What is retinoschisis
Retinoschisis is splitting into two layers of the neurosensory retina, the tissue lining the inside of the back of the eye that transmits visual signals to the optic nerve and brain. Retinoschisis can resemble the appearance of a retinal detachment. The part of the retina that is affected by retinoschisis will have suboptimal vision. This can occur in different layers of the retina, and for different reasons.
The two major causes of retinoschisis are:
- Genetic (typically occurring in younger patients)
- Degenerative (typically occurring in older patients)
The most common form of retinoschisis is acquired retinoschisis also called degenerative retinoschisis or senile retinoschisis, where the split is in the outer plexiform retina layer. The inferotemporal quadrant is most commonly involved. Most acquired retinoschisis remains stationary over many years, but some eyes do progress to rhegmatogenous retinal detachment.
Risk factors for retinoschisis
Retinal detachment can occur if the anchoring of the outer layer of the retina to the eye wall is impaired. Since retinoschisis patients are more susceptible to retinal detachment, they should have regular examinations with an ophthalmologist. When detected early, a complicating retinal detachment can be treated surgically; however, splitting or schisis of the retina itself cannot be corrected by medication or surgery.
Retinoschisis affects two primary aspects of vision:
- Central vision can be impaired, with visual acuity ranging from 20/30 to less than 20/200. Acuity loss is caused by the formation of tiny cysts between the separated layers of the retina. These cysts often form a “spoke-wheel” pattern which is very subtle and is usually detected only by a trained clinician. Since the nerve tissue is damaged by these cysts, visual acuity cannot be improved with glasses.
- Peripheral vision can also be lost if the inner layer of nerve cells split off from the outer layer of cells.
Retinoschisis can be confused with other eye diseases, such as amblyopia (lazy eye). If someone in your family has retinoschisis and you are diagnosed with amblyopia you should have a thorough exam by an ophthalmologist experienced in diagnosing this condition.
B-scan ultrasound is a useful adjunct to confirm splitting of the retinal layers in the periphery. Bilateral symmetry and an inferotemporal location are highly suggestive of retinoschisis. Also, A-scan measurements showing splitting of the retinal layers are helpful.
Retinoschisis vs Retinal detachment
Retinal detachment is a medical emergency in which a thin layer of tissue (the retina) at the back of the eye pulls away from its normal position. If not promptly treated, it can cause permanent vision loss. If you have any symptoms, see an eye care professional immediately. Treatment includes different types of surgery. Retinal detachment can occur at any age, but it is more common in people over age 40. Retinal detachment affects men more than women and whites more than African Americans. A retinal detachment is also more likely to occur in people who:
- Are extremely nearsighted
- Have had a retinal detachment in the other eye
- Have a family history of retinal detachment
- Have had cataract surgery
- Have other eye diseases or disorders
- Have had an eye injury
Retinal detachment itself is painless. But warning signs almost always appear before it occurs or has advanced, such as:
- The sudden appearance of many floaters — tiny specks that seem to drift through your field of vision
- Flashes of light in one or both eyes (photopsia)
- Blurred vision
- Gradually reduced side (peripheral) vision
- A curtain-like shadow over your visual field
Retinal detachment symptoms include an increase in the number of floaters, which are little “cobwebs” or specks that float about in your field of vision, and/or light flashes in the eye. It may also seem like there is a “curtain” over your field of vision.
Seek immediate medical attention if you are experiencing the signs or symptoms of retinal detachment. Retinal detachment is a medical emergency in which you can permanently lose your vision.
There are three different types of retinal detachment:
- Rhegmatogenous—A tear or break in the retina allows fluid to get under the retina and separate it from the retinal pigment epithelium (RPE), the pigmented cell layer that nourishes the retina. These types of retinal detachments are the most common.
- Tractional—In this type of detachment, scar tissue on the retina’s surface contracts and causes the retina to separate from the RPE. This type of detachment is less common.
- Exudative—Frequently caused by retinal diseases, including inflammatory disorders and injury/trauma to the eye. In this type, fluid leaks into the area underneath the retina, but there are no tears or breaks in the retina.
Retinoschisis causes
The two major causes of retinoschisis are:
- Juvenile X-linked Retinoschisis: Juvenile X-linked Retinoschisis is a genetic disease of the retina and affects primarily boys and young men. In this form of retinoschisis, a mutation or abnormal gene is carried on the X chromosome. Men have only one X chromosome, while women have two. Therefore, women can carry the condition, but because they almost always have another normal X chromosome, they typically retain normal vision, even as carriers. Men, on the other hand, will develop subnormal vision if they have an affected X chromosome. This condition typically presents during childhood and is estimated to affect one in 5,000 to 25,000 individuals. Affected males are usually identified in grade school, but occasionally are identified as young infants. Affected boys and men should have periodic eye examinations.
