Rasopathy
RASopathy is a group of genetic syndromes caused by germline mutations in genes that encode components or regulators of the Ras-mitogen activated protein kinase (MAPK) pathway 1. They’re called RASopathies because they’re caused by problems in the Ras-MAPK pathway, which is one way cells in the body communicate. The Ras-MAPK pathway plays an essential role in regulating the cell cycle and cellular growth, differentiation, and senescence, all of which are critical to normal development (Figure 1). Therefore, it is not surprising that Ras-MAPK pathway dysregulation has profound deleterious effects on both embryonic and later stages of development. The Ras/MAPK pathway has been well studied in cancer and is an attractive target for small-molecule inhibition to treat various malignancies. The use of these molecules to ameliorate developmental defects in the RASopathies is under consideration.
The individual RASopathies are rare, but as a group:
- They’re among the most common genetic conditions.
- RASopathies cause most genetic-related learning and development problems.
RASopathies include neurofibromatosis type 1 (NF1), Noonan syndrome, Noonan syndrome with multiple lentigines (formerly called LEOPARD syndrome), capillary malformation–arteriovenous malformation syndrome, Costello syndrome, cardio-facio-cutaneous syndrome, and Legius syndrome 1. Because of the common underlying Ras/MAPK pathway dysregulation, the RASopathies exhibit numerous overlapping phenotypic features. Rasopathy syndromes share many clinical features such as distinct facial features, developmental delays, cardiac defects, growth delays, neurologic issues, and gastrointestinal difficulties. While these individual syndromes are rare, as a group, the RASopathies are among the most common genetic conditions in the world.
The MAPK pathway is one of several critical downstream signaling cascades of Ras. Activated Ras leads to the activation of Raf (ARAF, BRAF, and/or CRAF), the first MAPK kinase kinase of the pathway. Raf phosphorylates and activates the MAPK kinases MEK1 and/or MEK2; these in turn phosphorylate and activate ERK1 and/or ERK2. ERK1 and ERK2 are the ultimate effectors and exert their function on a large number of downstream molecules, both nuclear and cytosolic. ERK1 and ERK2 substrates include nuclear components, transcription factors, membrane proteins, and protein kinases that in turn control vital cellular functions, including cell cycle progression, cellular differentiation, and cellular growth 2.
The Ras/MAPK pathway has been studied extensively in the context of oncogenesis because its somatic dysregulation is one of the primary causes of cancer. Ras is somatically mutated in approximately 20% of malignancies (8), and BRAF is somatically mutated in approximately 7% of malignancies 3. Because of this, the RASopathies are considered cancer syndromes, with the majority of associated mutations resulting in enhanced pathway activation or dysregulated signaling. However, biochemical studies have demonstrated that a large fraction of the novel germline mutations identified in the pathway are not as robustly activating as those associated with oncogenesis. This is likely due to the embryonic lethality arising from these germline mutations 1.
Figure 1. Ras/mitogen-activated protein kinase (MAPK) pathway
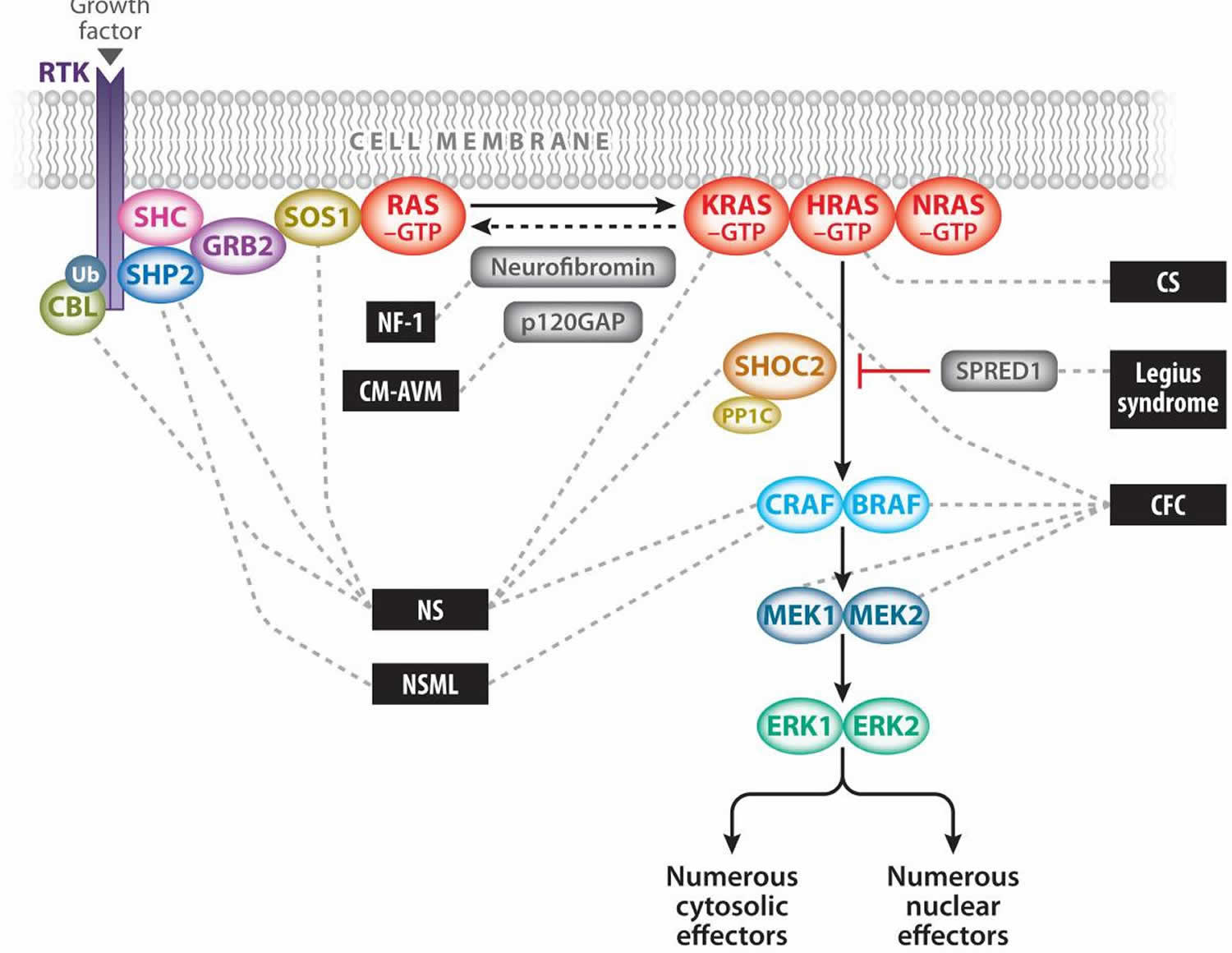
Footnote: The Ras/MAPK signal transduction pathway. The MAPK signaling pathway of protein kinases is critically involved in cellular proliferation, differentiation, motility, apoptosis, and senescence. The RASopathies are medical genetic syndromes caused by mutations in genes that encode components or regulators of the Ras/MAPK pathway (indicated by dashed lines). These disorders include neurofibromatosis type 1 (NF1), Noonan syndrome (NS), Noonan syndrome with multiple lentigines (NSML), capillary malformation–ateriovenous malformation syndrome (CM-AVM), Costello syndrome (CS), cardio-facio-cutaneous syndrome (CFC), and Legius syndrome.
[Source 1 ]Rasopathy genes
RAS genes constitute a multigene family that includes HRAS, NRAS, and KRAS 1. Ras proteins are small guanosine nucleotide-bound GTPases that function as a critical signaling hub within the cell. They are activated through growth factors binding to receptor tyrosine kinases (RTKs), G-protein-coupled receptors, cytokine receptors, and extracellular matrix receptors. Ras proteins cycle between an active GTP-bound form and an inactive GDP-bound form. Activation through RTKs occurs with the binding of a growth factor that causes RTK autophosphorylation and interaction with the adaptor protein growth factor receptor-bound protein 2 (GRB2). GRB2 is bound to son of sevenless (SOS), which is then recruited to the plasma membrane. SOS proteins are guanosine nucleotide exchange factors (GEFs) that increase the Ras nucleotide exchange rate of GDP for GTP, thereby increasing the level of active GTP-bound Ras.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Rasopathy syndrome
Each RASopathy exhibits a unique phenotype, but owing to the common mechanisms of Ras/MAPK pathway dysregulation, they share many overlapping characteristics, including craniofacial dysmorphology; cardiac malformations; cutaneous, musculoskeletal, and ocular abnormalities; neurocognitive impairment; hypotonia; and an increased cancer risk. Taken together, they are one of the largest known groups of malformation syndromes, affecting approximately 1 in 1,000 individuals. Neurofibromatosis type 1 (NF1) was the first syndrome identified as being caused by mutation of a gene in the Ras/MAPK pathway (NF1) 4, and numerous other syndromes have subsequently been identified. These disorders include:
- Noonan syndrome caused by activating mutations in PTPN11 5, SOS1 6, RAF1 7, KRAS 8, NRAS 9, SHOC2 10, and CBL 11;
- Noonan syndrome with multiple lentigines caused by mutations in PTPN11 12 and RAF1 13;
- Capillary malformation–arteriovenous malformation syndrome caused by haploinsufficiency of RASA1 14;
- Costello syndrome caused by activating mutations in HRAS 15;
- Cardio-facio-cutaneous syndrome caused by alteration of MAPK pathway activation by activating mutations in BRAF 16 and MAP2K1 (MEK1) or MAP2K2 (MEK2) 17;
- Legius syndrome caused by inactivating mutations in SPRED1 18.
Neurofibromatosis type 1
Neurofibromatosis type 1 also called von Recklinghausen’s disease, is a autosomal dominant genetic disorder characterized by changes in skin coloring (pigmentation) and the growth of tumors along nerves in the skin, brain, and other parts of the body. The signs and symptoms of neurofibromatosis type 1 vary widely among affected people. Neurofibromatosis type 1 occurs in 1 in 3,000 to 4,000 people worldwide 19.
Beginning in early childhood, almost all people with neurofibromatosis type 1 have multiple café-au-lait spots, which are flat patches on the skin that are darker than the surrounding area. These spots increase in size and number as the individual grows older. Freckles in the underarms and groin typically develop later in childhood.
Most adults with neurofibromatosis type 1 develop neurofibromas, which are noncancerous (benign) tumors that are usually located on or just under the skin. These tumors may also occur in nerves near the spinal cord or along nerves elsewhere in the body. Some people with neurofibromatosis type 1 develop cancerous tumors that grow along nerves. These tumors, which usually develop in adolescence or adulthood, are called malignant peripheral nerve sheath tumors. People with neurofibromatosis type 1 also have an increased risk of developing other cancers, including brain tumors and cancer of blood-forming tissue (leukemia).
During childhood, benign growths called Lisch nodules often appear in the colored part of the eye (the iris). Lisch nodules do not interfere with vision. Some affected individuals also develop tumors that grow along the nerve leading from the eye to the brain (the optic nerve). These tumors, which are called optic gliomas, may lead to reduced vision or total vision loss. In some cases, optic gliomas have no effect on vision.
Additional signs and symptoms of neurofibromatosis type 1 vary, but they can include high blood pressure (hypertension), short stature, an unusually large head (macrocephaly), and skeletal abnormalities such as an abnormal curvature of the spine (scoliosis). Although most people with neurofibromatosis type 1 have normal intelligence, learning disabilities and attention-deficit/hyperactivity disorder (ADHD) occur frequently in affected individuals.
Figure 2. Neurofibromatosis type 1
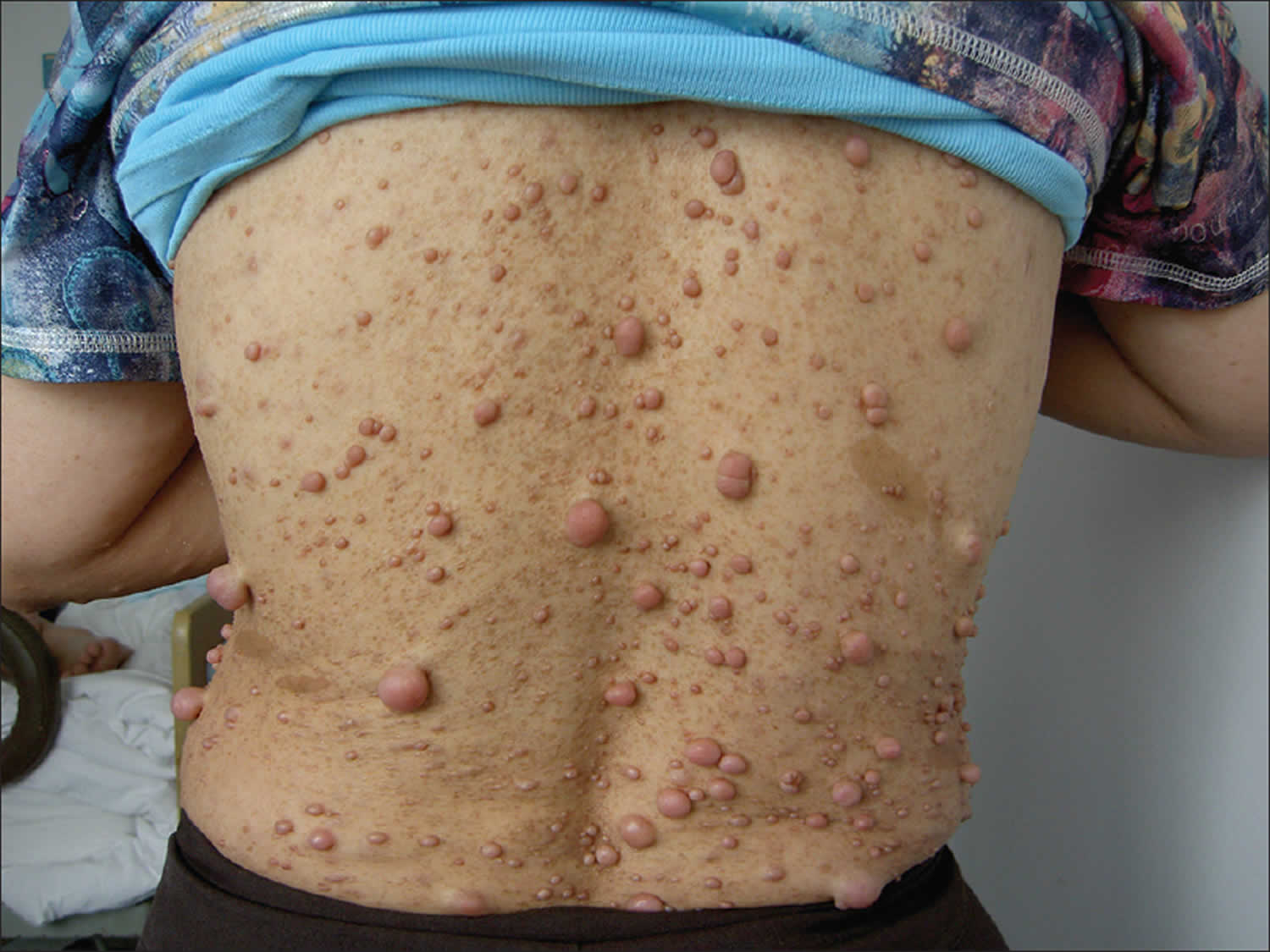
Footnote: Neurofibromas and multiple café-au-lait spots
Neurofibromatosis type 1 causes
Mutations in the NF1 gene cause neurofibromatosis type 1. The NF1 gene provides instructions for making a protein called neurofibromin. This protein is produced in many cells, including nerve cells and specialized cells surrounding nerves (oligodendrocytes and Schwann cells). Neurofibromin acts as a tumor suppressor, which means that it keeps cells from growing and dividing too rapidly or in an uncontrolled way. Mutations in the NF1 gene lead to the production of a nonfunctional version of neurofibromin that cannot regulate cell growth and division. As a result, tumors such as neurofibromas can form along nerves throughout the body. It is unclear how mutations in the NF1 gene lead to the other features of neurofibromatosis type 1, such as café-au-lait spots and learning disabilities.
