad cow disease cases in the United Kingdom have dropped sharply 2:
- 2 cases in 2015
- 11 cases in 2010
- 225 cases in 2005
- 1,443 cases in 2000
- 14,562 cases in 1995
Through August 2018, mad cow disease (bovine spongiform encephalopathy) surveillance has identified 26 cases in North America: 6 mad cow disease cases in the United States and 20 in Canada. Of the 6 cases identified in the United States, one was born in Canada; of the 20 cases identified in Canada, one was imported from the United Kingdom 3. Of the five U.S. cows found with bovine spongiform encephalopathy, four were atypical bovine spongiform encephalopathy.
Strains of bovine spongiform encephalopathy
There is increasing evidence that there are different strains of bovine spongiform encephalopathy (BSE): the typical or classic bovine spongiform encephalopathy strain responsible for the outbreak in the United Kingdom and two atypical strains (H and L strains).
Classic bovine spongiform encephalopathy strain
The bovine spongiform encephalopathy strain responsible for most of the mad cow disease cases in Canada is the same classic or typical strain linked to the outbreak in the United Kingdom. It is known to be preventable through elimination of bovine spongiform encephalopathy contaminated feed and has been causally linked to variant Creutzfeldt-Jakob disease (vCJD) in humans. This typical strain has not yet been identified in any U.S.-born cattle.
All five of the U.S.-born bovine spongiform encephalopathy cases and two of the 20 Canadian-born bovine spongiform encephalopathy cases were caused by atypical bovine spongiform encephalopathy strains. Of these seven North American cases, four were linked to an atypical bovine spongiform encephalopathy strain known as the H-type. and three were identified as the L-type. The strain type for the other three older North American cases, a 13-year-old bovine spongiform encephalopathy-infected Canadian cow, a 10-year-old bovine spongiform encephalopathy-infected U.S. cow, and an 11-year- old bovine spongiform encephalopathy-infected U.S. cow, have been identified as the L-type.
How does a cow get mad cow disease?
The parts of a cow that are not eaten by people are cooked, dried, and ground into a powder. The powder is then used for a variety of purposes, including as an ingredient in animal feed. A cow gets bovine spongiform encephalopathy by eating feed contaminated with parts that came from another cow that was sick with mad cow disease (bovine spongiform encephalopathy). The contaminated feed contains the abnormal prion, and a cow becomes infected with the abnormal prion when it eats the feed. If a cow gets mad cow disease (bovine spongiform encephalopathy), it most likely ate the contaminated feed during its first year of life. Remember, if a cow becomes infected with the abnormal prion when it is one-year-old, it usually will not show signs of mad cow disease (bovine spongiform encephalopathy) until it is five-years-old or older.
Can other animals get mad cow disease?
Sheep, goats, mink, deer, and elk can get sick with their own versions of bovine spongiform encephalopathy. Cats are the only common household pet known to have a version of bovine spongiform encephalopathy. It is called feline spongiform encephalopathy, and the same things that are being done to protect people and cows are also protecting cats. No cat in the U.S. has ever been found to have feline spongiform encephalopathy.
What causes mad cow disease in humans
There’s clear evidence that variant Creutzfeldt-Jakob disease (vCJD) is caused by the same strain of prions that causes bovine spongiform encephalopathy (mad cow disease).
In 2000, a UK government inquiry concluded that the prion was spread through cattle that were fed meat-and-bone mix containing traces of infected brains or spinal cords.
The prion then ended up in processed meat products, such as beef burgers, and entered the human food chain.
Strict controls have been in place since 1996 to prevent bovine spongiform encephalopathy entering the human food chain, and the use of meat-and-bone mix has been made illegal.
It appears not everyone who’s exposed to bovine spongiform encephalopathy-infected meat will go on to develop variant Creutzfeldt-Jakob disease (vCJD).
All definite cases of variant Creutzfeldt-Jakob disease (vCJD) occurred in people with a specific version (MM) of the prion protein gene, which affects how the body makes a number of amino acids.
It’s estimated up to 40% of the UK population have this version of the gene.
Cases of variant Creutzfeldt-Jakob disease (vCJD) peaked in the year 2000, in which there were 28 deaths from variant Creutzfeldt-Jakob disease (vCJD). There were no confirmed deaths in 2014.
Some experts believe that the food controls have worked and further cases of variant Creutzfeldt-Jakob disease (vCJD) will continue to decline, but this doesn’t rule out the possibility that other cases may be identified in the future.
It’s also possible for variant Creutzfeldt-Jakob disease (vCJD) to be transmitted by blood transfusion, although this is very rare and measures have been put in place to reduce the risk of it happening.
Experts don’t know how many people in the UK population could develop variant Creutzfeldt-Jakob disease (vCJD) in the future and how long it’ll take for symptoms to appear, if they ever will.
A study published in October 2013 that tested random tissue samples suggested around 1 in 2,000 people in the UK population may be infected with variant Creutzfeldt-Jakob disease (vCJD), but show no symptoms to date.
Since the link between variant Creutzfeldt-Jakob disease (vCJD) and bovine spongiform encephalopathy was discovered in 1996, strict controls have proved very effective in preventing meat from infected cattle entering the food chain.
But the average time it takes for the symptoms of variant Creutzfeldt-Jakob disease (vCJD) to occur after initial infection (the incubation period) is still unclear.
The incubation period could be very long (more than 10 years) in some people, so those exposed to infected meat before the food controls were introduced can still develop variant Creutzfeldt-Jakob disease (vCJD).