- Degenerative retinoschisis: Degenerative retinoschisis also called senile retinoschisis is the splitting of the retina as a result of aging. It can affect men and women. This is not a genetic condition.
X-linked juvenile retinoschisis
X-linked retinoschisis or X-linked juvenile retinoschisis is an X-linked recessive congenital malformation of the retina caused by a mutation in the RS1 gene on Xp22.1, which encodes encodes a 224-amino acid protein called retinoschisin, which is secreted by photoreceptors, a protein involved in intercellular adhesion 1. Retinoschisin protein is found throughout the retina, and is thought to be involved in cell-cell adhesion and intercellular matrix retinal architecture development through interactions with αβ crystallin and β2-laminin. On histopathological examination, the splitting in X-linked retinoschisis occurs predominantly in the nerve fiber layer 2.
X-linked retinoschisis results in splitting of the foveal and sometimes peripheral neurosensory retina. X-linked juvenile retinoschisis is characterized by symmetric bilateral macular involvement with onset in the first decade of life, in some cases as early as age three months 3. Affected males generally present with reduction in vision by early elementary school. Affected males typically have vision of 20/60 to 20/120 on first presentation. Estimates of the prevalence of X-linked juvenile retinoschisis vary from one in 5,000 to one in 25,000 4.
Visual acuity may deteriorate during the first and second decades of life but then remain relatively stable, with only very slowly progressive reduction from macular atrophy, until the fifth or sixth decade 5. Visual loss may progress to legal blindness (acuity <20/200) by the sixth or seventh decade. In individuals over age 50 years, macular pigmentary changes and some degree of atrophy of the retinal pigment epithelium (RPE) are common. The delayed B-wave onset seen in a study of 68 affected males suggests that photoreceptor synapse or bipolar cell dysfunction increases with age 6.
Variation in disease presentation and disease progression is observed even among members of the same family.
Appearance of foveal lesions varies from largely radial striations (3%), microcystic lesions (34%), honeycomb-like cysts (8%), or their combinations (31%) to non-cystic-appearing foveal changes including pigment mottling (8%), loss of the foveal reflex (8%), or an atrophic-appearing lesion (8%) 5.
X-linked juvenile retinoschisis progresses to retinal detachment in an estimated 5% to 22% of affected individuals. Retinal detachment can occur in infants with severe retinoschisis. About 4% to 40% of individuals with X-linked juvenile retinoschisis develop vitreous hemorrhage.
Fundus examination shows areas of retinoschisis (splitting of the nerve fiber layer of the retina) in the macula, sometimes giving the impression of a spoke wheel pattern (see Figure 1 below). Retinoschisis of the peripheral retina, predominantly inferotemporally, occurs in approximately 50% of individuals 3. Affected males typically have vision of 20/60 to 20/120. Visual acuity often deteriorates during the first and second decades of life but then remains relatively stable until the fifth or sixth decade.
Clinically, retinoschisis appears as cystic spaces and radial striae in the central macula, as seen in these photographs. The splitting is even more apparent on optical coherence tomography (OCT). Optical coherence tomography (OCT) is a high-resolution cross-sectional imaging technique that allows for in vivo quantification of the retinal nerve fiber layer and the optic disc features. Over time, patients with X-linked juvenile retinoschisis develop decreased acuity with a central scotoma as seen in these Goldmann visual fields.
X-linked retinoschisis, with a prevalence of about 1 in 15,000 to 30,000, is the main cause of juvenile macular degeneration 7. It is caused by a large variety of mutations in RS1 on XP22.1, which encodes retinoschisin. This gene is responsible for proteins that are involved in intercellular adhesion. Most affected individuals are males, as heterozygous females are rarely affected. However, retinoschisis has been reported in non-consanguinous females 8. The phenotype can be markedly variable even within the same genotype, and can extend into the erpiheral retina.
X-linked retinoschisis genetic counseling
X-linked juvenile retinoschisis is inherited in an X-linked manner. Males will pass the x-linked recessive mutation to all daughters, but not sons. Female carriers have a 50% chance of transmitting the mutation in each pregnancy: all sons with the mutation will be affected; females who inherit the pathogenic variant will most likely be asymptomatic carriers and will nearly always have normal visual function and electrophysiology. Affected males pass the pathogenic variant to all of their daughters and none of their sons. Carrier testing for at-risk female relatives and prenatal testing for pregnancies at increased risk are possible if the pathogenic variant in the family is known.
