Pallister Killian mosaic syndrome
Pallister-Killian syndrome (PKS) also called Pallister-Killian mosaic syndrome or isochromosome 12p syndrome, is a rare chromosome abnormality causing developmental disorder that affects many parts of the body. The signs and symptoms of Pallister-Killian syndrome vary from child to child and range in severity. Pallister-Killian mosaic syndrome is characterized by extremely weak muscle tone (hypotonia) in infancy and early childhood, intellectual disability, distinctive facial features, sparse hair, areas of unusual skin coloring (pigmentation), and other birth defects 1. PKS was first described in 1977 by Pallister in adults 2 and later in 1981 by Killian and Teschler Nicola in children 3 with mental retardation, severe dysmorphic features, and skin changes.
Most babies with Pallister-Killian syndrome are born with significant hypotonia, which can cause difficulty breathing and problems with feeding. Hypotonia also interferes with the normal development of motor skills such as sitting, standing, and walking. About 30 percent of affected individuals are ultimately able to walk without assistance. Additional developmental delays result from intellectual disability, which is usually severe to profound. Speech is often limited or absent in people with this condition.
Pallister-Killian syndrome is associated with a distinctive facial appearance that is often described as “coarse.” Characteristic facial features include a high, rounded forehead; a broad nasal bridge; a short nose; widely spaced eyes; low-set ears; rounded cheeks; and a wide mouth with a thin upper lip and a large tongue. Some affected children are born with an opening in the roof of the mouth (cleft palate) or a high arched palate.
Most children with Pallister-Killian mosaic syndrome have sparse hair on their heads, particularly around the temples. These areas may fill in as affected children get older. Many affected individuals also have streaks or patches of skin that are darker or lighter than the surrounding skin. These skin changes can occur anywhere on the body, and they may be apparent at birth or occur later in life.
Additional features of Pallister-Killian mosaic syndrome can include hearing loss, vision impairment, seizures, extra nipples, genital abnormalities, and heart defects. Affected individuals may also have skeletal abnormalities such as extra fingers and/or toes, large big toes (halluces), and unusually short arms and legs. About 40 percent of affected infants are born with a congenital diaphragmatic hernia, which is a hole in the muscle that separates the abdomen from the chest cavity (the diaphragm). This potentially serious birth defect allows the stomach and intestines to move into the chest, where they can crowd the developing heart and lungs.
The signs and symptoms of Pallister-Killian mosaic syndrome vary, although most people with this disorder have severe to profound intellectual disability and other serious health problems. The most severe cases involve birth defects that are life-threatening in early infancy. However, several affected people have had milder features, including mild intellectual disability and less noticeable physical abnormalities.
Pallister-Killian mosaic syndrome is caused by the presence of four copies of the short arm of chromosome 12 instead of the normal two. The extra two copies of the short arm of chromosome 12 (12p) usually appear as a single chromosome known as isochromosome or i(12p) and are sometimes present in some but not all cells examined (mosaicism). The chromosome abnormality in Pallister-Killian mosaic syndrome is limited to specific cell types. The mechanism and parental origin of the isochromosome 12p can usually not be determined. The extra genetic material disrupts the normal course of development and results in the signs and symptoms of this disorder.
Pallister-Killian mosaic syndrome is not inherited; Pallister Killian syndrome is the result of a random event during the formation of reproductive cells, it usually occurs in the mother. Typically, an error in cell division (nondisjunction) causes a reproductive cell to contain an isochromosome 12p.
Pallister-Killian mosaic syndrome appears to be a rare condition, although its exact prevalence is unknown. The prevalence has been estimated to be 1 in 20,000 4. In a population-based study in Britain estimated the birth incidence of Pallister-Killian syndrome to be 5.1 per million live births 5. Pallister Killian syndrome may be underdiagnosed because it can be difficult to detect in people with mild signs and symptoms. As a result, most diagnoses are made in children with more severe features of the disorder. More than 150 people with Pallister-Killian mosaic syndrome have been reported in the medical literature.
Currently, there is no cure for Pallister-Killian syndrome. Pallister Killian syndrome treatment depends upon the specific symptoms present in each individual. Treating medical and developmental problems early can help to optimize outcome.