The prion that causes variant Creutzfeldt-Jakob disease (vCJD) can also be transmitted by blood transfusion, although this has only happened 4 times in the UK.
In 2014, there were no recorded deaths from variant Creutzfeldt-Jakob disease (vCJD) in the UK.
How do humans get mad cow disease?
There is strong evidence indicates that classic mad cow disease (bovine spongiform encephalopathy) has been transmitted to people primarily in the United Kingdom, causing a new human prion disease called variant Creutzfeldt-Jakob disease (vCJD). The absence of confirmed cases of variant Creutzfeldt-Jakob disease (vCJD) in other geographic areas free of bovine spongiform encephalopathy supports a causal association. In addition, the interval between the most likely period for the initial extended exposure of the population to potentially bovine spongiform encephalopathy-contaminated food (1984-1986) and onset of initial variant Creutzfeldt-Jakob disease (vCJD) cases (1994-1996) is consistent with known incubation periods for Creutzfeldt-Jakob disease.
There is now strong scientific evidence that the agent responsible for the outbreak of prion disease in cows, bovine spongiform encephalopathy (mad cow disease), is the same agent responsible for the outbreak of variant Creutzfeldt-Jakob disease (vCJD) in humans.
- An experimental study reported in June 1996 showed that three cynomologus macaque monkeys inoculated with brain tissue obtained from cattle with bovine spongiform encephalopathy had clinical and neuropathological features strikingly similar to those of variant Creutzfeldt-Jakob disease (vCJD) 4.
- A study published in 1996 indicated that a Western blot analysis of infecting prions obtained from 10 variant Creutzfeldt-Jakob disease (vCJD) patients and bovine spongiform encephalopathy-infected animals had similar molecular characteristics that were distinct from prions obtained from patients with other types of Creutzfeldt-Jakob disease 5.
- An experimental study involving inoculation of a panel of inbred mice with the agents causing mad cow disease (bovine spongiform encephalopathy) and variant Creutzfeldt-Jakob disease (vCJD) substantially increased the strength of the scientific evidence for a causal association between variant Creutzfeldt-Jakob disease (vCJD) and bovine spongiform encephalopathy 6. In this study, groups of inbred mice and a group of cross-bred mice inoculated with brain homogenates from variant Creutzfeldt-Jakob disease (vCJD) cases were reported to have had latency periods and lesion profiles consistent with the bovine spongiform encephalopathy pattern.
- The latency period, neuropathology, and disease-causing prion protein (PrP) isoforms in transgenic mice expressing bovine prion protein (PrP) that were inoculated with variant Creutzfeldt-Jakob disease (vCJD), bovine spongiform encephalopathy, and scrapie brain extracts provided additional evidence supporting the link between bovine spongiform encephalopathy and variant Creutzfeldt-Jakob disease (vCJD) 7.
In the United Kingdom, where over 1 million cattle may have been infected with classic mad cow disease, a substantial species barrier appears to protect people from widespread illness. Since variant Creutzfeldt-Jakob disease (vCJD) was first reported in 1996, a total of only 231 patients with this disease, including 3 secondary, blood transfusion-related cases, have been reported worldwide 3. The risk to human health from mad cow disease (bovine spongiform encephalopathy) in the United States is extremely low 3.
Public health control measures, such as surveillance, culling sick animals, or banning specified risk materials, have been instituted in many countries, particularly in those with indigenous cases of confirmed mad cow disease (bovine spongiform encephalopathy), in order to prevent potentially bovine spongiform encephalopathy-infected tissues from entering the human food supply.
The most stringent control measures include a UK program that excludes all animals more than 30 months of age from the human food and animal feed supplies. The program appears to be highly effective.
In June 2000, the European Union Commission on Food Safety and Animal Welfare strengthened the European Union’s bovine spongiform encephalopathy control measures by requiring all member states to remove specified risk materials from animal feed and human food chains as of October 1, 2000; such bans had already been instituted in most member states. Other control measures include banning the use of mechanically recovered meat from the vertebral column of cattle, sheep, and goats for human food and bovine spongiform encephalopathy testing of all cattle more than 30 months of age destined for human consumption.
To reduce any risk of acquiring variant Creutzfeldt-Jakob disease (vCJD) from food, travelers to Europe or other areas with indigenous cases of mad cow disease (bovine spongiform encephalopathy) may consider either avoiding beef and beef products altogether or selecting beef or beef products, such as solid pieces of muscle meat (rather than brains or beef products like burgers and sausages), that might have a reduced opportunity for contamination with tissues that may harbor the bovine spongiform encephalopathy agent. Milk and milk products from cows are not believed to pose any risk for transmitting the bovine spongiform encephalopathy agent.
Blood transfusion
In the UK, there have been 4 cases where variant Creutzfeldt-Jakob disease has been transmitted by blood transfusion. In each case, the person received a blood transfusion from a donor who later developed variant Creutzfeldt-Jakob disease. However, it’s not 100 percent certain whether the blood transfusion was the cause of the infection, as those involved could have contracted variant Creutzfeldt-Jakob disease (vCJD) through dietary sources.