Neurofibromatosis type 1 inheritance pattern
Neurofibromatosis type 1 is considered to have an autosomal dominant pattern of inheritance. People with this condition are born with one mutated copy of the NF1 gene in each cell. In about half of cases, the altered gene is inherited from an affected parent. The remaining cases result from new mutations in the NF1 gene and occur in people with no history of the disorder in their family.
Unlike most other autosomal dominant conditions, in which one altered copy of a gene in each cell is sufficient to cause the disorder, two copies of the NF1 gene must be altered to trigger tumor formation in neurofibromatosis type 1 20. A mutation in the second copy of the NF1 gene occurs during a person’s lifetime in specialized cells surrounding nerves. Almost everyone who is born with one NF1 mutation acquires a second mutation in many cells and develops the tumors characteristic of neurofibromatosis type 1.
Neurofibromatosis type 1 signs and symptoms
The following list includes the most common signs and symptoms in people with neurofibromatosis type 1. These features may be different from person to person. Some people may have more symptoms than others and symptoms can range from mild to severe. This list also does not include every symptom or feature that has been described in this condition.
Neurofibromatosis type 1 signs and symptoms may include 21:
- Non-cancerous growths along the nerves under the skin (cutaneous neurofibromas)
- Large growths along nerves that may become cancerous (plexiform neuromas)
- Dark spots of skin (café au lait spots)
- Freckling, especially in the underarm and groin
- Pigment in the colored part of the eye (Lisch nodules)
- Learning disabilities
- Seizures
- Autism spectrum disorder
- High blood pressure
- Short stature
- Large head (macrocephaly)
- Curvature of the spine (scoliosis)
In many cases, the first symptom of neurofibromatosis type 1 (NF1) is multiple small dark colored birth marks known as café-au-lait spots. As they grow older, people with NF1 develop neurofibromas, benign tumors that can affect nearly any nerve in the body. These tumors usually grow on or just underneath the skin, but neurofibromas can also grow in other places in the body and may even affect multiple nerves. Some of these tumors can cause skin irritation, nerve damage, or affect a person’s appearance. In addition, some of these tumors may become cancerous. The most common type of cancerous tumors in people with NF1 are malignant peripheral nerve sheath tumors 22.
Neurofibromatosis type 1 diagnosis
The diagnosis of neurofibromatosis type 1 (NF1) is based on clinical examination and the presence of characteristic signs and symptoms. Diagnostic criteria have been published to help health care professionals make a diagnosis of neurofibromatosis type 1 23. Genetic testing for genetic changes (DNA variants) in the NF1 gene is available, and can help make the diagnosis or can help exclude other conditions that might look like neurofibromatosis type 1 24.
Neurofibromatosis type 1 treatment
The treatment of neurofibromatosis type 1 (NF1) is based on the signs and symptoms present in each person. Treatment may include surgery to remove neurofibromas that are disfiguring, irritating or cancerous. There is currently no way to prevent or stop the growth of the tumors associated with neurofibromatosis type 1. Guidelines have been published for taking care of children and adults with neurofibromatosis type 1 25.
Specialists involved in the care of someone with neurofibromatosis type 1 may include 22:
- Neurologist
- Neurosurgeon
- Ophthalmologist
- Genetics specialist
- Developmental specialist
Noonan syndrome
Noonan syndrome is a genetic condition that affects many areas of the body. Noonan syndrome is characterized by mildly unusual facial features, short stature, heart defects, bleeding problems, skeletal malformations, and many other signs and symptoms. Noonan syndrome occurs in approximately 1 in 1,000 to 2,500 people for severe phenotype 26, but mild cases may be as common as one in 100 live births 27.
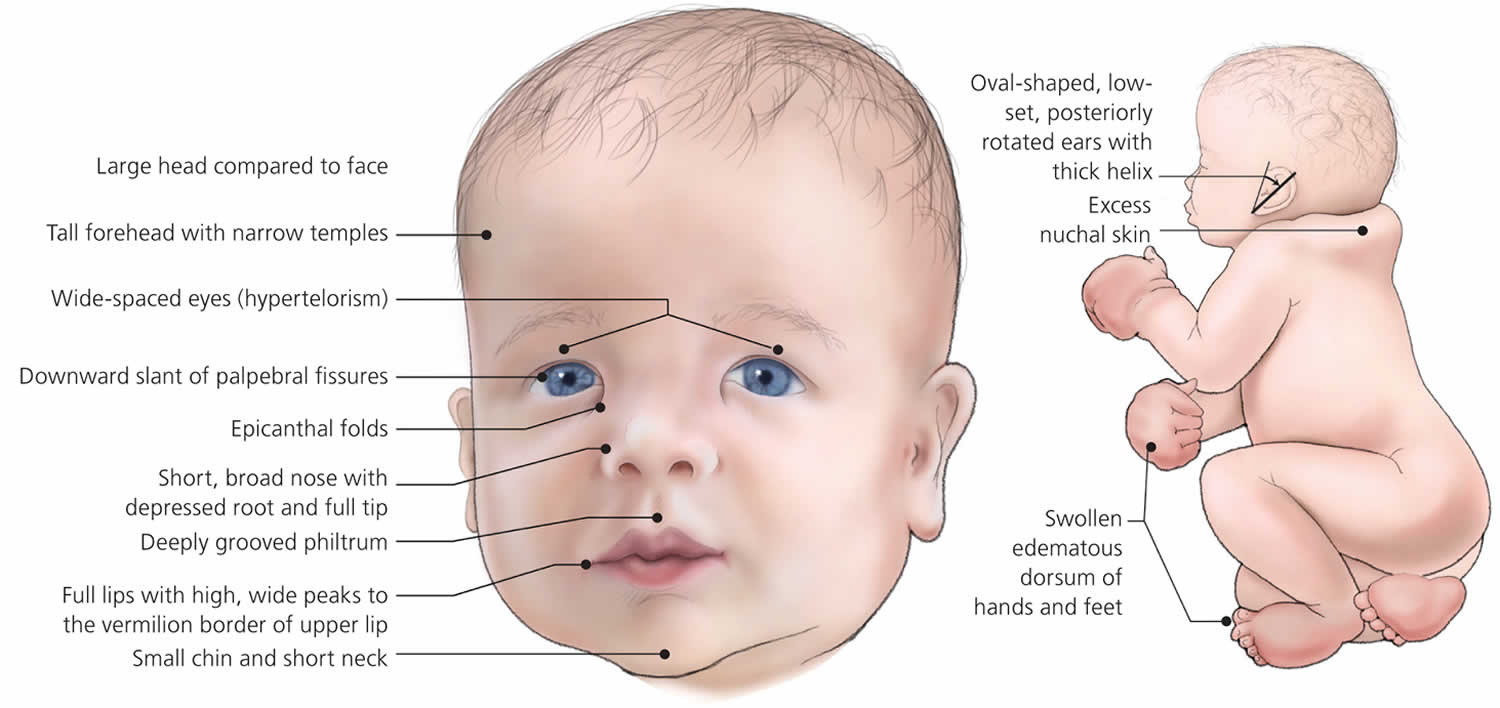
People with Noonan syndrome have distinctive facial features such as a deep groove in the area between the nose and mouth (philtrum), widely spaced eyes that are usually pale blue or blue-green in color, and low-set ears that are rotated backward. Affected individuals may have a high arch in the roof of the mouth (high-arched palate), poor teeth alignment, and a small lower jaw (micrognathia). Many children with Noonan syndrome have a short neck, and both children and adults may have excess neck skin (also called webbing) and a low hairline at the back of the neck.
Between 50 and 70 percent of individuals with Noonan syndrome have short stature. At birth, they are usually a normal length and weight, but growth slows over time. Abnormal levels of growth hormone, a protein that is necessary for the normal growth of the body’s bones and tissues, may contribute to the slow growth.
Individuals with Noonan syndrome often have either a sunken chest (pectus excavatum) or a protruding chest (pectus carinatum). Some affected people may also have an abnormal side-to-side curvature of the spine (scoliosis).
Most people with Noonan syndrome have some form of critical congenital heart disease. The most common heart defect in these individuals is a narrowing of the valve that controls blood flow from the heart to the lungs (pulmonary valve stenosis). Some have hypertrophic cardiomyopathy, which enlarges and weakens the heart muscle.
A variety of bleeding disorders have been associated with Noonan syndrome. Some affected individuals have excessive bruising, nosebleeds, or prolonged bleeding following injury or surgery. Rarely, women with Noonan syndrome who have a bleeding disorder have excessive bleeding during menstruation (menorrhagia) or childbirth.
Adolescent males with Noonan syndrome typically experience delayed puberty. They go through puberty starting at age 13 or 14 and have a reduced pubertal growth spurt that results in shortened stature. Most males with Noonan syndrome have undescended testes (cryptorchidism), which may contribute to infertility (inability to father a child) later in life. Females with Noonan syndrome can experience delayed puberty but most have normal puberty and fertility.
Noonan syndrome can cause a variety of other signs and symptoms. Most children diagnosed with Noonan syndrome have normal intelligence, but a few have special educational needs, and some have intellectual disability. Some affected individuals have vision or hearing problems. Affected infants may have feeding problems, which typically get better by age 1 or 2 years. Infants with Noonan syndrome may be born with puffy hands and feet caused by a buildup of fluid (lymphedema), which can go away on its own. Older individuals can also develop lymphedema, usually in the ankles and lower legs.
Some people with Noonan syndrome develop cancer, particularly those involving the blood-forming cells (leukemia). It has been estimated that children with Noonan syndrome have an eightfold increased risk of developing leukemia or other cancers over age-matched peers.
Figure 3. Noonan syndrome
Noonan syndrome causes
Mutations in multiple genes can cause Noonan syndrome. Mutations in the PTPN11 gene cause about half of all cases. SOS1 gene mutations cause an additional 10 to 15 percent, and RAF1 and RIT1 genes each account for about 5 percent of cases. Mutations in other genes each account for a small number of cases. The cause of Noonan syndrome in 15 to 20 percent of people with this disorder is unknown.
The PTPN11, SOS1, RAF1, and RIT1 genes all provide instructions for making proteins that are important in the RAS-MAPK cell signaling pathway, which is needed for cell division and growth (proliferation), the process by which cells mature to carry out specific functions (differentiation), and cell movement (migration). Many of the mutations in the genes associated with Noonan syndrome cause the resulting protein to be turned on (active) longer than normal, rather than promptly switching on and off in response to cell signals. This prolonged activation alters normal RAS-MAPK signaling, which disrupts the regulation of cell growth and division, leading to the characteristic features of Noonan syndrome.
Rarely, Noonan syndrome is associated with genes that are not involved in the RAS-MAPK cell signaling pathway. Researchers are working to determine how mutations in these genes can lead to the signs and symptoms of Noonan syndrome.
Noonan syndrome inheritance pattern
Noonan syndrome is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
Noonan syndrome signs and symptoms
Some of the signs and symptoms seen in people with Noonan syndrome are listed below 26. Please note that the list below does not include all possible symptoms of Noonan syndrome and that not all people with Noonan syndrome will have all of these the signs and symptoms. In general, the signs and symptoms of Noonan syndrome are more obvious early in life and become less obvious as individuals get older 28.
Head/Neck:
- Widely spaced eyes (hypertelorism)
- Large ears rotated back
- Short webbed neck
- Droopy eyelids (ptosis)
- Low hairline
- Multiple giant cell lesions: painless, benign growths in the jaw that can lead to dental or orthodontic issues
Heart:
- Pulmonary stenosis
- Aortic regurgitation
- Atrial septal defect
Skeletal:
- Short stature
- Concave chest (pectus excavatum)
- Bending or curvature of the finger (clinodactyly)
- Weak bones (generalized osteopenia)
Skin:
- Café au lait spots
- Hemangioma (raised red birthmark)
Neurological:
- Delayed milestones due to low muscle tone
- Developmental delay
- Learning disabilities or intellectual impairment
- Bleeding disorder
- Undescended testicles (cryptorchidism)
A syndrome named Noonan-like-multiple giant cell lesion syndrome used to be considered a separate condition from Noonan syndrome. It is now known that multiple giant cell lesions are one of the possible symptoms that can occur in people with Noonan syndrome 29.
Noonan syndrome diagnosis
Noonan syndrome is diagnosed on clinical grounds by observation of key features 28. Affected individuals have normal chromosome studies. Molecular genetic testing identifies a pathogenic variant in PTPN11 in 50% of affected individuals, SOS1 in approximately 13%, RAF1 and RIT1 each in 5%, and KRAS in fewer than 5%. Other reported genes – in which pathogenic variants have been found to cause Noonan syndrome in fewer than 1% of cases – include BRAF, LZTR1, MAP2K1, and NRAS. Several additional genes associated with a Noonan-syndrome-like phenotype in fewer than ten individuals have been identified.
Noonan syndrome treatment
Management of Noonan syndrome generally focuses on the specific signs and symptoms present in each person. Treatments for the complications of Noonan syndrome (such as cardiovascular problems) are generally standard and do not differ from treatment in the general population 28.
Developmental disabilities are addressed by early intervention programs. Some children with Noonan syndrome may need special help in school, including for example, an individualized educational program (IEP) 28.
Treatment for bleeding problems depends on the cause 28. Growth hormone (GH) therapy can increase the rate at which a child with Noonan syndrome grows in most cases. GH therapy during childhood and teen years may also increase final adult height slightly, often enough to reach the low normal range of average height 30.
Noonan syndrome prognosis
There is a wide range in the nature and severity of signs and symptoms that may be present in people with Noonan syndrome, so the long-term outlook (prognosis) and life expectancy may differ among affected people. Studies generally suggest that long-term outcome depends largely on the presence and severity of congenital heart defects. Death in affected people has been frequently associated with the presence of complex left ventricular disease 31. Studies have indicated that people with Noonan syndrome have a 3-fold higher mortality rate than those in the general population 32.
Some affected people have ongoing health problems due to congenital heart defects, lymphatic vessel dysplasia, urinary tract malformations, blood disorders, or other associated health issues 32. However, with special care and counseling, the majority of children with Noonan syndrome grow up and function normally as adults. Signs and symptoms tend to lessen with age, and new medical problems associated with the condition are generally not expected to appear in adulthood 33.
Noonan syndrome with multiple lentigines
Noonan syndrome with multiple lentigines formerly called LEOPARD syndrome, is a condition that affects many areas of the body. As the condition name suggests, Noonan syndrome with multiple lentigines is very similar to a condition called Noonan syndrome, and it can be difficult to tell the two disorders apart in early childhood. However, the features of these two conditions differ later in life. The characteristic features of Noonan syndrome with multiple lentigines include brown skin spots called lentigines that are similar to freckles, heart defects, widely spaced eyes (ocular hypertelorism), a sunken chest (pectus excavatum) or protruding chest (pectus carinatum), and short stature. These features vary, however, even among affected individuals in the same family. Not all individuals with Noonan syndrome with multiple lentigines have all the characteristic features of this condition.