Figure 1. Pallister Killian syndrome
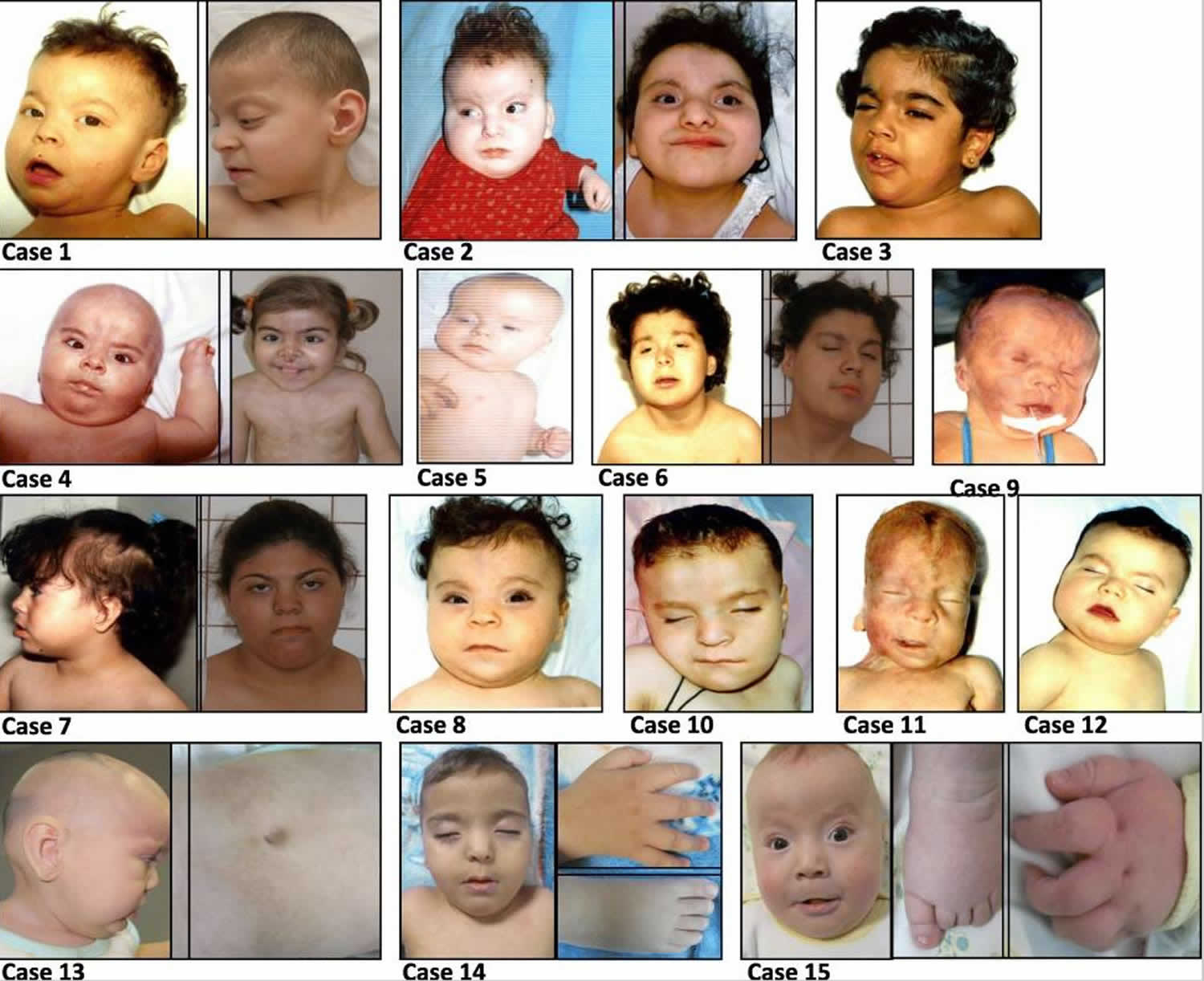
Footnote: Facial appearance of Pallister Killian syndrome patients. Alopecia (hair loss) or sparse temporal hair, hypo- or hyper pigmented areas and other stigmata such as epicanthic folds and slightly low-set ears, prominent forehead, hypertelorism, up- slanting palpebral fissures, anteverted nostrils, long philtrum, everted lower lip, short neck and short hands and toes.
[Source 6 ]Can Pallister Killian syndrome happen again?