In 2012, three cases of variant Creutzfeldt-Jakob disease (vCJD) have occurred in individuals in the UK who received non-leucodepleted red blood cells from asymptomatic UK donors who subsequently died from variant Creutzfeldt-Jakob disease (vCJD) after donation 8, 9. All 3 recipients were methionine homozygotes at codon 129 in the PRNP gene, with incubation periods ranging from 6.5 to 7.8 years between the date of the implicated transfusion and the onset of clinical disease. The clinical and neuropathological phenotypes of the disease in the recipients was similar to other cases of variant Creutzfeldt-Jakob disease (vCJD) 10. However, asymptomatic variant Creutzfeldt-Jakob disease (vCJD) infection was identified in an elderly patient who had undergone transfusion of one unit of non-leucodepleted red blood cells from another asymptomatic donor who subsequently developed variant Creutzfeldt-Jakob disease (vCJD) 11. The recipient had no signs or symptoms of variant Creutzfeldt-Jakob disease (vCJD) or any other neurological disorder and died of an unrelated illness 5 years after the transfusion. Analysis of the codon 129 polymorphism in the PRNP gene found that this recipient was heterozygous (methionine/valine). No neuropathological evidence of variant Creutzfeldt-Jakob disease (vCJD) was found in the brain and Western blot for PrPSc (abnormal prion) in the brain was negative. However, immunohistochemistry for PrPSc (abnormal prion) was positive in the spleen and a cervical lymph node, but not in the tonsil or the appendix, and Western blot analysis confirmed the presence of PrPSc (abnormal prion) in the spleen 11.
The identification of variant Creutzfeldt-Jakob disease (vCJD) infection in 4 individuals who had received red cell transfusions from variant Creutzfeldt-Jakob disease (vCJD)-infected donors strongly suggests that blood is infectious during the incubation period for variant Creutzfeldt-Jakob disease (vCJD). Since donations from asymptomatic donors who subsequently died from variant Creutzfeldt-Jakob disease (vCJD) were also used for plasma processing in the UK 9, these findings renewed concerns that variant Creutzfeldt-Jakob disease (vCJD) might be transmissible by plasma products. In anticipation of and response to these concerns, a range of precautionary measures was introduced in the UK to reduce the likelihood of transmission of variant Creutzfeldt-Jakob disease (vCJD) by blood and plasma products 12. None of the measures are likely to remove all risks, but it appears that leucodepletion may reduce levels of infectivity in blood 13.
A risk assessment was commissioned by the Department of Health in the UK in order to address the possible transmission of variant Creutzfeldt-Jakob disease (vCJD) by blood and blood products. The conclusions of this exercise were based on generally pessimistic assumptions made concerning the likely levels of infectivity in blood in variant Creutzfeldt-Jakob disease (vCJD), and the potential consequences of the processing steps used in the manufacture of plasma products. Accordingly, recipients of concentrates Factor VIII and IX were deemed likely to be at a sufficiently increased risk of variant Creutzfeldt-Jakob disease (vCJD) to require additional public health measures in order to minimize any risk of secondary transmission. Patients treated with UK-sourced pooled clotting factor concentrates between 1980 and 2001, including most of the adult hemophilia patients, were therefore informed that they had been assessed as being at an increased risk of infection with variant Creutzfeldt-Jakob disease (vCJD) 14. This approach was taken on the advice of the UK Haemophilia Centre Doctors Organisation (UKHCDO) and was endorsed by the UK Haemophilia Society.
Measures currently in place in the UK to reduce the potential risk of transmitting variant Creutzfeldt-Jakob disease (vCJD) via blood 14:
- Since October 1999, white blood cells, which may carry a risk of transmitting variant Creutzfeldt-Jakob disease (vCJD), have been reduced in all blood used for transfusion, by a process known as leucodepletion or leucoreduction.
- Since April 2004, following the report of the first presumed case of transmission of variant Creutzfeldt-Jakob disease (vCJD) by blood transfusion, individuals who had themselves received a transfusion of blood components since January 1980 were excluded from donating blood. In July 2004, this exclusion criterion for blood donation was extended to include two other groups who had received transfusions of blood components since 1980:
- Previously transfused platelet donors
- Donors who were unsure if they had previously had a blood transfusion.
- This measure applies to donors who have been transfused anywhere in the world.
- Clotting factors and immunoglobulin. Since 1999, plasma for the manufacture of fractionated plasma products, such as clotting factors and immunoglobulins, has been obtained from non-UK sources. Since 1998, synthetic (recombinant) clotting factor for the treatment of haemophilia has been provided to the under-16s, and since 2005 this measure has been extended to all patients for whom it is suitable.
- Plasma sourcing. Since 2004, fresh frozen plasma for treating those born on or after 1 January 1996 has been obtained from abroad (the USA or Austria).
- Platelets. In December 2014 Safety of Blood, Tissues and Organs (SaBTO) reviewed the evidence and confirmed removal of the requirement to produce at least 80% of platelets by apheresis, and recommended that platelet additive solution (PAS) should be used for the suspension of pooled platelets.
- Cryoprecipitate. From 2005, cryoprecipitate produced from methylene blue treated plasma imported from the USA is being used for children up to the age of 16.
Can people get mad cow disease?
People can get a version of mad cow disease called variant Creutzfeldt-Jakob disease (vCJD). As of December 4, 2017, 231 people worldwide are known to have become sick with variant Creutzfeldt-Jakob disease (vCJD) according to the University of Edinburgh’s National Creutzfeldt-Jakob Disease Research and Surveillance Unit 15. It is thought that they got the disease from eating food made from cows sick with bovine spongiform encephalopathy. Most of the people who have become sick with variant Creutzfeldt-Jakob disease (vCJD) lived in the United Kingdom at some point in their lives. Only four lived in the U.S., and most likely, these four people became infected when they were living or traveling overseas.