LEOPARD is an acronym for the characteristic abnormalities associated with the disorder: L stands for (L)entigines (multiple black or dark brown spots on the skin); (E)lectrocardiographic conduction defects (abnormalities of the electrical activity and the coordination of proper contractions of the heart); (0)cular hypertelorism (widely-spaced eyes); (P)ulmonary stenosis (obstruction of the normal outflow of blood from the right ventricle of the heart); (A)bnormalities of the genitals; (R)etarded growth resulting in short stature; and (D)eafness or hearing loss due to malfunction of the inner ear (sensorineural deafness). Some affected individuals may also exhibit mild intellectual disability, speech difficulties, and/or, in some cases, additional physical abnormalities.
The lentigines seen in Noonan syndrome with multiple lentigines typically first appear in mid-childhood, mostly on the face, neck, and upper body. Affected individuals may have thousands of small dark brown skin spots by the time they reach puberty. Unlike freckles, the appearance of lentigines has nothing to do with sun exposure. In addition to lentigines, people with this condition may have lighter brown skin spots called café-au-lait spots. Café-au-lait spots tend to develop before the lentigines, appearing within the first year of life in most affected people.
Of the people with Noonan syndrome with multiple lentigines who have heart defects, about 80 percent have hypertrophic cardiomyopathy, which is a thickening of the heart muscle that forces the heart to work harder to pump blood. The hypertrophic cardiomyopathy most often affects the lower left chamber of the heart (the left ventricle). Up to 20 percent of people with Noonan syndrome with multiple lentigines who have heart problems have a narrowing of the artery from the heart to the lungs (pulmonary stenosis).
People with Noonan syndrome with multiple lentigines can have a distinctive facial appearance. In addition to ocular hypertelorism, affected individuals may have droopy eyelids (ptosis), thick lips, and low-set ears. Affected individuals also usually have an abnormal appearance of the chest; they either have pectus excavatum or pectus carinatum.
At birth, people with Noonan syndrome with multiple lentigines are typically of normal weight and height, but in some, growth slows over time. This slow growth results in affected individuals being shorter than average, although less than half of people with Noonan syndrome with multiple lentigines have significantly short stature.
Other signs and symptoms of Noonan syndrome with multiple lentigines include hearing loss caused by abnormalities in the inner ear (sensorineural deafness), mild intellectual disability, and extra folds of skin on the back of the neck. Affected males often have genital abnormalities, which can include undescended testes (cryptorchidism) and a urethra that opens on the underside of the penis (hypospadias). These abnormalities may reduce the ability to have biological children (decreased fertility). Females with Noonan syndrome with multiple lentigines may have poorly developed ovaries and delayed puberty.
Noonan syndrome with multiple lentigines is thought to be a rare condition; approximately 200 cases have been reported worldwide 34.
Figure 4. Noonan syndrome with multiple lentigines
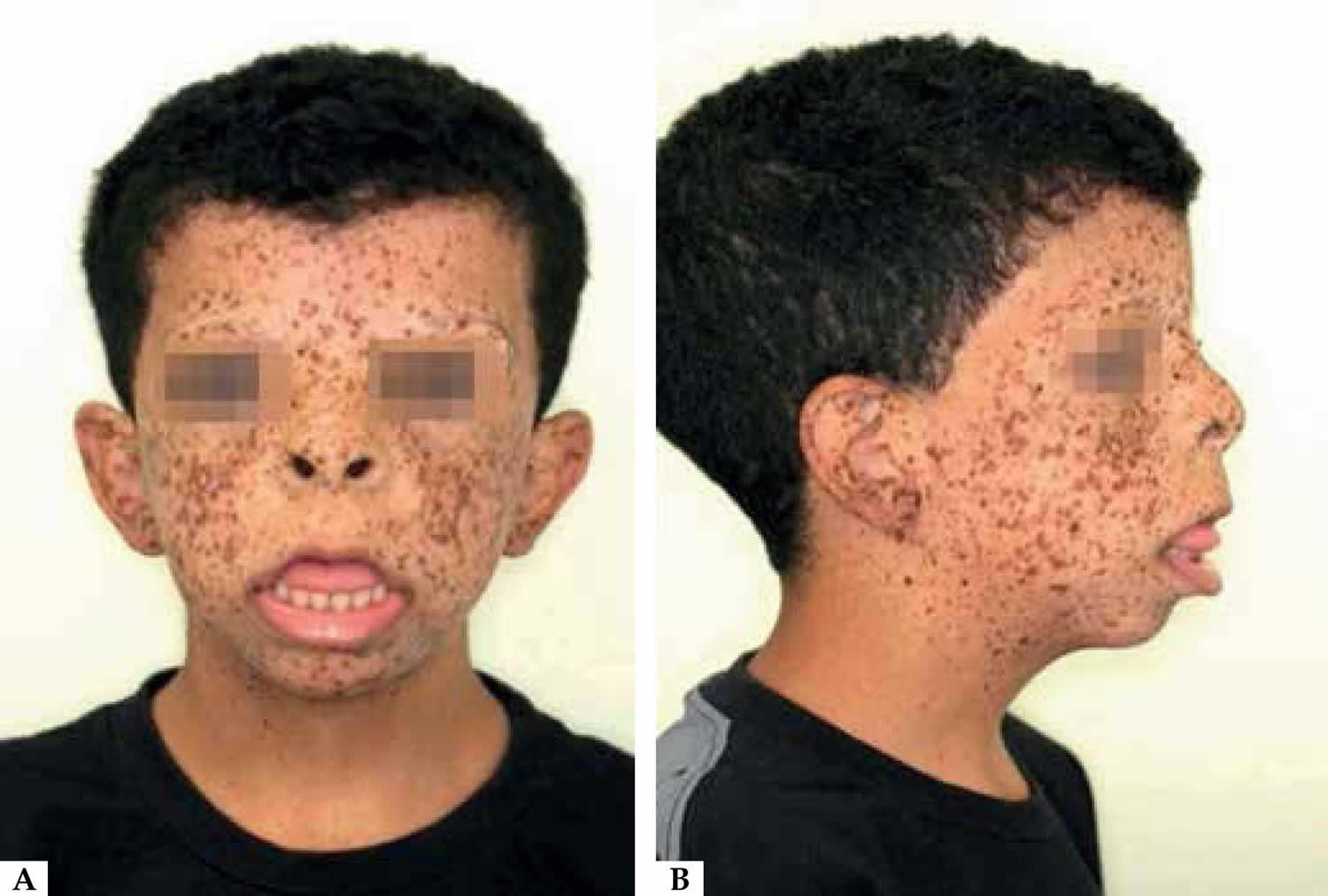
Footnote: Patient’s face (front view), showing lentigines, hypertelorism, and tongue protrusion. B. Patient’s face (profile view), showing lentigines, hypertelorism, and tongue protrusion.
[Source 35 ]Noonan syndrome with multiple lentigines causes
Mutations in one of several genes can cause Noonan syndrome with multiple lentigines. Approximately 85 percent of individuals with this condition have mutations in the PTPN11 gene. Another 10 percent have mutations in the RAF1 gene. In rare cases, mutations in the BRAF or MAP2K1 gene have been found to cause this condition. The remaining individuals with Noonan syndrome with multiple lentigines do not have an identified mutation in any of these four genes. In these individuals, the cause of the condition is unknown.
The PTPN11, RAF1, BRAF, and MAP2K1 genes all provide instructions for making proteins that are involved in important signaling pathways needed for the proper formation of several types of tissue during development. These proteins also play roles in the regulation of cell division, cell movement (migration), and cell differentiation (the process by which cells mature to carry out specific functions).
A mutation in the PTPN11, RAF1, BRAF, or MAP2K1 gene leads to the production of a protein that functions abnormally, which impairs the protein’s ability to respond to cell signals. A disruption in the regulation of systems that control cell growth and division leads to the characteristic features of Noonan syndrome with multiple lentigines.
Noonan syndrome with multiple lentigines inheritance pattern
Noonan syndrome with multiple lentigines is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
Noonan syndrome with multiple lentigines signs and symptoms
The symptoms and physical characteristics associated with Noonan syndrome with multiple lentigines are highly variable. However, most affected individuals tend to exhibit characteristic abnormalities of the skin, the structure and function of the heart, the head and facial (craniofacial) area, and/or the genitals.
Children with Noonan syndrome with multiple lentigines may exhibit numerous black or dark brown “freckle-like” spots on the skin (multiple lentigines). However, most individuals with Noonan syndrome with multiple lentigines do not exhibit lentigines during the first few years of life. Lentigines increase in number with age, usually until puberty. Many affected individuals may exhibit thousands of such lentigines. Although these small, flat discolorations (macules) resemble freckles, they tend to be darker, range from approximately one to five millimeters (mm) in size, and do not darken upon exposure to sunlight. The lentigines tend to be most numerous on the neck and upper chest area (trunk), be less concentrated below the knees, and involve only the skin, not the mucous membranes. However, they may appear anywhere on the skin of the body including the scalp, face, upper arms and/or upper legs, palms of the hands, soles of the feet, and/or genitals. In approximately 20 percent of affected individuals, larger, dark brown discolorations (café-au-lait spots) may also appear on the skin.
Many individuals with Noonan syndrome with multiple lentigines also have cardiac anomalies. Such heart defects, which are often apparent during infancy or early childhood, may include the abnormal transmission of electrical impulses that coordinate contractions of the heart (electrocardiographic conduction defects). In many cases of Noonan syndrome with multiple lentigines, there may be an interruption of the normal passage of electrical impulses (heart block) through the heart’s conducting system. As a result, although the two upper chambers of the heart (atria) may beat normally, the two lower chambers (ventricles) may contract less often or “fall behind” the atria.
The effects of such electrocardiographic conduction defects in individuals with Noonan syndrome with multiple lentigines may be highly variable, ranging from no apparent symptoms (asymptomatic) in some affected individuals to potentially serious complications in others. For example, those who exhibit prolonged P-R intervals may not exhibit any associated symptoms. Observable symptoms may also not occur in affected individuals who experience dropped beats. In more severe cases of heart block, however, if there is inadequate blood output from the ventricles, affected individuals may experience breathlessness due to the heart’s inability to pump blood effectively (heart failure), fatigue, or experience fainting episodes. If the ventricular beat slows dramatically or stops, affected individuals may black out, experience seizures, or exhibit life-threatening symptoms.
In addition, individuals with Noonan syndrome with multiple lentigines may also have structural (anatomical) malformations of the heart. The most common cardiac malformation appears to be overgrowth (hypertrophy) of the cardiac muscle in the ventricular wall(s) (hypertrophic obstructive cardiomyopathy), particularly the left ventricle. This condition may cause reduced cardiac output, potentially resulting in fatigue; fainting episodes (syncope), particularly during physical activity; and/or, in some cases, potentially life-threatening symptoms (e.g., arrhythmias, etc.)
The second most common abnormality is the obstruction of the normal outflow of blood from the lower right chamber (ventricle) of the heart to the lungs (isolated valvar pulmonary stenosis). Such obstruction may be due to abnormal narrowing (stenosis) of the pulmonary artery, which carries blood from the right ventricle to the lungs; stenosis of the pulmonary valve, the valve that controls the regular flow of deoxygenated blood through the pulmonary artery and on to the lungs; abnormal narrowing of the upper portion of the right ventricle; and/or other causes. In individuals with pulmonary stenosis, the heart must work harder to send blood to the lungs for oxygenation.
In most individuals with Noonan syndrome with multiple lentigines, pulmonary stenosis tends to be mild and symptoms may not occur (asymptomatic). In those cases when symptoms do occur, they often may not appear until later in childhood.
In some people, hypertrophic cardiomyopathy and pulmonary stenosis may be associated.
Some affected individuals with pulmonary stenosis may also exhibit abnormal narrowing (stenosis) of the aorta, the main artery of the body. Aortic stenosis may result in obstruction of blood flow from the left ventricle to the aorta and abnormal thickening of cardiac muscle in the wall of the left ventricle. As a result, aortic stenosis may contribute to such symptoms as fatigue, chest pain (angina pectoris) during exertion, breathlessness, and/or fainting episodes.
Many individuals with Noonan syndrome with multiple lentigines exhibit widely spaced eyes (ocular hypertelorism), with or without additional malformations of the head and facial (craniofacial) area. These may include a triangular-shaped face, drooping of the upper eyelids (ptosis), the presence of abnormal folds of skin over the inner corners of the eye (epicanthal folds), abnormal protrusion of the lower jaw (mandibular prognathism), and/or low-set, unusually prominent ears. Some affected individuals may also exhibit additional abnormalities including crossing of the eyes (strabismus) and/or mild webbing of the neck (pterygium colli).
Many individuals with Noonan syndrome with multiple lentigines also have genital abnormalities. Affected males may exhibit abnormal placement of the urinary opening (meatus) on the underside of the penis (hypospadias), unusual smallness of the penis (micropenis), and/or failure of one or both testes to descend into the scrotum (unilateral or bilateral cryptorchidism). Affected females may exhibit underdevelopment (hypoplasia) or absence (agenesis) of an ovary. Abnormally decreased function of the gonads (i.e., testes in males, ovaries in females) may result in delayed development of secondary sexual characteristics (puberty) in some affected males and females.
Individuals with Noonan syndrome with multiple lentigines may also exhibit growth retardation that results in short stature. Affected individuals may also have additional skeletal abnormalities. These may include malformations of the chest (thoracic deformity) such as abnormal depression of the bone forming the center of the chest (sternum), known as “funnel chest” or pectus excavatum, or abnormal protrusion of the sternum, known as “keeled chest” or pectus carinatum. Some individuals with Noonan syndrome with multiple lentigines may exhibit additional skeletal abnormalities such as unusually prominent shoulder blades (winged scapula), abnormal sideways curvature of the spine (scoliosis), and/or the development of abnormal front-to-back spinal curvature (kyphosis) during later life.
Some individuals with Noonan syndrome with multiple lentigines may also exhibit mild to severe hearing loss due to malfunction of the inner ears (sensorineural deafness). In some people, such hearing loss may be apparent at birth or during early childhood. However, other affected individuals may have normal hearing in early childhood yet eventually exhibit hearing loss at a later age. Hearing impairment may contribute to speech difficulties in many people. Although most affected individuals have normal intelligence, others may exhibit mild intellectual disability.
Noonan syndrome with multiple lentigines diagnosis
In some children, the diagnosis of Noonan syndrome with multiple lentigines may be suspected soon after birth due to the presence of pale tan or light brown discolorations on the skin (café-au-lait spots), characteristic facial features and hypertrophic cardiomyopathy. Multiple lentigines are usually not apparent before the age of five years. When they are seen in combination with two other related features in the LEOPARD acronym or when an individual has three related features plus a parent or sibling with multiple lentigines, the diagnosis of Noonan syndrome with multiple lentigines can be made.
Molecular genetic testing for the PTPN11 gene and the RAF1 gene is available to confirm the diagnosis and for prenatal diagnosis.
The diagnosis of certain specific abnormalities that may occur in association with Noonan syndrome with multiple lentigines may be confirmed by specialized imaging studies and/or additional tests. Examination of skin samples (biopsies) under a microscope that uses light (light microscopy) or an electron beam (electron microscopy) may confirm that pigmented areas of skin represent multiple lentigines as opposed to freckles.
Children who exhibit multiple lentigines should receive prompt, thorough cardiac evaluation. If no cardiac abnormalities are revealed during such evaluation, individuals should receive periodic reassessments to detect any heart abnormalities that may develop later.