Pallister Killian syndrome has only been known to occur sporadically, so that the affected couple and other family members (including siblings of those with mosaic tetrasomy 12p) appear to be no more likely to have another(a) child with Pallister Killian syndrome than anyone else in the population. However, there is a very small theoretical possibility that one parent’s ovaries or testes contain a proportion of cells with the additional 12p isochromosome. This would increase the chances of a recurrence and is the reason you may be offered prenatal testing in your next pregnancy. Anyone with mosaic tetrasomy 12p who is thinking about having children should also have a talk first with a genetic counseling service 7. As far as we are aware there has never been a report of a family having a second child with PKS.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Pallister-Killian mosaic syndrome causes
Pallister-Killian mosaic syndrome is usually caused by the presence of an abnormal extra chromosome called an isochromosome 12p or i(12p). An isochromosome is a chromosome with two identical arms. Normal chromosomes have one long (q) arm and one short (p) arm, but isochromosomes have either two q (long) arms or two p (short) arms. Isochromosome 12p is a version of chromosome 12 made up of two p (short) arms. It has been reported that the risk for isochromosome 12p (tetrasomy 12p) increases with advanced maternal age 8.
Cells normally have two copies of each chromosome, one inherited from each parent. In people with Pallister-Killian mosaic syndrome, cells have the two usual copies of chromosome 12, but some cells also have the isochromosome 12p. These cells have a total of four copies of all the genes on the p arm of chromosome 12. The extra genetic material from the isochromosome disrupts the normal course of development, causing the characteristic features of this disorder.
Although Pallister-Killian mosaic syndrome is usually caused by the presence of an isochromosome 12p, other, more complex chromosomal changes involving chromosome 12 are responsible for the disorder in rare cases.
Pallister Killian syndrome inheritance pattern
Pallister-Killian mosaic syndrome is not inherited. The chromosomal change responsible for Pallister-Killian mosaic syndrome typically occurs as a random event during the formation of reproductive cells (eggs or sperm) in a parent of the affected individual, usually the mother. Affected individuals have no history of the disorder in their families.
An error in cell division called nondisjunction likely results in a reproductive cell containing an isochromosome 12p. If this atypical reproductive cell contributes to the genetic makeup of a child, the child will have two normal copies of chromosome 12 along with an isochromosome 12p.
As cells divide during early development, some cells lose the isochromosome 12p, while other cells retain the abnormal chromosome. This situation is called mosaicism. Almost all cases of Pallister-Killian mosaic syndrome are caused by mosaicism for an isochromosome 12p. If all of the body’s cells contained the isochromosome, the resulting syndrome would probably not be compatible with life.
Pallister-Killian syndrome signs and symptoms
Pallister Killian syndrome has the following characteristics 9:
- Low muscle tone (hypotonia)
- Facial features that are common to Pallister Killian syndrome such as:
- Large forehead that is often broad
- Depressed or broad nasal bridge
- Widely-spaced eyes (orbital hypertelorism), which may include droopy eyelids (ptosis)
- Low-set ears with thickened helices (top rim of the ear)
- Cupid’s bow lip with extension of the phitral skin into the vermilion border (termed Pallister lip)
- Highly arched or cleft palate
- Thin hair on the head and sparse eyebrows
- Sparse scalp hair at birth
- High, arched palate
- Hypopigmented and/or hyperpigmented lesions (patches of lighter or darker skin)
- Extra nipples
- Intellectual disabilities. Although most PKS children have profound mental retardation, many children are only mildly handicapped 10.
- Developmental and speech delays.
- Congenital diaphragmatic hernia (a hole in the muscular wall separating the heart and lungs from the contents of the abdomen that is present at birth)
Other features of Pallister Killian syndrome:
- Heart condition
- Anal atresia (the anus may be closed or absent)
- Sacral appendage (bony outgrowth at the base of the spine)
- Short arms, legs, hands, feet, fingers or toes
- Exomphalos (a defect in the abdominal wall that causes the intestines, liver and occasionally other organs to remain outside of the abdomen in a sac)
- Polydactyly (extra fingers or toes)
- Hip dislocation
- Joints that are stiff or will not move (contractures)
- Anhydrosis or hypohydrosis (deficiency or absence of sweating)
- Seizures
- Hearing and vision problems
- Lung and breathing difficulties
- Abnormalities of the genitals and genitourinary system
Individuals with Pallister-Killian mosaic syndrome typically have low muscle tone at birth (hypotonia), sparse scalp hair, a high forehead, a coarse face, an abnormally wide space between the eyes, a broad nasal bridge, a highly arched palate, a fold of the skin over the inner corner of the eyes, and large ears with lobes that are thick and protrude outward.