X-linked retinoschisis symptoms
There is large variation in disease severity among patients, , ranging from normal vision to legal blindness, even among patients with the same genetic mutation. Boys typically present complaining of difficulty in school, although younger boys may present in infancy with nystagmus, strabismus, hyperopia, foveal ectopia, hemorrhage, or detachment. Visual acuity may range from 20/20 to blindness, and depends on the amount and area of schisis. The vision is often stable until middle-age.
X-linked retinoschisis may also present as spontaneous vitreous hemorrhage or retinal detachment. These complications occur commonly and are the main causes of complete vision loss 9.
Peripheral vision may be normal in the unaffected areas. Absolute scotoma may be present in areas of peripheral retinoschisis. Color vision is often normal as well.
On fundoscopic examination, 98-100% of patients have foveal schisis, noted as a spokewheel pattern radiating from the fovea and a domelike elevation of thin layer or retina. Schisis is most often in the macular, but extension into the periphery occurs in more than half of patients 9. The bullous retinoschisis may improve over time.
Subretinal linear fibrosis, pigmentation, white retinal flecks, vascular attenuation, and vascular sheathing may also be present.
Visual function may be severely limited with progression to retinal detachment, most often rhegmatogenous, in 5-20%. Vitreous hemorrhage is another common complication, and hemorrhage may also occur within the schisis cavity.
Other complications include intraretinal splitting, neovascular glaucoma, macular dragging, and optic atrophy.
X-linked retinoschisis diagnosis
The diagnosis of X-linked juvenile retinoschisis is based on fundus findings, results of electrophysiologic testing, and molecular genetic testing. RS1 is the only gene known to be associated with X-linked juvenile retinoschisis.
Diagnostic procedures:
- Digital fundus photography may help with examination of a child
- Red-free illumination may help to highlight the area of foveal schisis
- Fundus autofluorescence may also help to highlight areas of foveal schisis
- Optical coherence tomography (OCT) reveals schisis in the superficial neural retina. There are often large cystic-like spaces, especially large below the fovea. These cystic spaces may be present in any layer of the retina and reveal areas of schisis not visible on fundus examination.
Fluorescein angiography is not required for diagnosis. Unlike cystic macula edema, there may be pooling but no leakage of the cystic spaces.
Full-field electroretinogram (ffERG) is electronegative (Reduced b-wave with preserved a-wave). This is not diagnostic as the differential for electronegative ERG includes several other retinal disorders, and the a-wave may be reduced as the disease progresses.
Genetic testing can confirm the diagnosis.
Intravenous fluorescein angiogram appears normal in younger individuals, whereas older individuals may have atrophic changes in the retinal pigment epithelium (RPE).
Optical coherence tomography (OCT) shows small cystic-appearing spaces in the perifoveal region and larger cystic-like spaces within the fovea in most school-age individuals [Apushkin et al 2005]. Cystic spaces are not as evident after adolescence. OCT scans of older individuals may appear normal because of flattening of cysts with age.
Affected males
The diagnosis of X-linked juvenile retinoschisis is made in a young male with the following findings:
- Bilaterally reduced visual acuity, typically between 20/60 and 20/120
- The following findings on fundus examination:
- Areas of retinoschisis (splitting of the nerve fiber layer of the retina) in the macula, sometimes giving the impression of a spoke wheel pattern (Figure 1).