Neither variant Creutzfeldt-Jakob disease (vCJD) nor bovine spongiform encephalopathy is contagious. This means that it is not like catching a cold. A person (or a cow) cannot catch it from being near a sick person or cow. Also, research studies have shown that people cannot get bovine spongiform encephalopathy from drinking milk or eating dairy products, even if the milk came from a sick cow.
Prevention measures against mad cow disease spread
To prevent mad cow disease (bovine spongiform encephalopathy) from entering the United States, severe restrictions were placed on the importation of live ruminants, such as cattle, sheep, and goats, and certain ruminant products from countries where mad cow disease (bovine spongiform encephalopathy) was known to exist 16. These restrictions were later extended to include importation of ruminants and certain ruminant products from all European countries.
Because the use of ruminant tissue in ruminant feed was probably a necessary factor responsible for the mad cow disease outbreak in the United Kingdom and because of the current evidence for possible transmission of mad cow disease to humans, the U.S. Food and Drug Administration (FDA) instituted a ruminant feed ban in June 1997 that became fully effective as of October 1997 17. In April 2009, the FDA took additional steps to make sure the food in the U.S. stays safe. Certain high-risk cow parts are not allowed to be used to make any animal feed, including pet food. This prevents all animal feed from being accidentally contaminated with the abnormal prion. High-risk cow parts are those parts of the cow that have the highest chance of being infected with the abnormal prion, such as the brains and spinal cords from cows that are 30 months of age or older. By keeping the food that is fed to cows safe, the FDA is protecting people by making sure that the food they eat comes from healthy cows.
As of October 26, 2009, a regulation issued by FDA in April 2009 came into effect establishing an enhanced bovine spongiform encephalopathy-related feed ban in the U.S. This enhanced feed ban will further harmonize bovine spongiform encephalopathy feed control measures in the U.S. with those in Canada (see below). In addition, FDA continues to enforce its important 1997 mammalian-to-ruminant feed ban through its bovine spongiform encephalopathy inspection and bovine spongiform encephalopathy feed testing programs 17.
As of July 12, 2007, an enhanced bovine spongiform encephalopathy-related feed ban came into effect in Canada. Canadian Food Inspection Agency (CFIA) established this ban to more effectively prevent and quickly eliminate bovine spongiform encephalopathy from Canada. The enhanced ban prohibits most proteins, including potentially bovine spongiform encephalopathy infectious tissues known as “specified risk materials” from all animal feeds, pet foods, and fertilizers, not just from cattle feed as required by the ban instituted in 1997. The 1997 feed ban in Canada was similar to the feed ban instituted in the United States that same year. As recently reported by CFIA, removing “specified risk materials” from the entire animal feed system addresses risks associated with the potential contamination of cattle feed during production, distribution, storage, and use. Applying the same measure to pet food and fertilizer materials addresses the possible exposure of cattle and other susceptible animals to these products. With this ban in place, CFIA expects bovine spongiform encephalopathy should be eliminated from the Canadian cattle herd by about the year 2017.
The FDA also works with the U.S. Department of Agriculture (USDA) to keep cows in the U.S. healthy and free of bovine spongiform encephalopathy. The USDA prevents high-risk cows and cow products from entering the U.S. from other countries. The USDA also makes sure that high-risk cow parts, such as the brains and spinal cords, and cows that are unable to walk or that show other signs of disease are not used to make food for people.
The steps the FDA and USDA have taken to prevent cows in the U.S. from getting mad cow disease (bovine spongiform encephalopathy) are working very well. Only five cows with mad cow disease have been found in the U.S. Four of these cows were born in the U.S., and one was born in Canada. The USDA reports that the last cow with mad cow disease in the U.S. was found in July 2017.
Mad cow disease in humans
Variant Creutzfeldt-Jakob disease (vCJD) is a prion disease that was first described in 1996 in the United Kingdom. There is now strong scientific evidence that the agent responsible for the outbreak of prion disease in cows, mad cow disease (bovine spongiform encephalopathy), is the same agent responsible for the outbreak of variant Creutzfeldt-Jakob disease (vCJD) in humans.
Variant Creutzfeldt-Jakob disease (vCJD) is not the same disease as classic Creutzfeldt-Jakob disease (often simply called CJD) 18. Variant Creutzfeldt-Jakob disease (vCJD) has different clinical and pathologic characteristics from classic Creutzfeldt-Jakob disease. Each disease also has a particular genetic profile of the prion protein gene 18. Both disorders are invariably fatal brain diseases with unusually long incubation periods measured in years, and are caused by an unconventional transmissible agent called a prion.
The variant form of Creutzfeldt-Jakob disease (vCJD) should not be confused with the classic form of Creutzfeldt-Jakob disease (CJD) that is endemic throughout the world, including the United States. There are several important differences between these two forms of the disease. The median age at death of patients with classic Creutzfeldt-Jakob disease in the United States, for example, is 68 years, and very few cases occur in persons under 30 years of age. In contrast, the median age at death of patients with variant Creutzfeldt-Jakob disease (vCJD) in the United Kingdom is 28 years.
Variant Creutzfeldt-Jakob disease (vCJD) can be confirmed only through examination of brain tissue obtained by biopsy or at autopsy, but a “probable case” of variant Creutzfeldt-Jakob disease (vCJD) can be diagnosed on the basis of clinical criteria developed in the United Kingdom. The incubation period for variant Creutzfeldt-Jakob disease (vCJD) is unknown because it is a new disease. However, it is likely that ultimately this incubation period will be measured in terms of many years or decades. In other words, whenever a person develops variant Creutzfeldt-Jakob disease (vCJD) from consuming a bovine spongiform encephalopathy-contaminated product, he or she likely would have consumed that product many years or a decade or more earlier.