Infants that are born with significant hypotonia can experience problems with feeding, breathing, walking and standing. About seventy percent of affected individuals are unable to walk without assistance.
Additional features frequently found in affected individuals may include streaks of skin in which there is no color (hypopigmentation); extra nipples; seizures; droopy upper eyelids, crossed eyes (strabismus); joints that will not move (contractures); and delays in perceiving, recognizing, judging, sensing, reasoning or imagining (cognitive delays). Intellectual disability and difficulties with speech development often occur as well. In rare cases, affected children may experience hearing loss.
Congenital heart defects, hernias of the diaphragm, a narrowing of the external auditory canal (stenosis) and an abnormal opening in the anus have also been associated with Pallister-Killian mosaic syndrome. Some affected individuals may have an underdeveloped (hypoplastic) lung, abnormalities of the genitourinary system, and skeletal malformations. Symptoms may vary according to which tissue has the additional chromosomal material, and may also affect each side of the body unevenly.
Pallister Killian syndrome seizures
Seizures affect about half of the children with Pallister Killian syndrome, although this value varies greatly between reports and may be dependent on the selection method for each study and the age of the participants 5. Seizures may start in babyhood, in childhood or not until puberty or adulthood. Seizures types vary, with nocturnal seizures reported by some families. Families do not perceive a link between seizure activity and their child’s ability to learn. Some children with reasonably fluent speech also have seizures.
Pallister-Killian syndrome diagnosis
Chromosome disorders are usually diagnosed by examining DNA taken from white blood cells. However, children with mosaic tetrasomy 12p often appear to have normal chromosomes in their blood cells, so the diagnosis is usually made from skin cells or cells taken from inside the cheek (buccal mucosa).
At present fluorescence in situ hybridization (FISH) using chromosome 12 centromeric probes on buccal mucosa cells is a very useful, non-invasive, reliable, rapid and cost-effective test for the definite diagnosis of Pallister Killian syndrome 11. Pallister Killian syndrome diagnosis is usually made from a chromosome study of skin cells (fibroblasts) that reveals 47 chromosomes including an extra small chromosome that has two short (p) arms and no long (q) arm (isochromosome). Chromosomal microarray, the first tier analyses in malformation syndromes, can also reveal Pallister Killian syndrome, if the right tissue is selected. Blood chromosome analyses usually shows normal number of chromosomes, but some affected persons have some blood cells (lymphocytes) with an isochromosome 12p. Cells with high cell turnover such as blood may lose the additional chromosomal material over time, and thereby give a false negative result on blood. Therefore, a normal blood chromosomal analysis does not completely rule out Pallister-Killian mosaic syndrome.
Two individuals with Pallister-Killian mosaic syndrome have been reported with five copies (hexasomy) for chromosome 12p.
Pallister-Killian mosaic syndrome can also be diagnosed before birth (prenatally) by removing a small amount of fluid that is in the womb during pregnancy (amniocentesis) or by removing a small number of cells from outside the sac where the fetus develops (chorionic villous sampling). Early prenatal diagnosis helps families to choose between continuation and termination of pregnancy, as it poses significant emotional and financial burden. Families may be better equipped to cope with the pregnancy and the care of the infant after birth when they choose to continue pregnancy. However, cell cultures may yield a false negative and a normal chromosomal analysis does not completely rule out the condition 12. There are very few reported cases in which the isochromosome was diagnosed in peripheral lymphocytes 13. The detection rate is 0–2% in lymphocytes, 50–100% in fibroblasts, and 100% in amniocytes and bone marrow cells 14.
Chiesa et al. 15 in 1988 reported the first case diagnosed prenatally based on fetal blood cells after cordocentesis in the second trimester. FISH was used to identify the interphase or the metaphase cells with the isochromosome 15.
Additional imaging tests, such as X-rays, MRIs, CT scans and ultrasounds, may be done to address specific areas of concern for your child.
Pallister-Killian syndrome treatment
Currently, there is no cure for Pallister-Killian syndrome. Treatments offered to children with Pallister-Killian syndrome can help manage each child’s specific symptoms and developmental needs. The goal of all these options is to help children with Pallister-Killian mosaic syndrome live as normal a life as possible and maximize their potential.