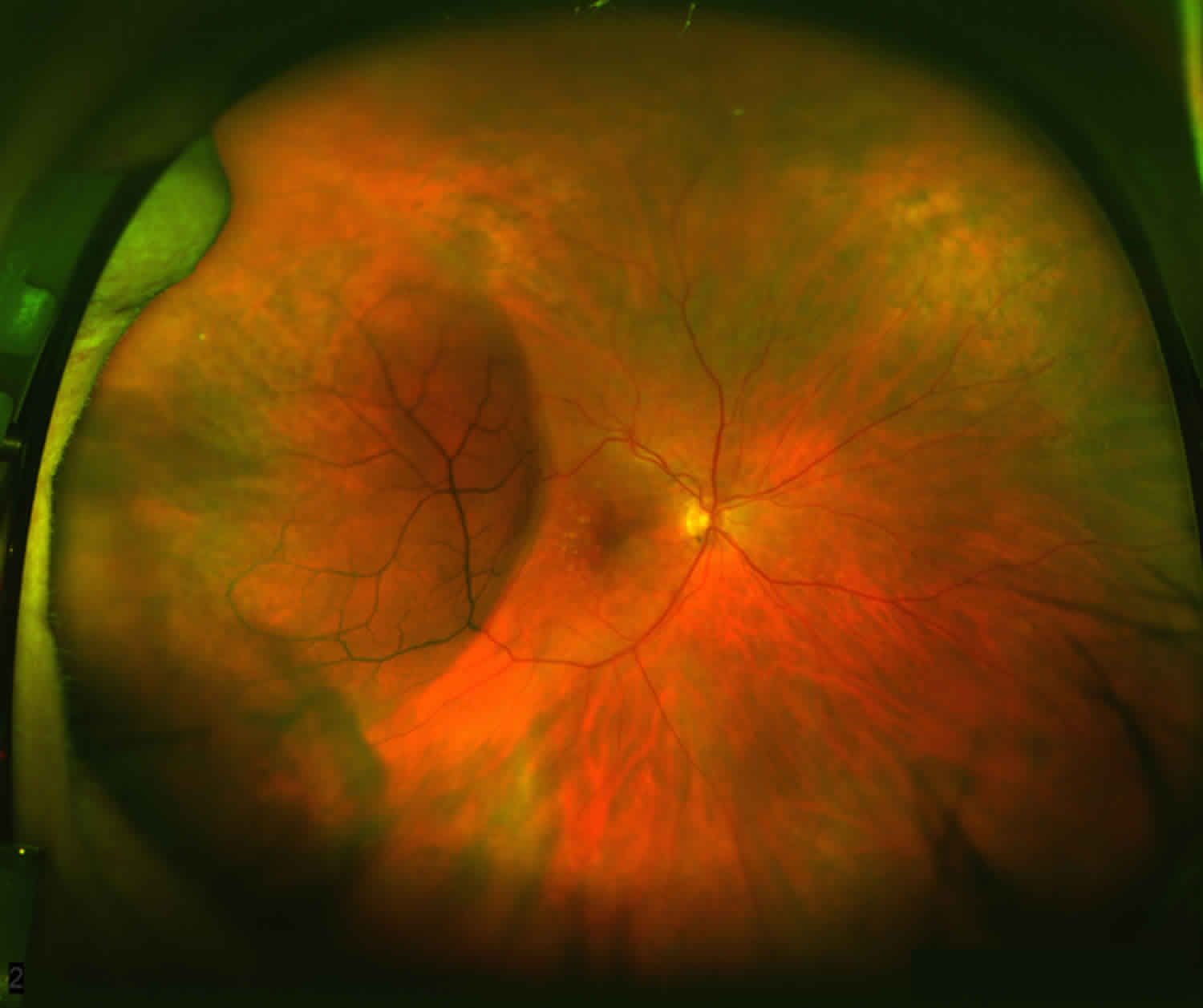
- Retinoschisis of the peripheral retina, predominantly inferotemporally, in approximately 50% of individuals 10. The associated elevation of the surface layer of the retina into the vitreous has been described as “vitreous veils” (Figure 2).
- On occasion, more severe involvement of the macula (Figure 3)
- On occasion, the Mizuo phenomenon, a color change in the retina after dark adaptation with the onset of light
- Spectral domain optical coherence tomography (SD-OCT) that reveals characteristic signs, such as foveal schisis and thinning of the retina. This is currently the major diagnostic technique for X-linked juvenile retinoschisis 11
- Increased fundus autofluorescence in the fovea, although this should be confirmed by SD-OCT 11
- A family history consistent with X-linked inheritance
- A pathogenic variant in RS1 gene
Carrier females
In most cases, carrier females cannot be identified by clinical examination. Carrier females nearly always have normal visual function and a normal electroretinogram (ERG). Rarely, examination of the peripheral retina may show white flecks or areas of retinoschisis.
- In a study of nine obligate carriers, two had areas of retinal dysfunction detected by multifocal electroretinogram (ERG) testing. Both had normal-appearing fundi 12.
- An Australian study 13 identified an obligate carrier female with abnormal rod ERG and multifocal ERG. The authors attribute this finding to either skewed X-inactivation or another underlying condition.