A variety of tests may be conducted to perform such a cardiac assessment. For example, the electrocardiographic conduction defects (e.g., varying degrees of heart block) and/or structural (anatomical) malformations of the heart (e.g., pulmonary and/or aortic stenosis, hypertrophic obstructive cardiomyopathy) may be confirmed by a thorough clinical examination and several specialized tests that allow physicians to evaluate the structure and function of the heart.
Clinical examination may include a physician’s evaluation of heart and lung sounds through use of a stethoscope. For example, in mild asymptomatic cases of pulmonary stenosis, the condition may initially be suspected through the detection of an abnormal heart murmur during such stethoscopic evaluation. Heart block may initially be identified by detection of a slow, regular heartbeat that fails to increase during exercise.
Specialized cardiac tests may include electrocardiography (EKG), echocardiography, cardiac MRI and/or cardiac catheterization. An EKG, which records the electrical activities of the heart muscle, may confirm the presence of abnormal electrical patterns such as those associated with varying degrees of heart block (e.g., prolonged P-R interval, left anterior hemiblock, widening of the QRS complex, complete heart block). During an echocardiogram, sound waves are directed toward the heart, enabling physicians to study cardiac function and motion. Echocardiography may play an essential role in helping to confirm hypertrophic obstructive cardiomyopathy. EKG, echocardiograms, cardiac catheterization, and/or other tests may help to clarify the underlying anatomical cause and/or severity of narrowing associated with pulmonary stenosis.
Sonographic and radiological techniques may be conducted to detect and/or confirm certain genital abnormalities in many individuals with Noonan syndrome with multiple lentigines (e.g., unilateral or bilateral cryptorchidism in affected males, hypoplastic ovaries in affected females). In addition, because deficiencies of certain hormones (gonadotrophin) may contribute to abnormally decreased function of the gonads (hypogonadism) and delayed puberty in some affected males and females, individuals with Noonan syndrome with multiple lentigines may be monitored closely to promptly detect such delays and laboratory tests may be conducted to screen for deficient levels of certain gonadotrophins in the blood.
In many individuals with Noonan syndrome with multiple lentigines, X-ray studies may also be used to confirm the presence of certain skeletal abnormalities suspected during clinical observation. Growth retardation may not become obvious until early childhood, when there may be an observable decline in the normal growth rate.
Thorough, regular hearing (audiological) evaluation should also be conducted in individuals with Noonan syndrome with multiple lentigines to promptly detect potential hearing impairment and help ensure early implementation of appropriate supportive measures. If hearing impairment is detected, a scanning procedure, such as computerized tomography or CT scan, may be performed to confirm the underlying cause (e.g., inner ear malformation) and characterize the type of hearing loss (e.g., sensorineural hearing impairment). During a CT scan, a computer and x-rays are used to create a film showing cross-sectional images of the structures of the inner ear.
Noonan syndrome with multiple lentigines treatment
The treatment of Noonan syndrome with multiple lentigines is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, surgeons, physicians who diagnose and treat disorders of the skin (dermatologists), cardiologists, specialists who diagnose and treat skeletal disorders (orthopedists), physicians who specialize in diagnosing and treating disorders of the glands (endocrinologists), specialists who assess and treat hearing problems (audiologists), speech pathologists, and/or other health care professionals may need to systematically and comprehensively plan an affected child’s treatment.
Specific therapies for Noonan syndrome with multiple lentigines are symptomatic and supportive. In affected individuals who exhibit mild forms of cardiac conduction abnormalities, treatment may not be required. However, in more severe cases when associated symptoms occur (e.g., fainting episodes) and in some cases of pulmonary stenosis, hypertrophic obstructive cardiomyopathy, and/or other structural heart abnormalities potentially associated with Noonan syndrome with multiple lentigines, treatment with certain medications, surgical intervention, and/or other techniques may be necessary. The surgical procedures performed will depend upon the location and severity of the anatomical abnormalities and their associated symptoms.
Other abnormalities potentially associated with Noonan syndrome with multiple lentigines may also be corrected surgically. These may include certain craniofacial, skeletal, genital, and/or other malformations. For example, in some affected males with cryptorchidism, hormone treatment may be administered; however, if this treatment is not successful, surgery may be performed to move the undescended testes into the scrotum and attach them in a fixed position to prevent retraction (orchiopexy).
In addition, if laboratory and additional tests demonstrate that deficient levels of gonadotrophin (hypogonadotropism) have contributed to abnormally decreased gonadal function (hypogonadism) and delayed puberty in affected males or females, sex hormone replacement therapy may be considered in some cases.
If individuals with Noonan syndrome with multiple lentigines demonstrate hearing impairment, hearing aids may be beneficial. Appropriate use of hearing aids, other supportive techniques, and speech therapy may help to prevent, improve, and/or correct some speech problems that may result from such hearing impairment.
Early intervention is important to ensure that children with Noonan syndrome with multiple lentigines reach their potential. Special services that may be beneficial to affected children include special remedial education, special social support, physical therapy, and/or other medical, social, and/or vocational services.
Genetic counseling is recommended for individuals with Noonan syndrome with multiple lentigines and their families.
Capillary malformation–arteriovenous malformation syndrome
Capillary malformation-arteriovenous malformation syndrome (CM-AVM) is a disorder of the vascular system, which is the body’s complex network of blood vessels. The vascular system consists of arteries, which carry oxygen-rich blood from the heart to the body’s various organs and tissues; veins, which carry blood back to the heart; and capillaries, which are tiny blood vessels that connect arteries and veins.
Capillary malformation-arteriovenous malformation syndrome is characterized by the presence of multiple small (1-2 cm in diameter) capillary malformations (CMs), which are composed of enlarged capillaries that increase blood flow near the surface of the skin. These malformations look like multiple small, round, pink or red spots on the skin. In most affected individuals, capillary malformations occur on the face, arms, and legs. These spots may be visible from birth or may develop during childhood. By themselves, capillary malformations usually do not cause any health problems.
In some people with capillary malformation-arteriovenous malformation syndrome, capillary malformations are the only sign of the disorder. However, other affected individuals also have more serious vascular abnormalities known as arteriovenous malformations (AVMs) and arteriovenous fistulas (AVFs). Arteriovenous malformations and arteriovenous fistulas are abnormal connections between arteries, veins, and capillaries that affect blood circulation. Depending on where they occur in the body, these abnormalities can be associated with complications including abnormal bleeding, migraine headaches, seizures, and heart failure. In some cases the complications can be life-threatening. In people with capillary malformation-arteriovenous malformation syndrome, complications of arteriovenous malformations (AVMs) and arteriovenous fistulas (AVFs) tend to appear in infancy or early childhood; however, some of these vascular abnormalities never cause any symptoms.
Some vascular abnormalities seen in capillary malformation-arteriovenous malformation syndrome are similar to those that occur in a condition called Parkes Weber syndrome (multiple micro-arteriovenous fistulas associated with a cutaneous capillary stain and excessive soft-tissue and skeletal growth of an affected limb). In addition to vascular abnormalities, Parkes Weber syndrome usually involves overgrowth of one limb. capillary malformation-arteriovenous malformation syndrome and some cases of Parkes Weber syndrome have the same genetic cause.
Capillary malformation-arteriovenous malformation syndrome is thought to occur in at least 1 in 100,000 people of northern European origin 36. The prevalence of the condition in other populations is unknown.
Figure 5. Capillary malformation-arteriovenous malformation syndrome
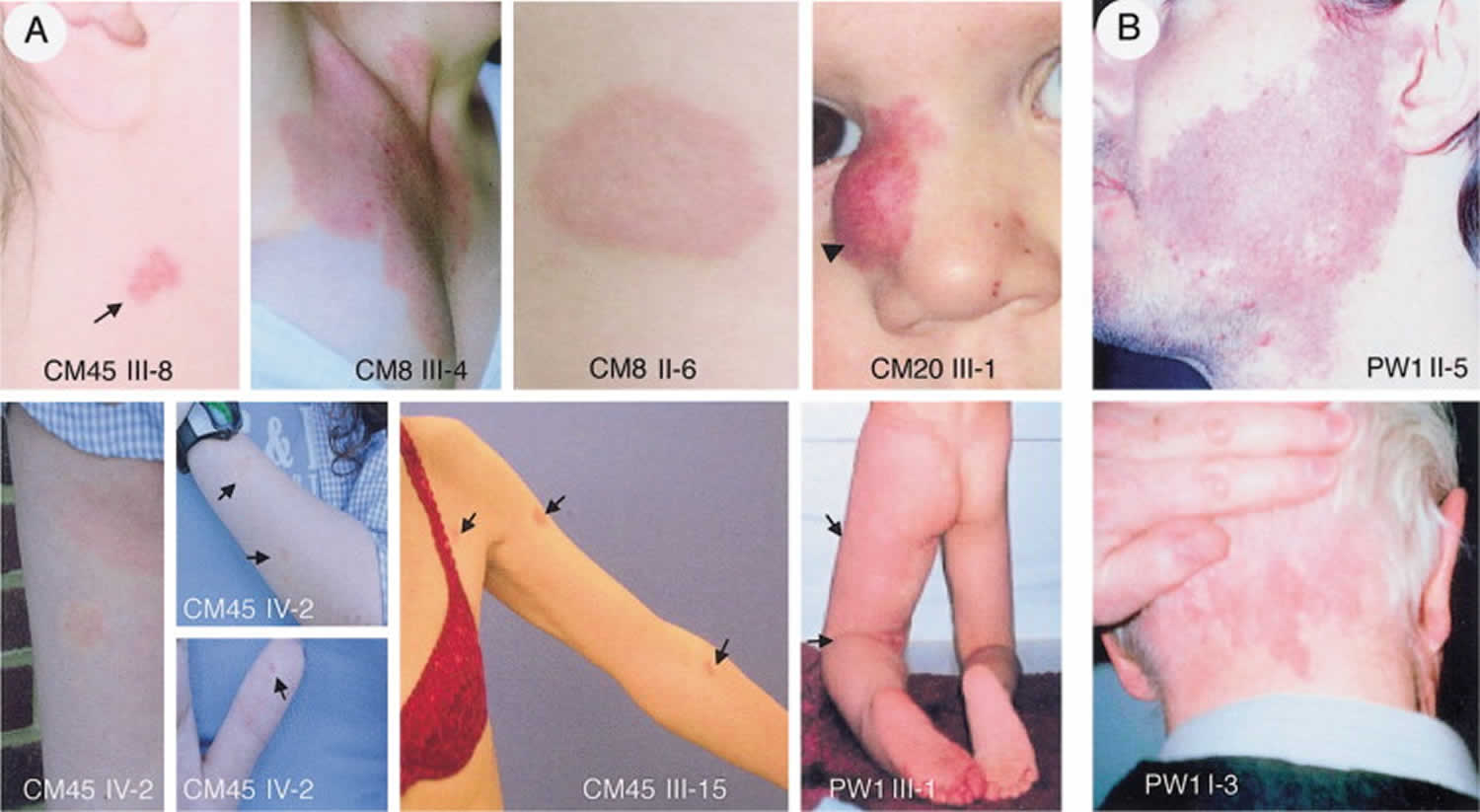
Footnote: Photographs of vascular malformations linked to RASA1 mutations. (A) Atypical inherited pink-to-red, round-to-oval capillary malformations (arrows) (patients CM45 III-8, CM45 III-15, and CM45 IV-2, CM8 III-4, and CM8 II-6); patient CM20 III-1, with a circumscribed nasal arteriovenous malformation and a cutaneous capillary blush (arrowhead); patient PW1 III-1, with Parkes Weber syndrome in the lower limb (arrows). (B) Photographs of capillary malformations not linked to RASA1 mutations. Typical red-purple facial capillary malformation (patient PW1 II-5); typical red capillary malformation of the neck (patient PW1 I-3).
[Source 37 ]Capillary malformation–arteriovenous malformation syndrome causes
Capillary malformation-arteriovenous malformation syndrome is caused by mutations in the RASA1 gene. This gene provides instructions for making a protein known as p120-RasGAP, which is involved in transmitting chemical signals from outside the cell to the nucleus. These signals help control several important cell functions, including cell growth and division (proliferation), the process by which cells mature to carry out specific functions (differentiation), and cell movement. The role of the p120-RasGAP protein is not fully understood, although it appears to be essential for the normal development of the vascular system.
Mutations in the RASA1 gene lead to the production of a nonfunctional version of the p120-RasGAP protein. A loss of this protein’s activity disrupts tightly regulated chemical signaling during development. However, it is unclear how these changes lead to the specific vascular abnormalities seen in people with capillary malformation-arteriovenous malformation syndrome.
Capillary malformation-arteriovenous malformation syndrome inheritance pattern
Capillary malformation-arteriovenous malformation syndrome is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
In most cases, an affected person inherits the mutation from one affected parent. Other cases result from new mutations in the gene and occur in people with no history of the disorder in their family.
Capillary malformation–arteriovenous malformation syndrome signs and symptoms
Capillary malformations
Capillary malformations are multiple round or oval pink lesions often with a blanched halo. capillary malformations can be present at birth and tend to increase in number over time.
Arterial flow with Doppler ultrasound has been reported over the capillary malformations 38 and is hypothesized to be a manifestation of an underlying arteriovenous malformation. It is unclear if arterial flow abnormalities associated with the capillary malformations can increase or develop over time.
Arteriovenous malformations and arteriovenous fistulas
Current data on long-term development of arteriovenous malformations or arteriovenous fistulas after initial screening are insufficient. Although it has been hypothesized that arteriovenous malformations or arteriovenous fistulas may develop over time 39, to date no individuals who had normal imaging screens have subsequently been reported to have developed
- Intracranial arteriovenous malformations or arteriovenous fistulas can manifest early in life 40. Vein of Galen aneurysmal malformation and other intracranial Arteriovenous malformations have led to seizures, hydrocephalus, migraine headaches, and cardiac failure 41. Vivanti et al 42 reported on a series of 19 children with vein of Galen malformation; 10% had EPHB4 pathogenic variants. Revencu et al 43 reported that most of the intracranial lesions were macrofistulas causing symptoms in infancy. However, this finding may be biased given that the identification of the Arteriovenous malformations/arteriovenous fistulas may be secondary to associated symptoms and that asymptomatic individuals may not have had the imaging studies needed to detect the lesions.
- Extracranial arteriovenous malformations and arteriovenous fistulas are typically reported in skin, muscle, and spine. Although approximately 50% of arteriovenous malformations/arteriovenous fistulas have been reported to be in the head/neck region 40, the frequency of Arteriovenous malformations/arteriovenous fistulas in this location may be an overestimate because it is likely that imaging is preferentially performed in this region. Symptomatic intraspinal arteriovenous malformations resulting in neurologic deficits have been reported; MRI has identified intraspinous lesions requiring endovascular/surgical treatment 44.
Arteriovenous malformations/arteriovenous fistulas have not been commonly reported in viscera 43, a distinguishing difference from hereditary hemorrhagic telangiectasia.
Parkes Weber syndrome
Limb overgrowth has been reported in both the upper and lower extremities in individuals with capillary malformation-arteriovenous malformation syndrome. The overgrowth is typically noticeable in infancy and can range in severity. Most individuals with limb overgrowth fulfill the findings of Parkes Weber syndrome, defined by Revencu et al 40 as the presence of a capillary stain, bony and soft tissue hyperplasia, and multiple arteriolovenular microfistulas throughout an upper or lower extremity 41.
Other
Bier spots, white spots on the skin surrounded by a pale halo of erythema, have been described in individuals with EPHB4-capillary malformation-arteriovenous malformation syndrome 45.