Depending on your child’s set of symptoms, treatment options may include:
- Surgery to address medical conditions such as congenital diaphragmatic hernia, heart anomalies, genitourinary conditions and cleft palate.
- Respiratory and breathing support, especially for younger children with weak muscle tone.
- Physical, occupational and recreational therapy to build muscle mass and address fine and gross motor skills.
- Enrollment in early intervention programs to address developmental delays, intellectual differences and issues with communication (such as speech, hearing and vision problems).
- Assisted communication devices and sign language training. Most children with Pallister-Killian mosaic syndrome speak later than other children and may develop only limited vocabulary throughout their lives.
- Bracing, casting or surgery to address bone issues. Some children with Pallister-Killian may develop a spine curvature (scoliosis or kyphosis) during childhood.
- Appropriate management of seizures, if present.
Pallister-Killian syndrome prognosis
The highly variable features of Pallister Killian syndrome remain problematic for determining the prognosis or the outcome of having mosaic tetrasomy 12p. Further studies and advanced techniques are needed to be established the correlation between mosaic ratio and clinical manifestations 16.
Children with Pallister-Killian syndrome will require coordinated, life-long medical care. Some babies born with Pallister Killian syndrome also face life-threatening birth defects in infancy and may not survive early childhood.
Babies with Pallister Killian syndrome may be vulnerable shortly after birth since they are often born prematurely and can have central nervous system anomalies and apnea (when they stop breathing). They are also prone to respiratory difficulties and may be born with a heart defect. One of the most severe problems that some babies with Pallister Killian mosaic syndrome can suffer is a diaphragmatic hernia and some do not cope with life outside the womb or the necessary surgery. However, if these possible early problems are overcome and later medical issues such as gastroesophageal reflux, intestinal problems and seizures are dealt with, it may be possible to manage health effectively and children with Pallister Killian mosaic syndrome could expect a long lifespan. The oldest patient described in the medical literature is 45 years old.A recent study of Pallister Killian syndrome families in the United Kingdom 5 identified eight individuals who had passed away. Ages ranged enormously between one hour (the baby was born at 21 weeks gestation) and 38 years. The youngest died due to prematurity, one adult died of aspiration, another of epilepsy and three children and one adult were described as having had respiratory infections at the time of death.
Most children will have learning difficulties and attend special schools where they can receive individualized instruction. Some — with milder forms of the disorder — can be educated in mainstream schools with the help of dedicated teachers and support workers.
In adulthood, many individuals with Pallister-Killian syndrome will remain dependent on parents and family members. There are some adults with milder forms of the condition who are able to have jobs and live more independent lives at home or in a group-home setting. Ongoing support, encouragement and monitoring will be needed.
Pallister Killian syndrome life span
Life expectancy in Pallister Killian syndrome has not been formally looked at but what scientists do know is this: there seems to be two or three main time points to consider. For the very severely affected newborn with a diaphragmatic hernia or severe congenital heart defect or other severe structural or functional anomaly this will have the most significant effect on morbidity and mortality and many newborns succumb early form these congenital differences. After that experts would say the most concerning issues that impact life expectancy are undiagnosed/untreated medical issues, such as reflux, malrotation of the intestine or seizures. Experts advocate for aggressive clinical evaluation’s for these issues and if present for aggressive treatment as this will not only improve health and life expectancy but also optimize learning and happiness for the children (any parent should understand the impact that untreated gastroesophageal reflux, for example, can have on a child resulting in constant/recurrent pain and discomfort, risk for aspiration and damage to the esophagus). For the child with PKS who has overcome these neonatal issues and is being managed effectively for the commonly associated medical issues, lifespan should be long or close to normal. For any individual with chronic medical issues like seizures or cognitive impairment there is an overall impact on life expectancy but not a large one. Experts know of some individuals with Pallister Killian syndrome in their 40s and 50s, and it is likely that there are many older individuals out there who have never been diagnosed since the type of testing available in the past decade or so is primarily used in the pediatric setting. Like most issues concerning rare diagnoses like Pallister Killian mosaic syndrome, the primary point of entry for scientists to learn about these issues is through the parents and support groups like PKS Kids (https://www.pkskids.com). It would be great for parents to report the ages of their kids as well as for parents who have lost a child with Pallister Killian syndrome to report how old their child was were when they passed away and what the cause of death was. This could form the initial database that can eventually be used to better answer this question as well as to help identify causes of death in individuals with Pallister Killian syndrome so that scientists can better identify those problems early and avoid bad outcomes.