Figure 1. X-linked juvenile retinoschisis
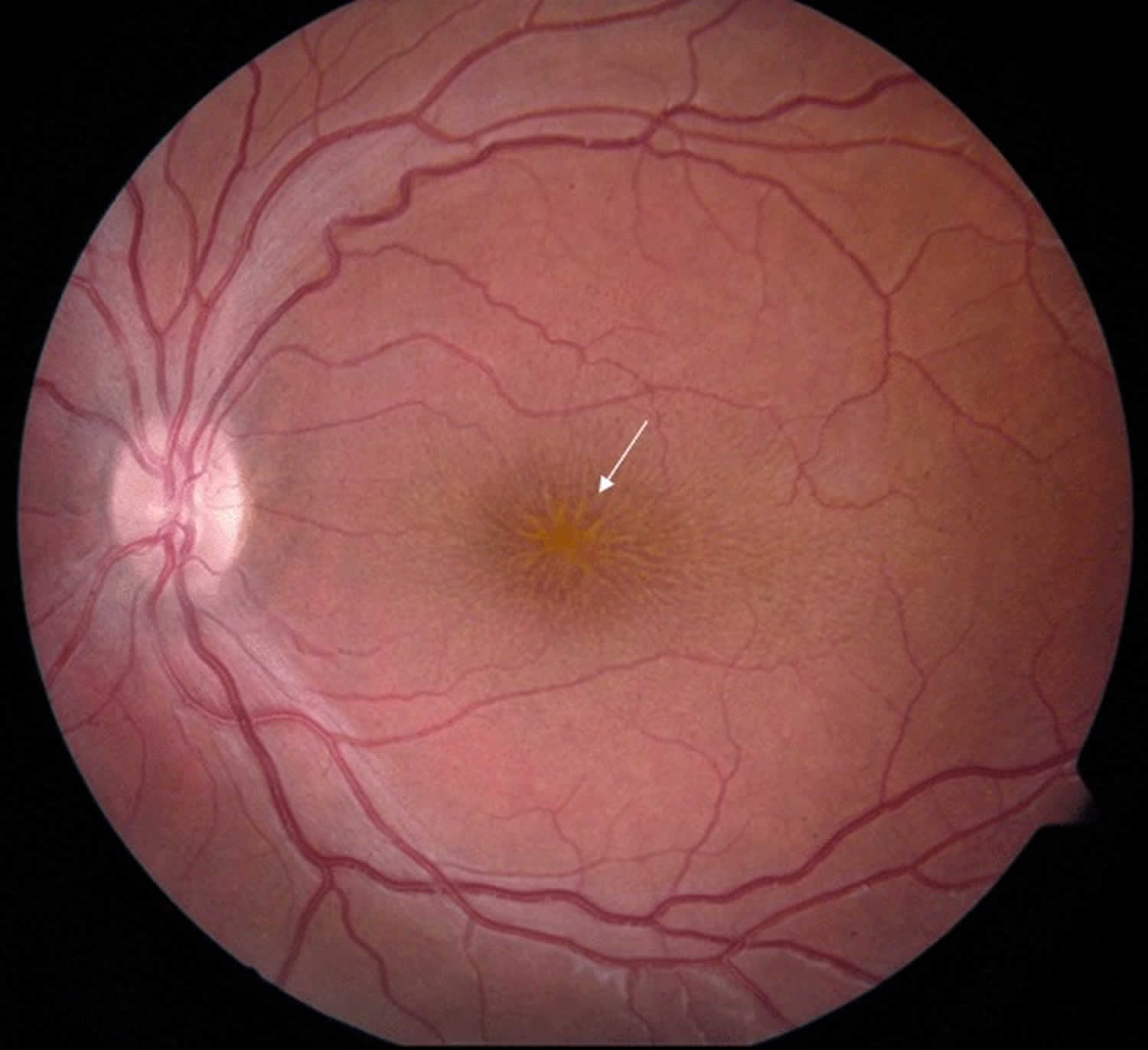
Footnote: Fundus photo of a male with juvenile retinoschisis. Arrow points to typical spoke-wheel pattern of foveal cysts.