Telangiectasia has been reported primarily in individuals with EPHB4-capillary malformation-arteriovenous malformation syndrome. The telangiectases were typically located on the lips, trunks, and/or arms/legs 45.
Epistaxis has been reported in individuals with EPHB4-capillary malformation-arteriovenous malformation syndrome 45.
Cardiac overload or failure is a potential complication in individuals with significant fast-flow lesions. In particular, one third of individuals with RASA1 Parkes Weber syndrome required cardiac follow up 43.
One infant with RASA1-capillary malformation-arteriovenous malformation syndrome had an arteriovenous fistula between the left carotid artery and jugular vein that caused cardiac overload requiring treatment 46.
One woman with RASA1-capillary malformation-arteriovenous malformation syndrome reported worsening of symptoms during pregnancy; she developed pulmonary and peripheral edema with concern for high-output heart failure that resolved after pregnancy 47.
Nonimmune hydrops fetalis due to an AVM has been reported in RASA1-capillary malformation-arteriovenous malformation syndrome 48.
Congenital heart defects have been reported in a few individuals with RASA1- and EPHB4-capillary malformation-arteriovenous malformation syndrome; however, this finding may be coincidental 49.
Lymphatic malformations have been reported in several individuals with EPHB4- and RASA1-capillary malformation-arteriovenous malformation syndrome 50. Lymphangiography and near-infrared fluorescence lymphatic imaging showed abnormally dilated collecting lymphatics with sluggish flow in the unaffected limb, and tortuous lymphatics of the affected limb with lymphocele-like vesicles in the groin of individuals with RASA1-capillary malformation-arteriovenous malformation syndrome 51. Whether these lymphatic abnormalities are progressive is not yet known. An EPHB4 pathogenic variant was identified in a four-generation pedigree with central conducting lymphatic anomaly 52. Hydrops fetalis was reported in individuals from two families with EPHB4-capillary malformation-arteriovenous malformation syndrome 49.
Tumors. Individuals with RASA1-capillary malformation-arteriovenous malformation syndrome may be at increased risk for tumor development, but review of the reported cases does not confirm this: Revencu et al 43 reported several different types of tumors (e.g., optic glioma, lipoma, superficial basal cell carcinoma, angiolipoma, non-small-cell lung cancer, and vestibular schwannoma) in 44 families; however, in their larger series of 138 individuals, the only tumors reported were two common basal cell carcinomas in two individuals from the same family 40. Whether the rate of tumors is increased compared to the general population is as yet unknown, but it is likely not dramatically increased.
Additional findings observed in a small number of individuals with RASA1-capillary malformation-arteriovenous malformation syndrome include seizures, headaches, hydrocephalus, neurogenic bladder, varicosities, and hemangiomas. It is not clear if these findings are primary manifestations of a germline heterozygous RASA1 pathogenic variant, secondary complications of Arteriovenous malformations/arteriovenous fistulas, or unrelated.
Capillary malformation–arteriovenous malformation syndrome diagnosis
Diagnostic criteria for capillary malformation-arteriovenous malformation syndrome have been proposed but not systematically evaluated 53.
Capillary malformation-arteriovenous malformation syndrome should be suspected in individuals who have any of the following:
- Capillary malformations (CMs), the hallmark of capillary malformation-arteriovenous malformation syndrome. Capillary malformations are generally:
- Multifocal, atypical pink-to-reddish brown, multiple, small (1-2 cm in diameter), round-to-oval lesions sometimes with a white halo;
- Composed of dilated capillaries in the papillary dermis 54;
- Mostly localized on the face and limbs;
- Seen in combination with arteriovenous malformations (AVMs) or arteriovenous fistulas (AVFs), but may be the only finding 40.
- Arteriovenous malformations (AVMs) or arteriovenous fistulas (AVFs) in soft tissue, bone, and brain that may be associated with overgrowth 46
- Parkes Weber syndrome phenotype, a cutaneous capillary malformation associated with underlying multiple micro-arteriovenous fistulas and soft-tissue and skeletal hypertrophy of the affected limb 55
The diagnosis of a capillary malformation-arteriovenous malformation syndrome is established in a proband with suggestive clinical findings and a heterozygous pathogenic variant in EPHB4 or RASA1 identified by molecular genetic testing.
Capillary malformation–arteriovenous malformation syndrome treatment
To establish the extent of disease and needs of an individual diagnosed with capillary malformation-arteriovenous malformation syndrome, the following evaluations (if not performed as part of the evaluation that led to the diagnosis) are recommended:
- Medical history and physical examination with a focus on symptoms and findings secondary to arteriovenous malformations and arteriovenous fistulas
- Brain imaging – if not already performed – to identify arteriovenous malformations/arteriovenous fistulas (e.g., vein of Galen aneurysms and other intracranial arteriovenous malformations) to allow early identification of macrofistulas that can be treated prior to the development of symptoms 43
- Consideration of spine imaging to identify and characterize arteriovenous malformations/arteriovenous fistulas. Currently no consensus protocols for radiographic evaluation of individuals with capillary malformation–arteriovenous malformation syndrome have been developed; therefore, discussion with a radiologist is recommended in order to develop an appropriate plan for imaging based on the patient’s age and the capabilities and experience of the imaging facility.
- Consideration of further imaging in individuals with evidence of cardiac overload, to look for causative arteriovenous malformations/arteriovenous fistulas
- Evaluation for evidence of epistaxis (nosebleeds), and if present, referral to otolaryngologist as appropriate. If epistaxis is present, consider complete blood count for evaluation of anemia.
- Consultation with a clinical geneticist and/or genetic counselor
Treatment of manifestations 56:
- For capillary malformations and telangiectases that are of cosmetic concern, referral to a dermatologist.
- For arteriovenous malformations and arteriovenous fistulas, the risks and benefits of intervention (embolization vs surgery) must be considered, usually with input from a multidisciplinary team (e.g., specialists in interventional radiology, neurosurgery, surgery, cardiology, and dermatology).
- For cardiac overload, referral to a cardiologist.
- For hemihyperplasia and/or leg-length discrepancy, referral to an orthopedist. Lymphangiography to evaluate for lymphatic malformations may be considered; compression stockings for those with evidence of lymphedema; epistaxis treatment includes humidification, nasal lubricants, referral to otolaryngologist, and complete blood count for evaluation of anemia.
Costello syndrome
Costello syndrome is a rare genetic disorder that affects many parts of the body. This condition is characterized by delayed development and intellectual disability, loose folds of skin (which are especially noticeable on the hands and feet), unusually flexible joints, and distinctive facial features including a large mouth with full lips. Heart problems are common, including an abnormal heartbeat (arrhythmia), structural heart defects, and a type of heart disease that enlarges and weakens the heart muscle (hypertrophic cardiomyopathy). Infants with Costello syndrome may be larger than average at birth, but most have difficulty feeding and grow more slowly than other children. People with this condition have relatively short stature and may have reduced growth hormone levels. Other signs and symptoms of Costello syndrome can include tight Achilles tendons (which connect the calf muscles to the heel), weak muscle tone (hypotonia), a structural abnormality of the brain called a Chiari I malformation, skeletal abnormalities, dental problems, and problems with vision.
Beginning in early childhood, people with Costello syndrome are at an increased risk of developing certain cancerous and noncancerous tumors. The most common noncancerous tumors associated with this condition are papillomas, which are small, wart-like growths that usually develop around the nose and mouth or near the anus. The most common cancerous tumor associated with Costello syndrome is a childhood cancer called rhabdomyosarcoma, which begins in muscle tissue. Neuroblastoma, a tumor that arises in developing nerve cells, also has been reported in children and adolescents with this syndrome. In addition, some teenagers with Costello syndrome have developed transitional cell carcinoma, a form of bladder cancer that is usually seen in older adults.
The signs and symptoms of Costello syndrome overlap significantly with those of two other genetic conditions, cardiofaciocutaneous syndrome and Noonan syndrome. In affected infants, it can be difficult to tell the three conditions apart based on their physical features. However, the conditions can be distinguished by their genetic cause and by specific patterns of signs and symptoms that develop later in childhood.
Costello syndrome is very rare; it probably affects 200 to 300 people worldwide. Reported estimates of Costello syndrome prevalence range from 1 in 300,000 to 1 in 1.25 million people 57.
Figure 6. Costello syndrome
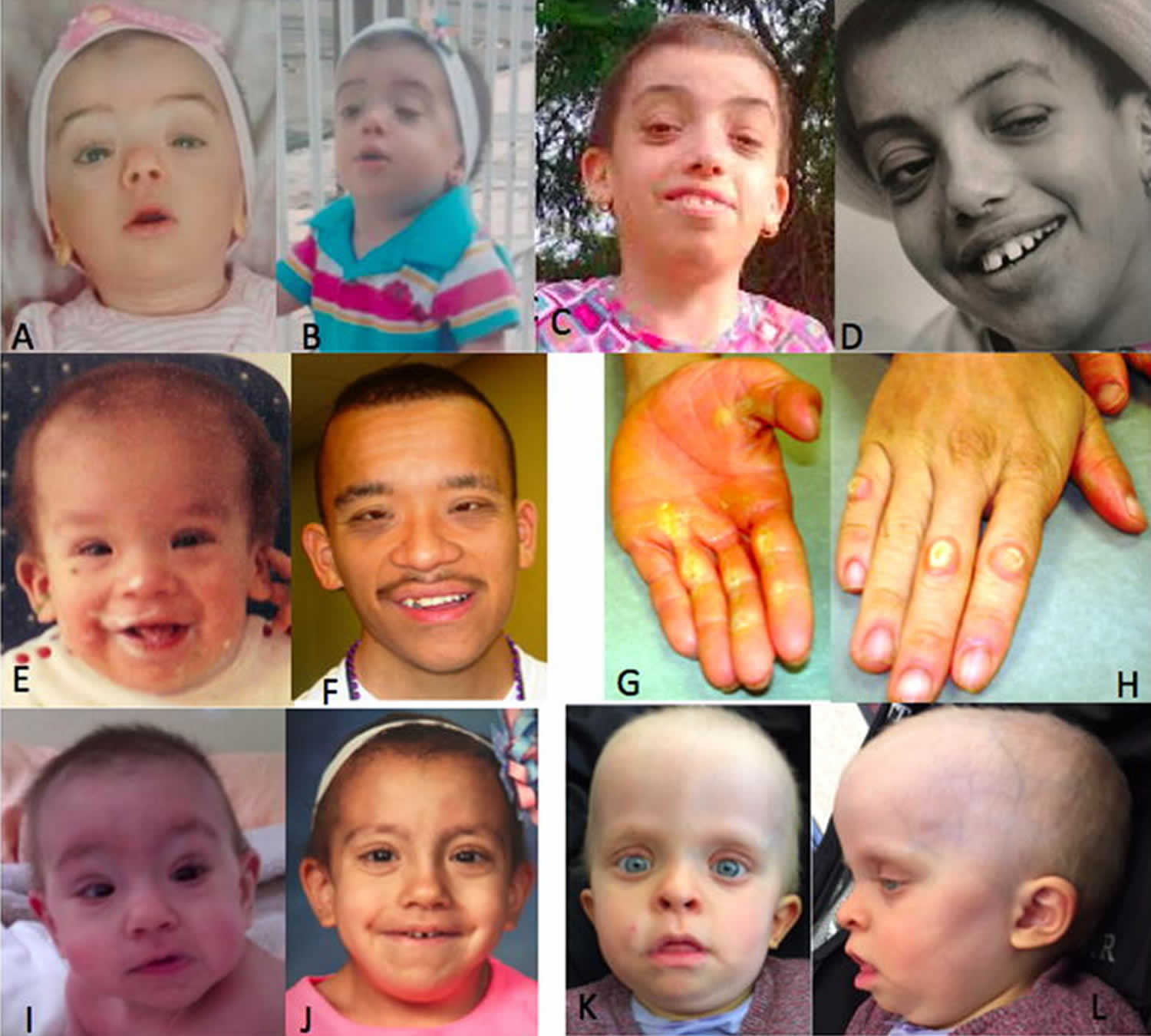
Footnote: Clinical features of Costello syndrome patients #1-4. Facial features at different ages observed in patients 1 (A – infancy, B – childhood and C and D – adolescence), 2 (E – infancy and F – adulthood) and 3 (I – infancy and J – childhood). Frontal and lateral view of patient 4 is depicted in K and L. Note the short, sparse hair with abnormal texture, especially in individuals 1 and 3, perinasal papilloma in patient 2(D), and the prominent hyperkeratosis in the hands of patient 2 (G,H).
[Source 58 ]Costello syndrome causes
Mutations in the HRAS gene cause Costello syndrome. This gene provides instructions for making a protein called H-Ras, which is part of a pathway that helps control cell growth and division. Mutations that cause Costello syndrome lead to the production of an H-Ras protein that is abnormally turned on (active). The overactive protein directs cells to grow and divide constantly, which can lead to the development of cancerous and noncancerous tumors. It is unclear how mutations in the HRAS gene cause the other features of Costello syndrome, but many of the signs and symptoms probably result from cell overgrowth and abnormal cell division.
Some people with signs and symptoms like those of Costello syndrome do not have an identified mutation in the HRAS gene. These individuals may actually have cardiofaciocutaneous syndrome or Noonan syndrome, which are caused by mutations in related genes. The proteins produced from these genes interact with one another and with the H-Ras protein as part of the same cell growth and division pathway. These interactions help explain why mutations in different genes can cause conditions with overlapping signs and symptoms.
Costello syndrome inheritance pattern
Costello syndrome is considered to be an autosomal dominant condition, which means one copy of the altered gene in each cell is sufficient to cause the disorder. Almost all reported cases have resulted from new gene mutations and have occurred in people with no history of the disorder in their family.
Costello syndrome signs and symptoms
Infants with Costello syndrome typically have a normal or high birth weight, but show poor sucking ability, have swallowing difficulties, and fail to grow and gain weight at the expected rate (failure to thrive). Growth delay after birth typically results in short stature during childhood and adulthood. Affected children may have developmental delay or mild to moderate intellectual disability. In some individuals, speech development and/or the ability to walk is significantly delayed. Children with Costello syndrome generally have warm, sociable personalities.
Individuals with Costello syndrome typically have loose skin (cutis laxa) on the neck, palms, fingers, and soles. The skin in these areas may lack elasticity and hang loosely; in addition, the skin may appear wrinkled and thickened. In some cases, certain areas of the skin may become unusually dark (hyperpigmentation). In addition, most patients with this disorder develop dry hardened patches of skin (hyperkeratosis) with unusually deep creases on the palms and soles. Some affected individuals may also have skeletal abnormalities such as dislocated hips, abnormally flexible (hyperextensible) joints of the fingers, wrists bent toward the little finger (ulnar deviation) and/or unusual tightening of the fibrous cords on the back of the heels (Achilles tendon). Additional skeletal abnormalities include side-to-side curvature of the spine (scoliosis), front-to-back curvature of the spine (kyphosis), and reduced range of motion in the shoulder and elbows.
Children with Costello syndrome usually devlelop papillomata around the mouth and nostrils. Papillomata may develop as early as two years of age or at older ages. In some cases, these wart-like (verrucal) lesions may be found near the anus. Papillomata usually become more apparent with age. Other benign tumors have also been reported.
Children with Costello syndrome have a distinctive facial appearance. Characteristic facial features may include an abnormally large head (macrocephaly); low-set ears with large, thick lobes; unusually thick lips; a large, depressed nasal bridge; abnormally wide nostrils (nares); and a coarse facial appearance. In addition, affected children may have unusually curly hair and/or sparse, thin hair on the front (anterior) of the head. Some children have folds of skin over the inner corners of the eyes (epicanthal folds).