In contrast to classic Creutzfeldt-Jakob disease (CJD), variant Creutzfeldt-Jakob disease (vCJD) in the United Kingdom predominantly affects younger people, has atypical clinical features, with prominent psychiatric or sensory symptoms at the time of clinical presentation and delayed onset of neurologic abnormalities, including ataxia within weeks or months, dementia and myoclonus late in the illness, a duration of illness of at least 6 months, and a diffusely abnormal non-diagnostic electroencephalogram.
The characteristic neuropathologic profile of variant Creutzfeldt-Jakob disease (vCJD) includes, in both the cerebellum and cerebrum, numerous kuru-type amyloid plaques surrounded by vacuoles and prion protein (PrP) accumulation at high concentration indicated by immunohistochemical analysis.
The median age at death from variant Creutzfeldt-Jakob disease (vCJD) in the United Kingdom has been 28 years and almost all cases have been in persons under age 55 years 19. The reasons for this age distribution are not well understood but it suggests that through the oral route of exposure, older adults are much less susceptible to variant Creutzfeldt-Jakob disease (vCJD) than children and young adults. By year of onset, the incidence of variant Creutzfeldt-Jakob disease (vCJD) in the UK appears to have peaked in 1999 and to have been declining thereafter. In contrast, the number of reported cases in France has been increasing since the beginning of 2005. However, the future pattern of these ongoing epidemics remains uncertain. In 2004, a prevalence study of asymptomatic variant Creutzfeldt-Jakob disease (vCJD) infections in the UK identified three positive appendices out of a sample of 12,674 surgically removed tonsils and appendices that were satisfactory for analysis. Genetic studies completed on two of the appendices regarded as positive for variant Creutzfeldt-Jakob disease (vCJD) revealed that both had a different polymorphism at codon 129 of the prion protein gene than any of the patients with clinical variant Creutzfeldt-Jakob disease (vCJD) tested to date, indicating that more people are genetically susceptible to variant Creutzfeldt-Jakob disease (vCJD) infection, although not necessarily to the disease, than had been previously determined 20.
Recently published data indicate that the epidemic of variant Creutzfeldt-Jakob disease (vCJD) in the United Kingdom may have already reached a peak. A listing of monthly updated numbers of Creutzfeldt-Jakob disease and variant Creutzfeldt-Jakob disease (vCJD) cases in the United Kingdom can be found here (https://www.cjd.ed.ac.uk/surveillance-1).
Variant Creutzfeldt-Jakob disease (vCJD) characteristics, as compared to classic Creutzfeldt-Jakob disease (CJD), are presented in the table below.
Table 1. Clinical and Pathologic Characteristics Distinguishing Classic Creutzfeldt-Jakob disease (CJD) from variant Creutzfeldt-Jakob disease (vCJD)
| Characteristic | Classic CJD | Variant CJD |
|---|---|---|
| Median age at death | 68 years | 28 years |
| Median duration of illness | 4-5 months | 13-14 months |
| Clinical signs and symptoms | Dementia; early neurologic signs | Prominent psychiatric/behavioral symptoms; painful dyesthesiasis; delayed neurologic signs |
| Periodic sharp waves on electroencephalogram | Often present | Often absent |
| “Pulvinar sign” on MRI* | Not reported | Present in >75% of cases |
| Presence of “florid plaques” on neuropathology | Rare or absent | Present in large numbers |
| Immunohitochemical analysis of brain tissue | Variable accumulation | Marked accumulation of protease-resistance prion protein |
| Presence of agent in lymphoid tissue | Not readily detected | Readily detected |
| Increased glycoform ratio on immunoblot analysis of protease-resistance prion protein | Not reported | Marked accumulation of protease-resistance prion protein |
*An abnormal signal in the posterior thalami on T2- and diffusion-weighted images and fluid-attenuated inversion recovery sequences on brain magnetic resonance imaging (MRI); in the appropriate clinical context, this signal is highly specific for variant Creutzfeldt-Jakob disease (vCJD).
[Source 21]Mad cow disease in humans diagnosis
A diagnosis of Creutzfeldt-Jakob disease (CJD) is usually based on medical history, symptoms and a series of tests.
A neurologist (a doctor who specialises in conditions of the nervous system) will carry out the tests to rule out other conditions with similar symptoms, such as Alzheimer’s disease, Parkinson’s disease, or a brain tumour.
- The only way to confirm a diagnosis of Creutzfeldt-Jakob disease is to examine the brain tissue by carrying out a brain biopsy or, more commonly, after death in a post-mortem examination of the brain.
Specialist services at the National Creutzfeldt-Jakob disease Research and Surveillance Unit in Edinburgh and the National Prion Clinic in London advise local teams when making a diagnosis.
Tests for Creutzfeldt-Jakob disease
A clinical neurologist will rule out other conditions with similar symptoms.