Figure 2. Juvenile retinoschisis
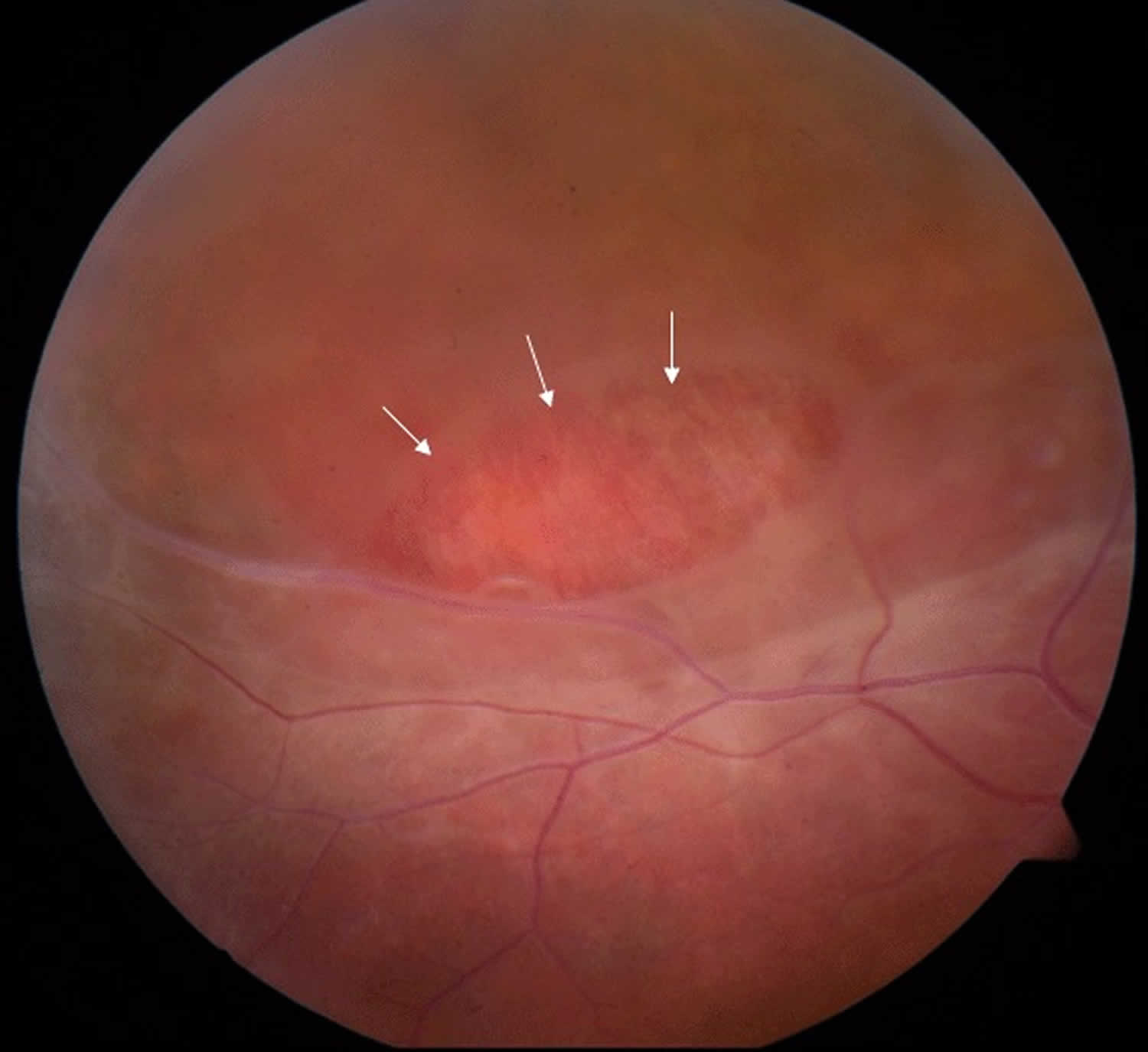
Footnote: Fundus photo of the peripheral retina of a male with juvenile retinoschisis. Area marked with arrows shows a partial-thickness retinal hole.
Figure 3. X-linked retinoschisis (severe)
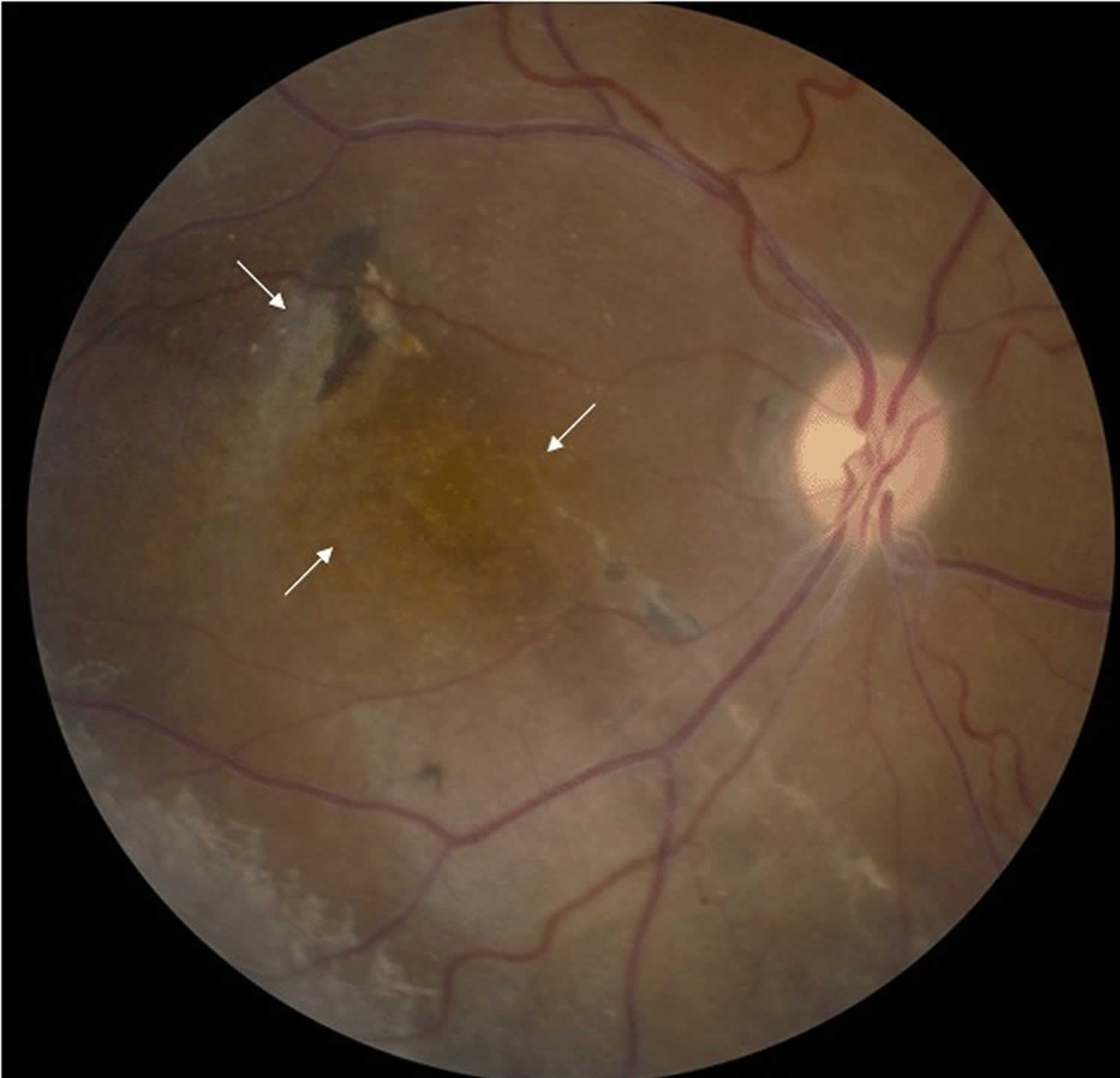
Footnote: Fundus photo of a male with juvenile retinoschisis showing atypical, more severe findings with arrows pointing to atrophic macular changes.