In early childhood relative overgrowth of the hindbrain compared to the space available in the posterior fossa of skull cavity can result in crowding and neurologic problems. Because severe crowding requires surgical intervention, screening with brain and cervical spine MRI has been suggested.
Eye and vision changes are common and include nystagmus (rapid eye movements) in younger individuals, strabismus and rarely in older individuals keratoconus (abnormal thickening of the cornea).
Children with Costello syndrome often have certain heart abnormalities. These may include structural malformations of the heart that are present at birth (congenital heart defects); abnormal thickening of the muscular walls of the left lower chamber of the heart (hypertrophic cardiomyopathy); leakage of the valve between the left upper (atrial) and lower (ventricular) heart chambers (mitral valve prolapse); and/or other cardiac defects. Associated symptoms and findings may include abnormal heart sounds (heart murmurs) that may be detected by a physician through use of a stethoscope; shortness of breath, particularly upon exertion; faintness; chest pain; abnormal heart rhythms (arrhythmias); and/or other findings that may potentially lead to life-threatening complications without appropriate treatment.
Affected individuals have an approximately 15% lifetime risk to develop malignant tumors such as a cancer of the muscle tissue (rhabdomyosarcoma), a cancer of the nerve cells (neuroblastoma), and transitional cell carcinoma of the bladder.
In some cases, the symptoms and findings of Costello syndrome overlap with two similar disorders known as Noonan syndrome and cardiofaciocutaneous syndrome which are caused by mutations in different genes.
Costello syndrome diagnosis
Costello syndrome is diagnosed by clinical examination and specific diagnostic criteria have been developed. Molecular genetic testing for mutations in the HRAS gene is available to confirm the diagnosis. Most clinically affected individuals have an identifiable HRAS mutation. Experts in the field suggested that individuals without an identifiable HRAS mutation should not be diagnosed with Costello syndrome, as they most likely have a related condition such as Noonan or cardiofaciocutaneous syndrome. The specific mutation in the HRAS gene, often referred to by the resulting amino acid change, is important to identify. While most individuals with Costello syndrome share a mutation that results in a change of the amino acid glycine in position 12 to serine, a number of changes have been seen. The prognosis for a patient is affected by the specific mutation, as some present with more severe medical problems than others.
Costello syndrome treatment
The treatment of Costello syndrome is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, physicians who diagnose and treat abnormalities of the heart (cardiologists), physicians who diagnose and treat skeletal abnormalities (orthopedists), orthopedic surgeons, specialists who diagnose and treat abnormalities of the skin (dermatologists), speech pathologists, dietitians, and other health care professionals may need to systematically and comprehensively plan an affected child’s treatment.
Individuals with cardiac abnormalities, such as hypertrophic cardiomyopathy, may be treated with certain medications (e.g., beta-blockers or calcium channel blockers, antiarrhythmic medications), surgical intervention, and/or other measures may be necessary. The specific surgical procedures performed will depend upon the severity and location of the anatomical abnormalities, their associated symptoms, and other factors.
Bracing, occupational and physical therapy may be used to treat ulnar deviation of the wrists. Surgery may be used to lengthen Achilles tendons. Facial papillomata can be removed with dry ice.
Early intervention is important to ensure that children with Costello syndrome reach their potential. Services that may be beneficial include special remedial education, speech therapy, special social support, and other medical, social, and/or vocational services.
Genetic counseling is recommended for affected individuals and their families. Other treatment for this disorder is symptomatic and supportive.
Cardiofaciocutaneous syndrome
Cardiofaciocutaneous syndrome is a very rare genetic disorder that affects many parts of the body, particularly the heart (cardio-), facial features (facio-), and the skin and hair (cutaneous). People with cardiofaciocutaneous syndrome also have delayed development and intellectual disability, usually ranging from moderate to severe. Cardiofaciocutaneous syndrome is a very rare condition whose incidence is unknown. Researchers estimate that 200 to 300 people worldwide have cardiofaciocutaneous syndrome 59.
Heart defects occur in most people with cardiofaciocutaneous syndrome. The heart problems most commonly associated with this condition include malformations of one of the heart valves that impairs blood flow from the heart to the lungs (pulmonic stenosis), a hole between the two upper chambers of the heart (atrial septal defect), and a form of heart disease that enlarges and weakens the heart muscle (hypertrophic cardiomyopathy).
Cardiofaciocutaneous syndrome is also characterized by distinctive facial features. These include a high forehead that narrows at the temples, a short nose, widely spaced eyes (ocular hypertelorism), outside corners of the eyes that point downward (down-slanting palpebral fissures), droopy eyelids (ptosis), a small chin, and low-set ears. Overall, the face is broad and long, and the facial features are sometimes described as “coarse.”
Skin abnormalities occur in almost everyone with cardiofaciocutaneous syndrome. Many affected people have dry, rough skin; dark-colored moles (nevi); wrinkled palms and soles; and a skin condition called keratosis pilaris, which causes small bumps to form on the arms, legs, and face. People with cardiofaciocutaneous syndrome also tend to have thin, dry, curly hair and sparse or absent eyelashes and eyebrows.
Infants with cardiofaciocutaneous syndrome typically have weak muscle tone (hypotonia), feeding difficulties, and a failure to grow and gain weight at the normal rate (failure to thrive). Additional features of this disorder in children and adults can include an unusually large head (macrocephaly), short stature, problems with vision, and seizures.
The signs and symptoms of cardiofaciocutaneous syndrome overlap significantly with those of two other genetic conditions, Costello syndrome and Noonan syndrome. The three conditions are distinguished by their genetic cause and specific patterns of signs and symptoms; however, it can be difficult to tell these conditions apart, particularly in infancy. Unlike Costello syndrome, which significantly increases a person’s cancer risk, cancer does not appear to be a major feature of cardiofaciocutaneous syndrome.
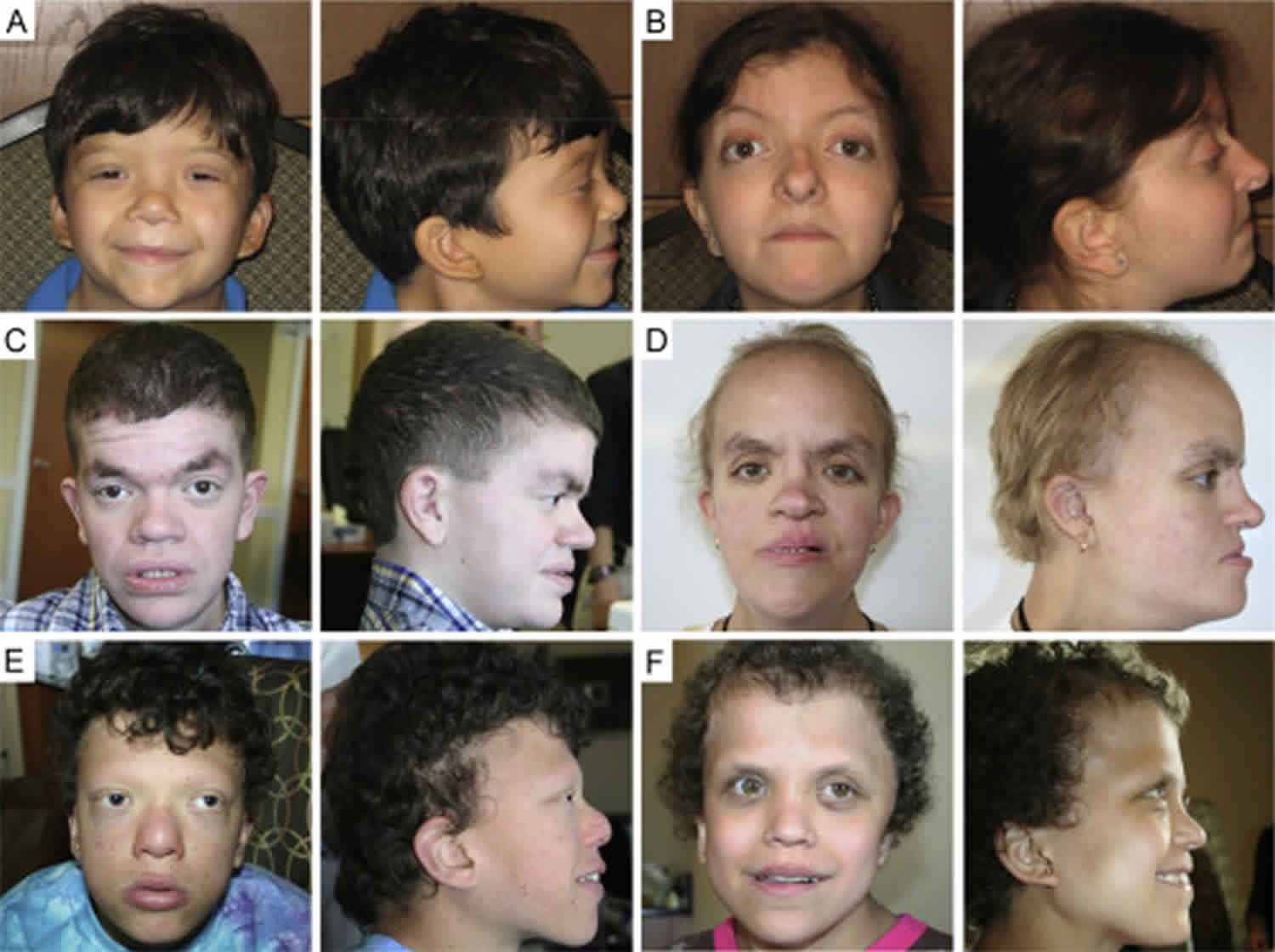
Figure 7. Cardiofaciocutaneous syndrome
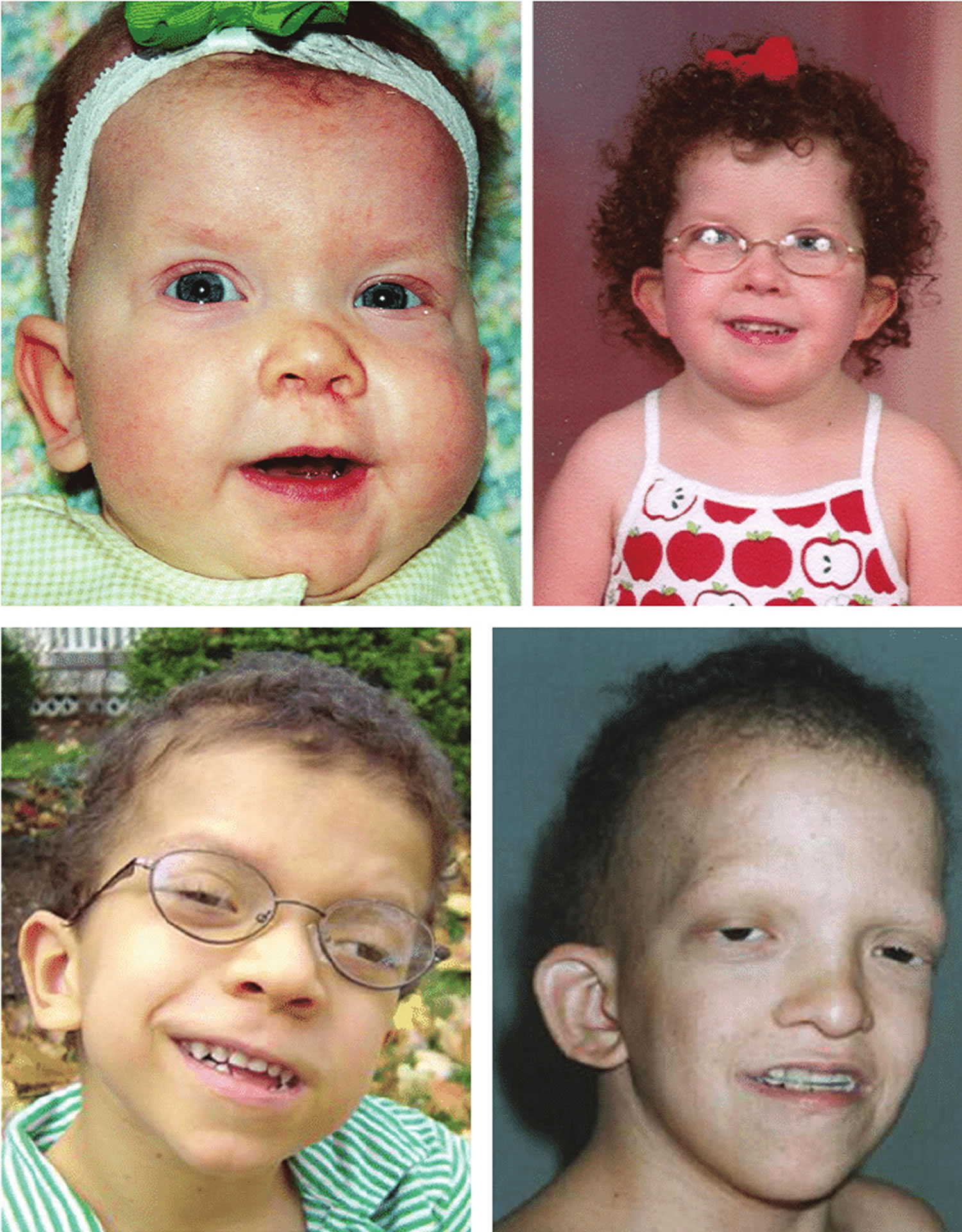
Footnote: Three patients of various ages with the cardiofaciocutaneous syndrome. The face is typical, with a broad forehead, bulbous tip of the nose, low-set ears, sparse scalp hair and absent eyebrows with ulerythema ophryogenes. The upper panel shows the same child at 10 months (left) and 6 years (right), describing the evolution of the phenotype. All of these children carry a BRAF gene mutation.
[Source 60 ]Cardiofaciocutaneous syndrome causes
Cardiofaciocutaneous syndrome can be caused by mutations in several genes. Mutations in the BRAF gene are most common, accounting for 75 to 80 percent of all cases. Another 10 to 15 percent of cases result from mutations in one of two similar genes, MAP2K1 and MAP2K2. Fewer than 5 percent of cases are caused by mutations in the KRAS gene.
The BRAF, MAP2K1, MAP2K2, and KRAS genes provide instructions for making proteins that work together to transmit chemical signals from outside the cell to the cell’s nucleus. This chemical signaling pathway, known as the RAS/MAPK pathway, is essential for normal development before birth. It helps control the growth and division (proliferation) of cells, the process by which cells mature to carry out specific functions (differentiation), cell movement, and the self-destruction of cells (apoptosis).
Mutations in any of these genes can result in the characteristic features of cardiofaciocutaneous syndrome. The protein made from the mutated gene is overactive, which alters tightly regulated chemical signaling during development. The altered signaling interferes with the development of many organs and tissues, leading to the signs and symptoms of cardiofaciocutaneous syndrome.
Some people with the signs and symptoms of cardiofaciocutaneous syndrome do not have an identified mutation in the BRAF, MAP2K1, MAP2K2, or KRAS gene. In these cases, affected individuals may actually have Costello syndrome or Noonan syndrome, which are also caused by mutations in genes involved in RAS-MAPK signaling. The proteins produced from these genes are all part of the same chemical signaling pathway, which helps explain why mutations in different genes can cause conditions with such similar signs and symptoms.
Cardiofaciocutaneous syndrome inheritance pattern
Cardiofaciocutaneous syndrome is considered to be an autosomal dominant condition, which means one copy of an altered gene in each cell is sufficient to cause the disorder.