They’ll also check for some common signs of Creutzfeldt-Jakob disease by carrying out the following tests:
- MRI brain scan – uses strong magnetic fields and radio waves to produce a detailed image of the brain, and can show up abnormalities particular to Creutzfeldt-Jakob disease
- Electroencephalogram (EEG) – records brain activity and may pick up abnormal electrical patterns seen in sporadic Creutzfeldt-Jakob disease
- Lumbar puncture – a procedure where a needle is inserted into the lower part of the spine to draw out a sample of cerebrospinal fluid (which surrounds your brain and spinal cord) so it can be tested for a certain protein that indicates you may have Creutzfeldt-Jakob disease
a prototype blood test for variant Creutzfeldt-Jakob disease has also been developed by the prion unit at the Medical Research Council (MRC) and is available through the National Prion Clinic - Tonsil biopsy – a small piece of tissue can be taken from the tonsils and checked for the abnormal prions found in variant Creutzfeldt-Jakob disease (they’re not present in other types of Creutzfeldt-Jakob disease)
- Genetic test – a simple blood test to find out whether you have a mutation (fault) in the gene that produces normal protein; a positive result may indicate familial (inherited) prion disease
Brain biopsy
During a brain biopsy, a surgeon drills a tiny hole into the skull and removes a small piece of brain tissue using a very thin needle.
It’s carried out under general anaesthetic, which means the person will be unconscious during the procedure.
As a brain biopsy carries the risk of causing brain damage or seizures (fits), it’s only performed in a few cases where there’s a concern that someone doesn’t have Creutzfeldt-Jakob disease but some other treatable condition.
Diagnostic Criteria
Definite variant Creutzfeldt-Jakob disease (vCJD)
Neuropathologic examination of brain tissue is required to confirm a diagnosis of variant Creutzfeldt-Jakob disease (vCJD). The following confirmatory features should be present.
- Numerous widespread kuru-type amyloid plaques surrounded by vacuoles in both the cerebellum and cerebrum – florid plaques.
- Spongiform change and extensive prion protein deposition shown by immunohistochemistry throughout the cerebellum and cerebrum.
Suspected variant Creutzfeldt-Jakob disease (vCJD)
- Current age or age at death <55 years (a brain autopsy is recommended, however, for all physician-diagnosed Creutzfeldt-Jakob disease cases).
- Psychiatric symptoms at illness onset and/or persistent painful sensory symptoms (frank pain and/or dysesthesia).
- Dementia, and development ≥4 months after illness onset of at least two of the following five neurologic signs: poor coordination, myoclonus, chorea, hyperreflexia, or visual signs. (If persistent painful sensory symptoms exist, ≥4 months delay in the development of the neurologic signs is not required).
- A normal or an abnormal EEG, but not the diagnostic EEG changes often seen in classic Creutzfeldt-Jakob disease.
- Duration of illness of over 6 months.
- Routine investigations of the patient do not suggest an alternative, non-Creutzfeldt-Jakob disease diagnosis.
- No history of receipt of cadaveric human pituitary growth hormone or a dura mater graft.
- No history of Creutzfeldt-Jakob disease in a first degree relative or prion protein gene mutation in the patient.
Note
- If a patient has the typical bilateral pulvinar high signal on MRI scan, a suspected diagnosis of variant Creutzfeldt-Jakob disease (vCJD) requires the presence of a progressive neuropsychiatric disorder, d, e, f and g of the above criteria, and four of the following five criteria: 1) early psychiatric symptoms (anxiety, apathy, delusions, depression, withdrawal); 2) persistent painful sensory symptoms (frank pain and/or dysesthesia); 3) ataxia; 4) myoclonus or chorea or dystonia; and 5) dementia.
- A history of possible exposure to bovine spongiform encephalopathy (BSE) such as residence or travel to a bovine spongiform encephalopathy-affected country after 1980 increases the index of suspicion for a variant Creutzfeldt-Jakob disease (vCJD) diagnosis.
Variant Creutzfeldt-Jakob disease (vCJD) Cases Reported in the US
Four cases of variant Creutzfeldt-Jakob disease (vCJD) have been reported from the United States 22. By convention, variant Creutzfeldt-Jakob disease (vCJD) cases are ascribed to the country of initial symptom onset, regardless of where the exposure occurred. There is strong evidence that suggests that two of the four cases were exposed to the bovine spongiform encephalopathy agent in the United Kingdom and that the third was exposed while living in Saudi Arabia. The specific overseas country where the fourth patient’s infection occurred is less clear.
The first patient was born in the United Kingdom in the late 1970’s and lived there until a move to Florida in 1992. The patient had onset of symptoms in November 2001 and died in June of 2004. The patient never donated or received blood, plasma, or organs, never received human growth hormone, nor did the patient ever have major surgery other than having wisdom teeth extracted in 2001. Additionally, there was no family history of Creutzfeldt-Jakob disease.
The second patient resided in Texas during 2001-2005. Symptoms began in early 2005 while the patient was in Texas. He then returned to the United Kingdom, where his illness progressed, and a diagnosis of variant Creutzfeldt-Jakob disease (vCJD) was made. The diagnosis was confirmed neuropathologically at the time of the patient’s death. While living in the United States, the patient had no history of hospitalization, of having invasive medical procedures, or of donation or receipt of blood and blood products. The patient almost certainly acquired the disease in the United Kingdom. He was born in the United Kingdom and lived there throughout the defined period of risk (1980-1996) for human exposure to the agent of bovine spongiform encephalopathy. His stay in the United States was too brief relative to what is known about the incubation period for variant Creutzfeldt-Jakob disease (vCJD).