Figure 4. Retinoschisis OCT
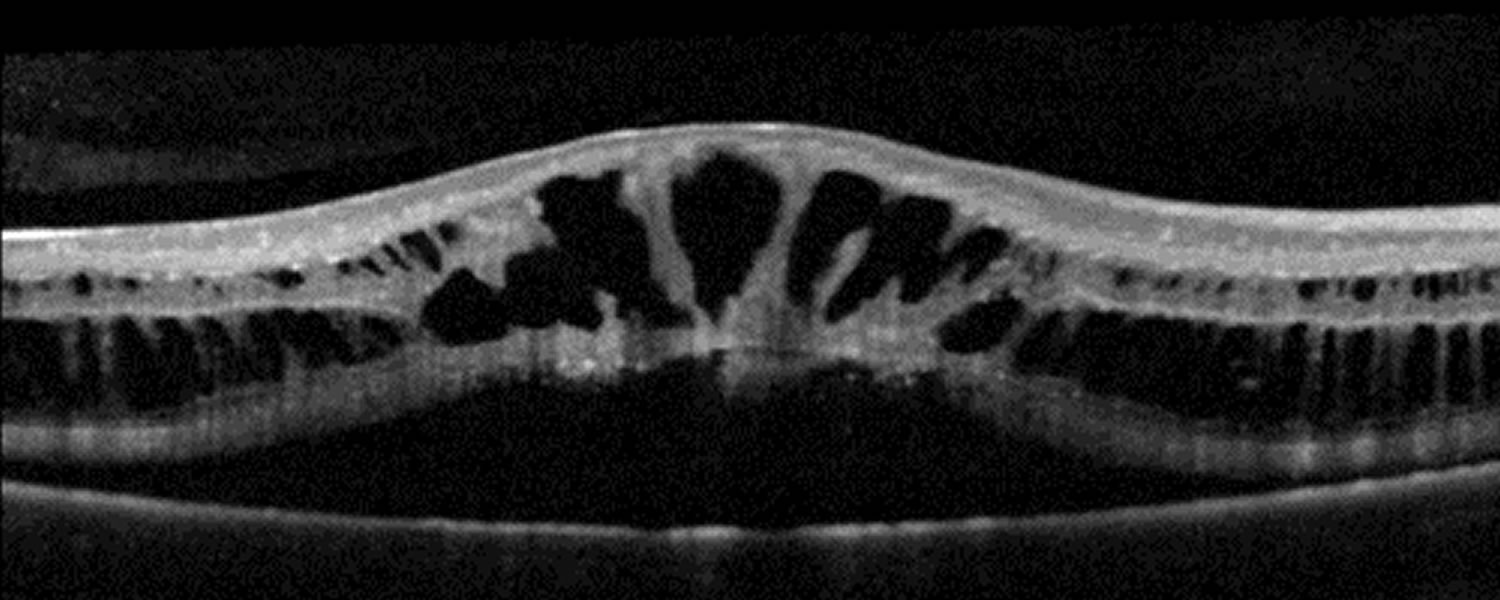
X-linked retinoschisis treatment
- Treatment of manifestations: Low-vision aids such as large-print textbooks; preferential seating in the front of the classroom; and use of handouts with high contrast. Surgery may be required to address the infrequent complications of vitreous hemorrhage and full-thickness retinal detachment.