Cardiofaciocutaneous syndrome usually results from new gene mutations (de novo mutation) and occurs in people with no history of the disorder in their family. In a few reported cases, an affected person has inherited the condition from an affected parent.
Cardiofaciocutaneous syndrome signs and symptoms
Most individuals are initially referred because of feeding difficulties (poor suck) and failure to thrive. Later, cognitive developmental delay and other clinical manifestations may be observed.
Facial appearance
Affected individuals may have a relatively large head (macrocephaly) when compared to their height, a high forehead and abnormal narrowing of the sides of the forehead (bitemporal narrowing), causing the head to appear “box-like” in shape. The ears are abnormally angulated towards the back of the head and low set (posteriorly angulated) with the ear lobes occasionally having creasing. The nose is short, bulbous and with anteverted nostrils and a depressed bridge. There is also an underdevelopment (hypoplasia) of the ridges of the bone above the eyes (supraorbital ridges); widely spaced eyes (ocular hypertelorism); downslant of eyelid openings and drooping of one or both upperlids (ptosis).
Skin, hair and nails
Most patients have some kind of ectodermal abnormality, either of skin, hair or nails. Children with cardiofaciocutaneous syndrome usually have sparse, slow growing, fine or thick, curly scalp hair that is abnormally dry and brittle. They also have absent or sparse eyebrows and eyelashes. In some children, the nails are dystrophic, with broad flat nails, and/or fast growing. Skin involvement ranges from dry skin to the skin disease known as hyperkeratosis. Pigmented nevi are very distinct to cardiofaciocutaneous syndrome and help define the syndrome. Other typical skin manifestations include small hard bumps (keratosis pilaris), facial skin lesions around the eyebrows (ulerythema ophryogenes), and benign vascular tumors (infantile hemangiomas).
Heart
Congenital heart defects are present in over 75% of patients, with the most common heart defects being pulmonic stenosis and atrial or ventricular septal defects. There may also be hypertrophic cardiomyopathy (thickening of the heart muscle) and rhythm disturbances. These defects may be diagnosed at birth or later in life.
Intellectual disability
There is some form of cognitive or neurologic delay in nearly all patients with cardiofaciocutaneous syndrome. Most individuals fall in the range of moderate intellectual disability. Global developmental delay including gross motor and language delay is very common. Autism and other sensory behavioral issues have been reported in some individuals with cardiofaciocutaneous syndrome.
Ocular
Symptoms affecting the eyes can effect both their appearance: ocular hypertelorism (increased distance between eyes), strabismus (uneven alignment of the eyes) and their function; involuntary eye movements, astigmatism, nearsightedness and/or farsightedness. Optic nerve hypoplasia, cortical blindness, and cataracts have been described. Although most individuals with cardiofaciocutaneous syndrome have ocular symptoms, some have a normal ophthalmologic examination.
Feeding and gastrointestinal
Severe feeding problems manifest as gastroesophageal reflux (GER), aspiration, vomiting, and affected individuals will avoid eating (avoidance of eating can lead to growth delays). Often individuals with gastrointestinal symptoms will require a feeding tube that may persist into early childhood. Other gastrointestinal problems include dysmotility, intestinal malrotation (abnormal development of the intestine), hernia, and/or constipation. Some individuals have inflammation of the spleen and/or liver. Most children have malnutrition secondary to avoidance of eating. Fatty liver and anal stenosis (tightening of the anal sphincter) have also been reported.
Endocrine abnormalities
Affected individuals can have growth hormone deficiency and early onset puberty.
Growth delays
Growth may be normal with appropriate birth weight and length; however, weight and length may drop to below the fifth percentile during early infancy while head circumference remains within the normal range, which gives the appearance of macrocephaly.
Additional abnormalities
Additional abnormalities that are present in some but not all patients include short stature; webbed neck; abnormal shape of the thorax (pectus carinatum); joint hyperextension; hypotonia (reduced muscle tone), neoplasia (typically lymphyoblastic leukemia), especially during the first years of life; urogenital anomalies; seizures; and undescended testes (cryptorchidism) of boys.
Cardiofaciocutaneous syndrome diagnosis
In most cases, cardiofaciocutaneous syndrome is diagnosed during infancy based upon a thorough clinical evaluation, characteristic physical findings, and specialized tests. Congenital heart defects that may occur in association with cardiofaciocutaneous syndrome (e.g., pulmonary stenosis and/or atrial septal defects) may be detected and/or confirmed by a thorough clinical examination and specialized tests that allow physicians to evaluate the structure and function of the heart.
Clinical examination may include a physician’s evaluation of heart and lung sounds through use of a stethoscope. For example, in mild asymptomatic cases of pulmonary stenosis, the condition may initially be detected through an abnormal heart murmur heard during such stethoscopic evaluation.
Specialized cardiac tests may include x-ray studies, electrocardiography (EKG), echocardiography, and/or cardiac catheterization. X-ray studies may reveal abnormal enlargement of the heart (cardiomegaly) or malformation of certain heart structures. An EKG, which records the electrical activities of the heart muscle, may reveal abnormal electrical patterns. During an echocardiogram, sound waves are directed toward the heart, enabling physicians to study cardiac function and motion. During cardiac catheterization, a small hollow tube (catheter) is inserted into a large vein and threaded through the blood vessels leading to the heart. This procedure allows physicians to determine the rate of blood flow through the heart, measure the pressure within the heart, and/or thoroughly identify anatomical abnormalities.
Physicians may also closely evaluate the respiratory (ventilatory) capabilities of affected individuals with pulmonary stenosis and/or other heart abnormalities since associated cardiac defects may result in inadequate blood supply to the lungs and breathlessness.
Additional specialized tests may also be conducted to help identify and/or confirm the presence of other abnormalities that may occur in some cases of cardiofaciocutaneous syndrome. For example, according to the medical literature, computerized tomography (CT) scanning may help confirm the presence of hydrocephalus or, in some cases, degeneration of the outer layer of the brain (cortical atrophy) in some individuals with the disorder. During CT scanning, a computer and X-rays are used to create a film showing cross-sectional images of an organ’s tissue structure. In addition, electroencephalography (EEG), which records the brain’s electrical impulses, may reveal brain wave patterns that are characteristic of certain types of seizure activity. Examination with an instrument that visualizes the interior of the eye (ophthalmoscopy), other specialized imaging techniques, and/or other tests may also be used to diagnose and/or confirm certain eye abnormalities that may be associated with cardiofaciocutaneous syndrome.
Molecular genetic testing is available for mutations in the four genes known to cause cardiofaciocutaneous syndrome. Testing can be done by using a multi-gene panel that screens all of the genes currently known to cause a RASopathy since there is a considerable overlap among them or whole exome sequencing can be considered.
Clinical testing and work-up
Individuals known to have or suspected to have cardiofaciocutaneous syndrome may have the following evaluations. a complete physical examination including measurement growth parameters; cardiac evaluation including echocardiogram and electrocardiogram; neurologic evaluation; MRI of the brain to detect any structural changes; electroencephalogram if seizures are suspected; full abdominal ultrasound examination to evaluate for renal and urogenital anomalies; psychomotor developmental evaluation; endocrine evaluation if growth delay is suspected; ophthalmologic examination; audiologic examination; nutrition and feeding evaluation, consider swallow study; and dermatologic evaluation. Consensus guidelines for the management of individuals with cardiofaciocutaneous were published in 2012 and should be referenced in the care of any patient with this diagnosis.
Cardiofaciocutaneous syndrome treatment
The treatment of cardiofaciocutaneous syndrome is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians; physicians who diagnose and treat skin disorders (dermatologists), heart abnormalities (cardiologists), eye disorders (ophthalmologists), and/or neurological abnormalities (neurologists); and/or other health care professionals may need to systematically and comprehensively plan an affected child’s treatment.
Specific therapies for cardiofaciocutaneous syndrome are symptomatic and supportive. In some individuals with congenital heart defects such as pulmonary stenosis and/or atrial septal defects, treatment with certain medications, surgical intervention, and/or other techniques may be necessary. In such cases, the surgical procedures performed will depend upon the location, severity, and/or combination of anatomical abnormalities and their associated symptoms.
In individuals with cardiofaciocutaneous syndrome, respiratory infections should be treated promptly and vigorously. Because of the potentially increased risk of bacterial infection of the lining of the heart (endocarditis) and the heart valves, individuals with atrial septal defects may be given antibiotic drugs before any surgical procedure, including dental procedures such as tooth extractions.
In affected individuals with hydrocephalus, shunts may be implanted to drain excess cerebrospinal fluid away from the brain, relieving pressure. In addition, in some cases, treatment with anticonvulsant drugs may help prevent, reduce, or control seizures.
In individuals affected by certain ocular abnormalities, corrective glasses, contact lenses, and/or surgery may be used to help improve vision.
Oftentimes, children who are failing to thrive will require a nasogastic or gastrostomy tube (feeding tubes). An increased caloric intake may also be beneficial in conjunction with increase fiber if the affected individual suffers from constipation.
In addition, to help alleviate skin abnormalities, physicians may recommend certain lubricating lotions or ointments, such as petroleum jelly. Applying such lubricants may be particularly effective after bathing while the skin is moist. In affected individuals with hemangiomas, treatment may not be required in some cases. In other cases, physicians may recommend removal of hemangiomas, depending upon severity, location, the occurrence of associated bleeding, and/or other associated symptoms or difficulties (e.g., obstruction of vision due to location on an eyelid). Various removal techniques may be used (e.g., laser surgery, cryosurgery, plastic surgery).
Early intervention may be beneficial in helping children with cardiofaciocutaneous syndrome reach their potential. Special services that may be of assistance may include special remedial education, speech therapy, occupational therapy, physical therapy, and/or other medical, social, and/or vocational services.
Genetic counseling is recommended for affected individuals and their families. Other treatment for the disorder is symptomatic and supportive.
Legius syndrome
Legius syndrome also known as neurofibromatosis type 1-like syndrome, is a rare genetic skin pigmentation condition characterized by changes in skin coloring (pigmentation). Almost all affected individuals have multiple cafe-au-lait spots, which are flat light-brown patches on the skin that are darker than the surrounding area (see Figure 1 below). Another pigmentation change, freckles in the armpits and groin, may occur in some affected individuals. Legius syndrome may occur in anyone who has at least one biological parent with genetically confirmed Legius syndrome 61.
Other signs and symptoms of Legius syndrome may include an abnormally large head (macrocephaly) and unusual facial characteristics. Although most people with Legius syndrome have normal intelligence, some affected individuals have been diagnosed with learning disabilities, attention-deficit disorder (ADD), or attention-deficit-hyperactivity disorder (ADHD) 61.
Many of the signs and symptoms of Legius syndrome also occur in a similar disorder called neurofibromatosis type 1 (NF1). It can be difficult to tell the two disorders apart in early childhood. However, the features of the two disorders differ later in life.
Legius syndrome was first described in 2007 by Brems and colleagues 62, who identified that a heterozygous mutation in SPRED1 gene was responsible of this mild neurofibromatosis phenotype. After 2007, more than 200 cases have been reported 63 and it has been demonstrated that up to 2% of patients fulfilling diagnostic criteria for neurofibromatosis type 1 (NF1), without NF1 gene mutations, have instead SPRED1 mutations 64. SPRED1 acts as a negative regulator of RAS pathway and interacts with neurofibromin, the product of the NF1 gene 65. Up to date, many different allelic variants have been found, but no genotype-phenotype correlation has been noted 66.
The prevalence of Legius syndrome is not known. Fewer than 300 cases have been reported to date. Many individuals with Legius syndrome are likely misdiagnosed because the signs and symptoms of Legius syndrome are similar to those of neurofibromatosis type 1 (NF1) 67. The incidence of neurofibromatosis type 1 is reported to be 1 per 3000, and about 2% of patients fulfilling diagnostic criteria for neurofibromatosis type 1 are found to have the genetic mutation underlying Legius syndrome (SPRED1).
Figure 8. Cafe-au-lait spots
Legius syndrome causes
Mutations in the SPRED1 gene on chromosome 15q14 cause Legius syndrome. Nearly 100 different mutations in this gene have been identified. The SPRED1 gene provides instructions for making the Spred-1 protein. This protein controls (regulates) an important cell signaling pathway called RAS-MAPK signal transduction pathway that is involved in the growth and division of cells (proliferation), the process by which cells mature to carry out specific functions (differentiation), cell movement, and the self-destruction of cells (apoptosis). Mutations in the SPRED1 gene lead to a nonfunctional protein that can no longer regulate the pathway, resulting in overactive signaling. It is unclear how mutations in the SPRED1 gene cause the signs and symptoms of Legius syndrome.
The proportion of cases related to de novo mutations (spontaneous mutations where no one in the family has it before and it appears to be a new thing in the family) is not yet known. No genotype-phenotype correlations have been found.
Legius syndrome inheritance pattern
Legius syndrome is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
Legius syndrome signs and symptoms
The clinical features of Legius syndrome vary significantly in nature and severity from one person to another. The clinical presentation of Legius syndrome is very similar to that of neurofibromatosis type 1 (NF1). Almost all patients with Legius syndrome present with multiple café-au-lait spots sometimes associated with intertriginous freckling, but lack Lisch nodules, optic pathway gliomas, bone abnormalities, neurofibromas or other tumor manifestations 61. The number of these café-au-lait spots tends to increase with age during childhood 68. Other less common manifestations include short stature, macrocephaly, Noonan-like facies, pectus excavatum/carinatum, lipomas, hypopigmented macules, vascular lesions, learning disabilities, attention deficit/hyperactivity disorder (ADHD), and developmental delay.
Other common skin features may include:
- Freckles in the armpit and groin regions
- Lipomas 61.
Non-skin features may include:
- Macrocephaly
- Unusual facial characteristics, similar to Noonan syndrome
- Short stature
- Pectus excavatum (sunken breastbone) or pectus carinatum (protruding breastbone) 61.
Neuropsychiatric features may include:
- Learning difficulties
- Developmental delay
- Attention-deficit hyperactivity disorder (ADHD) 61.
The intellectual disabilities are generally less severe in patients with Legius syndrome compared to patients with neurofibromatosis type 1 69.
Importantly, Legius syndrome does not cause neurofibromas and Lisch nodules, which are typically seen in neurofibromatosis type 1 61.
Legius syndrome diagnosis
Legius syndrome is difficult to diagnose on a clinical basis alone given its similar cutaneous clinical presentation to other disorders with multiple café-du-lait spots.
Where Legius syndrome is suspected, genetic testing can be used to confirm the diagnosis. The diagnosis of Legius syndrome is established with suggestive findings and a heterozygous pathogenic variant in SPRED1 identified by molecular genetic testing. This involves:
- Sequence analysis of SPRED1
- Deletion/duplication analysis (if sequence analysis is unremarkable)
- A multi-gene panel for SPRED1 and other genes of interest 61.
Suspicious findings that may warrant genetic testing for Legius syndrome include:
- Café-au-lait macules without other clinical features, suggesting neurofibromatosis type 1
- A parent with café-au-lait macules without clinical features, again suggesting neurofibromatosis type 1 61.
Legius syndrome treatment
Children with Legius syndrome should be under regular screening and evaluation for developmental delay, cognitive impairment, and behavioral issues 61.
The treatment of Legius syndrome is primarily supportive and should be focused on the specific problems of the affected individual.
Appropriate management, if indicated, may include:
- Physical, speech, and/or occupational therapy
- Behavioral-modification therapy and pharmacological therapy (ie, for ADHD) 61.