The third patient was born and raised in Saudi Arabia and has lived in the United States since late 2005. The patient occasionally stayed in the United States for up to 3 months at a time since 2001 and there was a shorter visit in 1989. The patient’s onset of symptoms occurred in Spring 2006. In late November 2006, the Clinical Prion Research Team at the University of California San Francisco Memory and Aging Center confirmed the variant Creutzfeldt-Jakob disease (vCJD) clinical diagnosis by pathologic study of adenoid and brain biopsy tissues. The patient has no history of receipt of blood, a past neurosurgical procedure, or residing in or visiting countries of Europe. Based on the patient’s history, the occurrence of a previously reported Saudi case of variant Creutzfeldt-Jakob disease (vCJD) attributed to likely consumption of bovine spongiform encephalopathy-contaminated cattle products in Saudi Arabia, and the expected greater than 7 year incubation period for food-related variant Creutzfeldt-Jakob disease (vCJD), this U.S. case-patient was most likely infected from contaminated cattle products consumed as a child when living in Saudi Arabia. The patient has no history of donating blood and the public health investigation has identified no known risk of transmission to U.S. residents from this patient.
The fourth patient was a US citizen born outside of the United States. The investigation by CDC and the Texas Department of State Health Services indicated that the patient’s exposure to the bovine spongiform encephalopathy agent most likely occurred before he moved to the United States; the patient had resided in Kuwait, Russia and Lebanon. The completed investigation did not support the patient’s having had extended travel to European countries, including the United Kingdom, or travel to Saudi Arabia. The specific overseas country where this patient’s infection occurred is less clear than those for the 3 previously reported US cases largely because the investigation did not definitely link him to a country where other known variant Creutzfeldt-Jakob disease (vCJD)
cases likely had been infected. The patent’s illness first manifested in late 2012 and death occurred 18 months later. The variant Creutzfeldt-Jakob disease (vCJD)
diagnosis was confirmed on the basis of a biochemical analysis of a urine sample collected late in the patient’s illness and by histopathologic examination of brain tissue obtained at autopsy.
Risk for Travelers
The current risk of acquiring variant Creutzfeldt-Jakob disease (vCJD) from eating beef (muscle meat) and beef products produced from cattle in countries with at least a possibly increased risk of bovine spongiform encephalopathy cannot be determined precisely. If public health measures are being well implemented the current risk of acquiring variant Creutzfeldt-Jakob disease (vCJD) from eating beef and beef products from these countries appears to be extremely small, although probably not zero. A rough estimate of this risk for the UK in the recent past, for example, was about 1 case per 10 billion servings 19. Among many uncertainties affecting such risk determinations are 1) the incubation period between exposure to the infective agent and onset of illness, 2) the appropriate interpretation and public health significance of the prevalence estimates of asymptomatic human variant Creutzfeldt-Jakob disease (vCJD) infections, 3) the sensitivities of each country’s surveillance for mad cow disease (bovine spongiform encephalopathy) and variant Creutzfeldt-Jakob disease (vCJD), 4) the compliance with and effectiveness of public health measures instituted in each country to prevent bovine spongiform encephalopathy contamination of human food, and 5) details about cattle products from one country distributed and consumed elsewhere. As of August 2006, despite the apparent exceedingly low risk of contracting variant Creutzfeldt-Jakob disease (vCJD) through consumption of food in Europe, the US blood donor deferral criteria focuses on the time (cumulatively 5 years or more) that a person lived in continental Europe from 1980 through the present. In addition, these deferral criteria apply to persons who lived in the United Kingdom from 1980 through 1996.
Variant Creutzfeldt-Jakob disease (vCJD) symptoms
In variant Creutzfeldt-Jakob disease (vCJD), symptoms that affect a person’s behavior and emotions (psychological symptoms) will usually develop first.
These are then followed by neurological symptoms around 4 months later, which get worse over the following few months.
Initial psychological symptoms
Initial psychological symptoms of variant Creutzfeldt-Jakob disease (vCJD) can include:
- severe depression
- intense feelings of despair
- withdrawal from family, friends and the world around you
- anxiety
- irritability
- difficulty sleeping (insomnia)
Advanced neurological symptoms
Advanced neurological symptoms of all forms of Creutzfeldt-Jakob disease can include:
- loss of physical co-ordination, which can affect a wide range of functions, such as walking, speaking and balance (ataxia)
- muscle twitches and spasms
- loss of bladder control and bowel control
- blindness
- swallowing difficulties (dysphagia)
- loss of speech
- loss of voluntary movement
Advanced psychological symptoms
Advanced psychological symptoms of all forms of Creutzfeldt-Jakob disease include:
- loss of memory, which is often severe
- problems concentrating
- confusion
- feeling agitated
- aggressive behavior
- loss of appetite, which can lead to weight loss
- paranoia
- unusual and inappropriate emotional responses
Final stages
As the condition progresses to its final stages, people with all forms of Creutzfeldt-Jakob disease will become totally bedridden.
They often become totally unaware of their surroundings and require around-the-clock care.
They also often lose the ability to speak and can’t communicate with their carers.
Death will inevitably follow, usually either as a result of an infection, such as pneumonia (a lung infection), or respiratory failure, where the lungs stop working and the person is unable to breathe.
Nothing can be done to prevent death in these circumstances.
Advancements in palliative care (the treatment of incurable conditions) mean that people with Creutzfeldt-Jakob disease often have a peaceful death.
Mad cow disease treatment
There’s no proven cure for Creutzfeldt-Jakob disease (CJD), but clinical studies are underway at the National Prion Clinic to investigate possible treatments.
At present, treatment involves trying to keep the person as comfortable as possible and reducing symptoms with medicines.
For example, psychological symptoms of Creutzfeldt-Jakob disease, such as anxiety and depression, can be treated with sedatives and antidepressants, and muscle jerks or tremors can be treated with medicines like clonazepam and sodium valproate.