References- Pallister-Killian mosaic syndrome. https://medlineplus.gov/genetics/condition/pallister-killian-mosaic-syndrome
- Pallister PD, Meisner LF, Elejalde BR, Francke U, Herrmann J, Spranger J, Tiddy W, Inhorn SL, Opitz JM. The pallister mosaic syndrome. Birth Defects Orig Artic Ser. 1977;13(3B):103-110.
- Teschler-Nicola M, Killian W. Case report 72: Mental retardation, unusual facial appearance, abnormal hair. Synd Ident. 1981;7:6–7.
- Pallister Killian syndrome. https://www.chop.edu/conditions-diseases/pallister-killian-syndrome
- Blyth, M., Maloney, V., Beal, S., Collinson, M., Huang, S., Crolla, J., Temple, I. K., Baralle, D.( 2015) .Pallister-Killian syndrome: a study of 22 British patients. J. Med. Genet. 52: 454-464.
- Karaman B, Kayserili H, Ghanbari A, et al. Pallister-Killian syndrome: clinical, cytogenetic and molecular findings in 15 cases. Mol Cytogenet. 2018;11:45. Published 2018 Aug 17. doi:10.1186/s13039-018-0395-z https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6098576
- Bérénice Doray, Françoise Girard-Lemaire, Bernard Gasser, Jean-Jacques Baldauf, Bernard De Geeter, Michèle Spizzo, Charles Zeidan, Elisabeth Flori. Pallister-Killian syndrome: difficulties of prenatal diagnosis. Prenatal diagnosis. 2002 Jun;22(6):470-7.
- Wenger SL, Boone LY, Steele MW. Mosaicism in Pallister i(12p) syndrome. Am J Med Genet. 1990;35(4):523–525. doi: 10.1002/ajmg.1320350416
- What is PKS? https://www.pkskids.com/copy-of-about-pks
- Bielanska MM, Khalifa MM, Duncan AM. Pallister-Killian syndrome: a mild case diagnosed by fluorescence in situ hybridization. Review of the literature and expansion of the phenotype. Am J Med Genet. 1996 Oct 16;65(2):104-8. doi: 10.1002/(SICI)1096-8628(19961016)65:2<104::AID-AJMG4>3.0.CO;2-S
- Manasse BF, et al. The Pallister-Killian syndrome is reliably diagnosed by FISH on buccal mucosa. Clin Dysmorphol. 2000;9(3):163–165. doi: 10.1097/00019605-200009030-00002
- Choo S, Teo SH, Tan M, Yong MH, Ho LY. Tissue-limited mosaicism in Pallister-Killian syndrome — a case in point. J Perinatol. 2002 Jul-Aug;22(5):420-3. doi: 10.1038/sj.jp.7210712
- Conlin LK, Kaur M, Izumi K, Campbell L, Wilkens A, Clark D, Deardorff MA, Zackai EH, Pallister P, Hakonarson H, Spinner NB, Krantz ID. Utility of SNP arrays in detecting, quantifying, and determining meiotic origin of tetrasomy 12p in blood from individuals with Pallister-Killian syndrome. Am J Med Genet A. 2012 Dec;158A(12):3046-53. doi: 10.1002/ajmg.a.35726
- Jamuar S, Lai A, Unger S, Nishimura G. Clinical and radiological findings in Pallister-Killian syndrome. Eur J Med Genet. 2012 Mar;55(3):167-72. doi: 10.1016/j.ejmg.2012.01.019
- Chiesa J, Hoffet M, Rousseau O, Bourgeois JM, Sarda P, Mares P, Bureau JP. Pallister-Killian syndrome [i(12p)]: first pre-natal diagnosis using cordocentesis in the second trimester confirmed by in situ hybridization. Clin Genet. 1998 Oct;54(4):294-302. doi: 10.1034/j.1399-0004.1998.5440406.x
- Li L, Huang L, Huang X, Lin S, He Z, Fang Q. Prenatal diagnosis of Pallister-Killian syndrome in one twin. Clin Case Rep. 2018;6(8):1470-1473. Published 2018 Jun 13. doi:10.1002/ccr3.1624 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6099054





{kind=link}