Legius syndrome prognosis
Patients with Legius syndrome have a generally good prognosis with appropriate problem-based management.
An affected individual must consider the 50% risk of genetic transmission to each child.
References- Rauen KA. The RASopathies. Annu Rev Genomics Hum Genet. 2013;14:355-369. doi:10.1146/annurev-genom-091212-153523 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4115674
- Yoon S, Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors. 2006;24:21–44.
- Pritchard C, Carragher L, Aldridge V, Giblett S, Jin H, et al. Mouse models for BRAF-induced cancers. Biochem Soc Trans. 2007;35:1329–33.
- Cawthon RM, O’Connell P, Buchberg AM, Viskochil D, Weiss RB, et al. Identification and characterization of transcripts from the neurofibromatosis 1 region: the sequence and genomic structure of EVI2 and mapping of other transcripts. Genomics. 1990;7:555–65.
- Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29:465–68.
- Tartaglia M, Pennacchio LA, Zhao C, Yadav KK, Fodale V, et al. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet. 2007;39:75–79.
- Razzaque MA, Nishizawa T, Komoike Y, Yagi H, Furutani M, et al. Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat Genet. 2007;39:1013–17.
- Schubbert S, Zenker M, Rowe SL, Boll S, Klein C, et al. Germline KRAS mutations cause Noonan syndrome. Nat Genet. 2006;38:331–36.
- Cirstea IC, Kutsche K, Dvorsky R, Gremer L, Carta C, et al. A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat Genet. 2010;42:27–29.
- Cordeddu V, Di Schiavi E, Pennacchio LA, Ma’ayan A, Sarkozy A, et al. Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat Genet. 2009;41:1022–26.
- Martinelli S, De Luca A, Stellacci E, Rossi C, Checquolo S, et al. Heterozygous germline mutations in the CBL tumor-suppressor gene cause a Noonan syndrome-like phenotype. Am J Hum Genet. 2010;87:250–57.
- Digilio MC, Conti E, Sarkozy A, Mingarelli R, Dottorini T, et al. Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am J Hum Genet. 2002;71:389–94.
- Pandit B, Sarkozy A, Pennacchio LA, Carta C, Oishi K, et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. 2007;39:1007–12.
- Eerola I, Boon LM, Mulliken JB, Burrows PE, Dompmartin A, et al. Capillary malformation–arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240–49.
- Aoki Y, Niihori T, Kawame H, Kurosawa K, Ohashi H, et al. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet. 2005;37:1038–40.
- Niihori T, Aoki Y, Narumi Y, Neri G, Cavé H, et al. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat Genet. 2006;38:294–96.
- Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, et al. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 2006;311:1287–90.
- Brems H, Chmara M, Sahbatou M, Denayer E, Taniguchi K, et al. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1–like phenotype. Nat Genet. 2007;39:1120–26.
- Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Neurofibromatosis type 1 revisited. Pediatrics. 2009;123:124–33.
- Neurofibromatosis type 1. https://ghr.nlm.nih.gov/condition/neurofibromatosis-type-1
- Ly KI, Blakeley JO. The Diagnosis and Management of Neurofibromatosis Type 1. Med Clin North Am. 2019;103(6):1035-1054. doi:10.1016/j.mcna.2019.07.004
- Friedman JM. Neurofibromatosis 1. 1998 Oct 2 [Updated 2019 Jun 6]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1109
- National Institutes of Health Consensus Development Conference Statement: neurofibromatosis. Bethesda, Md., USA, July 13-15, 1987. Neurofibromatosis. 1988;1(3):172-178.
- Giugliano T, Santoro C, Torella A, et al. Clinical and Genetic Findings in Children with Neurofibromatosis Type 1, Legius Syndrome, and Other Related Neurocutaneous Disorders. Genes (Basel). 2019;10(8):580. Published 2019 Jul 31. doi:10.3390/genes10080580
- Miller DT, Freedenberg D, Schorry E, et al. Health Supervision for Children With Neurofibromatosis Type 1. Pediatrics. 2019;143(5):e20190660. doi:10.1542/peds.2019-0660
- Noonan syndrome. https://ghr.nlm.nih.gov/condition/noonan-syndrome
- Mendez HM, Opitz JM. Noonan syndrome: a review. Am J Med Genet. 1985;21(3):493–506.
- Allanson JE, Roberts AE. Noonan Syndrome. 2001 Nov 15 [Updated 2019 Aug 8]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1124
- Karbach J, Coerdt W, Wagner W, Bartsch O. Case report: Noonan syndrome with multiple giant cell lesions and review of the literature. Am J Med Genet A. 2012;158A(9):2283-2289. doi:10.1002/ajmg.a.35493
- Dahlgren J. GH therapy in Noonan syndrome: Review of final height data. Horm Res. 2009;72 Suppl 2:46-48. doi:10.1159/000243779
- Binder G. et al. Health and quality of life in adults with Noonan syndrome. J. Pediatr. September 2012; 161(3):501-505.
- Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet. January 26, 2013; 381(9863):333-342.
- van der Burgt, I. Noonan syndrome. Orphanet J Rare Dis 2, 4 (2007). https://doi.org/10.1186/1750-1172-2-4
- Noonan syndrome with multiple lentigines. https://ghr.nlm.nih.gov/condition/noonan-syndrome-with-multiple-lentigines
- Cançado, Flávio Heleno da Silva Queiroz, Silva, Luis Candido Pinto da, Taitson, Paulo Franco, Andrade, Ana Carolina Dias Viana de, Pithon, Matheus Melo, & Oliveira, Dauro Douglas. (2017). Do you know this syndrome? Leopard syndrome. Anais Brasileiros de Dermatologia, 92(1), 127-129. https://doi.org/10.1590/abd1806-4841.20174505
- Capillary malformation-arteriovenous malformation syndrome. https://ghr.nlm.nih.gov/condition/capillary-malformation-arteriovenous-malformation-syndrome
- Capillary Malformation–Arteriovenous Malformation, a New Clinical and Genetic Disorder Caused by RASA1 Mutations. Am. J. Hum. Genet. 73:1240–1249, 2003 https://doi.org/10.1086/379793
- Kim C, Ko CJ, Baker KE, Antaya RJ. Histopathologic and ultrasound characteristics of cutaneous capillary malformations in a patient with capillary malformation-arteriovenous malformation syndrome. Pediatr Dermatol. 2015;32:128–31.
- Orme CM, Boyden LM, Choate KA, Antaya RJ, King BA. Capillary malformation–arteriovenous malformation syndrome: review of the literature, proposed diagnostic criteria, and recommendations for management. Pediatr Dermatol. 2013;30:409–15.
- Revencu N, Boon LM, Mendola A, et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum Mutat. 2013;34(12):1632-1641. doi:10.1002/humu.22431
- Amyere M, Revencu N, Helaers R, et al. Germline Loss-of-Function Mutations in EPHB4 Cause a Second Form of Capillary Malformation-Arteriovenous Malformation (CM-AVM2) Deregulating RAS-MAPK Signaling. Circulation. 2017;136(11):1037-1048. doi:10.1161/CIRCULATIONAHA.116.026886
- Vivanti A, Ozanne A, Grondin C, Saliou G, Quevarec L, Maurey H, Aubourg P, Benachi A, Gut M, Gut I, Martinovic J, Sénat MV, Tawk M, Melki J. Loss of function mutations in EPHB4 are responsible for vein of Galen aneurysmal malformation. Brain. 2018;141:979–88.
- Revencu N, Boon LM, Mulliken JB, Enjolras O, Cordisco MR, Burrows PE, Clapuyt P, Hammer F, Dubois J, Baselga E, Brancati F, Carder R, Quintal JM, Dallapiccola B, Fischer G, Frieden IJ, Garzon M, Harper J, Johnson-Patel J, Labrèze C, Martorell L, Paltiel HJ, Pohl A, Prendiville J, Quere I, Siegel DH, Valente EM, Van Hagen A, Van Hest L, Vaux KK, Vicente A, Weibel L, Chitayat D, Vikkula M. Parkes Weber syndrome, vein of Galen aneurysmal malformation, and other fast-flow vascular anomalies are caused by RASA1 mutations. Hum Mutat. 2008;29:959–65.
- Thiex R, Mulliken JB, Revencu N, Boon LM, Burrows PE, Cordisco M, Dwight Y, Smith ER, Vikkula M, Orbach DB. A novel association between RASA1 mutations and spinal arteriovenous anomalies. AJNR Am J Neuroradiol. 2010;31:775–9.
- Wooderchak-Donahue WL, Akay G, Whitehead K, Briggs E, Stevenson DA, O’Fallon B, Velinder M, Farrell A, Shen W, Bedoukian E, Skrabann CM, Antaya RJ, Henderson K, Pollak J, Treat J, Day R, Jacher JE, Hannibal M, Bontempo K, Marth G, Bayrak-Toydemir P, McDonald J. Phenotype of CM-AVM2 caused by variants in EPHB4: how much overlap with hereditary hemorrhagic telangiectasia (HHT)? Genet Med. 2019;21:2007–14.
- Eerola I, Boon LM, Mulliken JB, Burrows PE, Dompmartin A, Watanabe S, Vanwijck R, Vikkula M. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;2003;73:1240–9.
- Durrington HJ, Firth HV, Patient C, Belham M, Jayne D, Burrows N, Morrell NW, Chilvers ER. A novel RASA1 mutation causing capillary malformation-arteriovenous malformation (CM-AVM) presenting during pregnancy. Am J Med Genet A. 2013;161A:1690–4.
- Overcash RT, Gibu CK, Jones MC, Ramos GA, Andreasen TS. Maternal and fetal capillary malformation-arteriovenous malformation (CM-AVM) due to a novel RASA1 mutation presenting with prenatal non-immune hydrops fetalis. Am J Med Genet A. 2015;167A:2440–3.
- Martin-Almedina S, Martinez-Corral I, Holdhus R, Vicente A, Fotiou E, Lin S, Petersen K, Simpson MA, Hoischen A, Gilissen C, Jeffery H, Atton G, Karapouliou C, Brice G, Gordon K, Wiseman JW, Wedin M, Rockson SG, Jeffery S, Mortimer PS, Snyder MP, Berland S, Mansour S, Makinen T, Ostergaard P. EPHB4 kinase-inactivating mutations cause autosomal dominant lymphatic-related hydrops fetalis. J Clin Invest. 2016;126:3080–8.
- Macmurdo CF, Wooderchak-Donahue W, Bayrak-Toydemir P, Le J, Wallenstein MB, Milla C, Teng JM, Bernstein JA, Stevenson DA. RASA1 somatic mutation and variable expressivity in capillary malformation/arteriovenous malformation (CM/AVM) syndrome. Am J Med Genet A. 2016;170:1450–4.
- Burrows PE, Gonzalez-Garay ML, Rasmussen JC, Aldrich MB, Guilliod R, Maus EA, Fife CE, Kwon S, Lapinski PE, King PD, Sevick-Muraca EM. Lymphatic abnormalities are associated with RASA1 gene mutations in mouse and man. Proc Natl Acad Sci U S A. 2013;110:8621–6.
- Li D, Wenger TL, Seiler C, March ME, Gutierrez-Uzquiza A, Kao C, Bhoj E, Tian L, Rosenbach M, Liu Y, Robinson N, Behr M, Chiavacci R, Hou C, Wang T, Bakay M, Pellegrino da Silva R, Perkins JA, Sleiman P, Levine MA, Hicks PJ, Itkin M, Dori Y, Hakonarson H. Pathogenic variant in EPHB4 results in central conducting lymphatic anomaly. Hum Mol Genet. 2018;27:3233–45.
- Weitz NA, Lauren CT, Behr GG, Wu JK, Kandel JJ, Meyers PM, Sultan S, Anyane-Yeboa K, Morel KD, Garzon MC. Clinical spectrum of capillary malformation-arteriovenous malformation syndrome presenting to a pediatric dermatology practice: a retrospective study. Pediatr Dermatol. 2015;32:76–84.
- Hershkovitz D, Bergman R, Sprecher E. A novel mutation in RASA1 causes capillary malformation and limb enlargement. Arch Dermatol Res. 2008b;300:385–8.
- Mulliken JB, Young AE, eds. Vascular Birthmarks: Hemangiomas and Vascular Malformations. Philadelphia, PA: WB Saunders Co; 1988.
- Bayrak-Toydemir P, Stevenson D. Capillary Malformation-Arteriovenous Malformation Syndrome. 2011 Feb 22 [Updated 2019 Sep 12]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK52764
- https://ghr.nlm.nih.gov/condition/costello-syndrome
- Bertola D, Buscarilli M, Stabley DL, et al. Phenotypic spectrum of Costello syndrome individuals harboring the rare HRAS mutation p.Gly13Asp. American Journal of Medical genetics. Part A. 2017 May;173(5):1309-1318. DOI: 10.1002/ajmg.a.38178
- Cardiofaciocutaneous syndrome. https://ghr.nlm.nih.gov/condition/cardiofaciocutaneous-syndrome
- Roberts, A & Allanson, J & Jadico, S & Kavamura, Maria & Noonan, Jacqueline & Opitz, John & Young, T & Neri, Giovany. (2006). Cardiofaciocutaneous syndrome. Journal of medical genetics. 43. 833-42. 10.1136/jmg.2006.042796
- Legius E, Stevenson D. Legius Syndrome. 2010 Oct 14 [Updated 2020 Aug 6]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK47312
- Brems H, Chmara M, Sahbatou M, Denayer E, Taniguchi K, Kato R, Somers R, Messiaen L, De Schepper S, Fryns JP, Cools J, Marynen P, Thomas G, Yoshimura A, Legius E. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nat Genet. 2007;39:1120–1126. doi: 10.1038/ng2113
- Brems H, Legius E. Legius syndrome, an Update. Molecular pathology of mutations in SPRED1. Keio J Med. 2013;62(4):107–112. doi: 10.2302/kjm.2013-0002-RE
- Messiaen L, Yao S, Brems H, Callens T, Sathienkijkanchai A, Denayer E, Spencer E, Arn P, Babovic-Vuksanovic D, Bay C, Bobele G, Cohen BH, Escobar L, Eunpu D, Grebe T, Greenstein R, Hachen R, Irons M, Kronn D, Lemire E, Leppig K, Lim C, McDonald M, Narayanan V, Pearn A, Pedersen R, Powell B, Shapiro LR, Skidmore D, Tegay D, et al. Clinical and mutational spectrum of Neurofibromatosis type 1-like syndrome. JAMA. 2009;302(19):2111–8. doi: 10.1001/jama.2009.1663
- Stowe IB, Mercado EL, Stowe TR, Bell EL, Oses-Prieto JA, Hernández H, Burlingame AL, McCormick F. A shared molecular mechanism underlies the human rasopathies Legius syndrome and Neurofibromatosis-1. Genes Dev. 2012;26(13):1421–1426. doi: 10.1101/gad.190876.112
- Benelli E, Bruno I, Belcaro C, Ventura A, Berti I. Legius syndrome: case report and review of literature. Ital J Pediatr. 2015;41:8. Published 2015 Feb 8. doi:10.1186/s13052-015-0115-9 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4323213
- Legius syndrome. https://ghr.nlm.nih.gov/condition/legius-syndrome
- Legius syndrome. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=137605
- Benelli E, Bruno I, Belcaro C, Ventura A, Berti I. Legius syndrome: case report and review of literature. Ital J Pediatr. 2015;41:8. Published 2015 Feb 8. doi:10.1186/s13052-015-0115-9





{kind=link}