Any pain experienced can be relieved using powerful opiate-based painkillers.
Advance directive
Many people with Creutzfeldt-Jakob disease draw up an advance directive (also known as an advance decision).
An advance directive is where a person makes their treatment preferences known in advance in case they can’t communicate their decisions later because they’re too ill.
Issues that can be covered by an advance directive include:
- whether a person with Creutzfeldt-Jakob disease wants to be treated at home, in a hospice, or in a hospital once they reach the final stages of the condition
- what type of medications they’d be willing to take in certain circumstances
- whether they’d be willing to have a feeding tube if they were no longer able to swallow food and liquid
- whether they’re willing to donate any of their organs for research after they die (the brains of people with Creutzfeldt-Jakob disease are particularly important for ongoing research)
- if they lose lung function, whether they’d be willing to be resuscitated by artificial means – for example, by having a breathing tube inserted into their neck
Your care team can provide more advice about making an advance directive.
Specialist team
In United Kingdom, if a person is thought to have Creutzfeldt-Jakob disease, they’re referred to the National Care Team for Creutzfeldt-Jakob disease at the National Creutzfeldt-Jakob disease Research and Surveillance Unit in Edinburgh, or the National Prion Clinic in London, for diagnosis and care.
A doctor and nurse from these services will be assigned to liaise with local services, including the person’s doctor, social worker, physiotherapist and occupational therapist.
Specialist teams are available for diagnosis and to offer clinical and emotional support to patients and their families, and work alongside the local care team.
A local care team may include doctors and nurses, occupational therapists, dietitians, continence advisers and social workers.
Care and support in the advanced stages of Creutzfeldt-Jakob disease
As Creutzfeldt-Jakob disease progresses, people with the condition will need significant nursing care and practical support.
As well as help with feeding, washing and mobility, some people may also need help peeing. A tube inserted into the bladder to drain urine (a catheter) is often required.
Many people will also have problems swallowing, so they may have to be given nutrition and fluids through a feeding tube.
It may be possible to treat someone with Creutzfeldt-Jakob disease at home, depending on the severity and progression of their condition.
Caring for someone with Creutzfeldt-Jakob disease can be distressing and difficult to cope with, so many carers prefer to use the specialist services of a hospital or hospice.
References- Bovine Spongiform Encephalopathy (BSE), or Mad Cow Disease. https://www.cdc.gov/prions/bse/
- Bovine Spongiform Encephalopathy (BSE), or Mad Cow Disease. https://www.cdc.gov/prions/bse/about.html
- BSE in North America. https://www.cdc.gov/prions/bse/bse-north-america.html
- Nature 1996;381:743-4
- Nature 1996;383:685-90
- Nature 1997;389:498-501
- Proc Natl Acad Sci 1999;96:15137-42
- Wroe SJ, Pal S, Siddique D, Hyare H, Macfarlane R, Joiner S, Linehan JM, Brandner S, Wadsworth JD, Hewitt P, Collinge J. Clinical presentation and pre-mortem diagnosis of Creutzfeldt-Jakob disease associated with blood transfusion. Lancet 2006; 368: 2061-2067.
- Hewitt PE, Llewelyn CA, Mackenzie J, Will RG. Creutzfeldt-Jakob disease and blood transfusion: results of the UK Transfusion Medicine Epidemiological Review study. Vox Sang 2006; 91: 221-230.
- Head MW, Yull H, Ritchie DL, Bishop MT, Ironside JW. Pathological investigation of the first donor and recipient pair linked by blood transfusion-associated variant Creutzfeldt-Jakob disease infection. Neuropathol Appl Neurobiol 2009; 35: 433-436.
- Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet 2004; 364: 527-9.
- Millar C, Connor N, Dolan G, Lee CA, Makris M, Wilde J, Winter M, Ironside JW, Gill N, Hill FG. Risk reduction strategies for variant Creutzfeldt-Jakob disease transmission by UK plasma products and their impact on patients with bleeding disorders. Haemophilia 2010; 16: 305-315.
- Gregori L, McCombie N, Palmer D, Birch P, Sowemimo-Coker SO, Giulivi A, Rohwer RG. Effectiveness of leucoreduction for removal of infectivity of transmissible spongiform encephalopathies from blood. Lancet 2004; 364: 529-531.
- Current measures to reduce the risk of vCJD transmission by blood. https://www.gov.uk/government/publications/current-measures-to-reduce-the-risk-of-vcjd-transmission-by-blood
- University of Edinburgh’s National CJD Research & Surveillance Unit. https://www.cjd.ed.ac.uk/
- Prevention Measures against BSE Spread. https://www.cdc.gov/prions/bse/prevention.html
- All About BSE (Mad Cow Disease). https://www.fda.gov/AnimalVeterinary/ResourcesforYou/AnimalHealthLiteracy/ucm136222.htm
- About vCJD. https://www.cdc.gov/prions/vcjd/about.html
- Risk for Travelers. https://www.cdc.gov/prions/vcjd/risk-travelers.html
- Ironside JW, Bishop MT, Connolly K, Hegazy D, Lowrie S, Le Grice M, Ritchie DL, McCardle LM, Hilton DA. BMJ 2006; 332:1186-1188.
- Belay E., Schonberger L. Variant Creutzfeldt-Jakob Disease and Bovine Spongiform Encephalopathy. Clin Lab Me. 2002;22:849-862
- vCJD Cases Reported in the US. https://www.cdc.gov/prions/vcjd/vcjd-reported.html





{kind=link}