- Prevention of secondary complications: Ambylopia prevention therapy is indicated following surgical intervention for vitreous hemorrhage or retinal detachment or in cases of severe retinoschisis or hypermetropia.
- Surveillance: Annual evaluation of children under age ten years by a pediatric ophthalmologist or retina specialist; patient education and close follow up may allow for early identification and treatment of vision-threatening complications.
- Agents/circumstances to avoid: Head trauma and high-contact sports to reduce risk of retinal detachment and vitreous hemorrhage.
Medical therapy
Carbonic anhydrase inhibitors may help to improve the retinoschisis cavities seen on OCT. Topical dorzolamide or systemic acetazolamide have both been reported to be beneficial in improving the cystic-appearing spaces on OCT 14. Clinical improvement can be monitored with visual acuity improvements and reduction in the cystic fluid on OCT.
Gene therapy with intra-ocular RS1 in knockout mice has restored b-wave function and there are several human clinical trials that are recruiting subjects including NCT02317887 and NCT02416622.
Surgery
- Complications such as retinal detachment and vitreous hemorrhage may require surgical intervention.
- Laser photocoagulation may prevent detachment. However, it may also induce detachment.
- External drainage has also been attempted 15.
X-linked juvenile retinoschisi prognosis
Vision is typically stable until the 5th or 6th decade.
Degenerative retinoschisis
Degenerative retinoschisis also known as acquired retinoschisis or senile retinoschisis, is an acquired, idiopathic retinoschisis characterized by gradual, peripheral splitting of retinal layers. This produces a well-circumscribed, transparent dome-shaped elevation of the inner retina that extends anteriorly towards the ora serrata 16. Most degenerative retinoschisis remains stationary over many years, but some eyes do progress to rhegmatogenous retinal detachment. Degenerative retinoschisis is found in ~4% of population and about 30% of affected individuals have bilateral involvement. Some studies have demonstrated prevalence rates of 3.9% in persons aged 60 to 80 years old 17. In a previously reported case series, incidence of retinoschisis was found to be 3.7% in patients 10 years or older and 7% for patients 40 years of age or older, with 82% of cases being bilateral 18. Men and women from all ethnicities seem to be affected equally 17. Retinoschisis lesions are most commonly found in the inferotemporal quadrant of the peripheral retina 19.
Retinoschisis rarely produces symptoms in the absence of a retinal detachment and is typically found on routine dilated fundoscopic exam 20.
Two histological forms, typical and reticular, have been previously described 21:
Typical retinoschisis
- Appearance: a bubbly, round appearance best visualized with scleral depression; the retinal splitting typically extends 2-3mm posterior to the ora
- Etiology: the retina splits in the outer plexiform layer, allowing the visualization of the footplates of the Muller cells, seen as white dots
- Complications: it is very uncommon for the retinal splitting to extend posteriorly towards the macula or form outer layer holes. Thus the risk for retinal detachment is lower than that of the reticular subtype
Reticular retinoschisis
- Appearance: an oval shaped, bullous elevation with a finely stippled internal surface and sclerotic looking retinal vessels; typically located posterior to and continuous with typical peripheral cystoid degeneration
- Etiology: the retina splits in the nerve fiber layer, similar to Juvenile X linked retinoschisis
- Complications: retinal holes in the outer wall occur in 23% of cases; posterior extension toward the macula is more common when compared to the typical subtype 22.
Degenerative retinoschisis causes
Although the exact cause is unknown, there is no evidence to suggest a genetic, vascular, or nutritional etiologic component 23. The degenerative (acquired) form of retinoschisis is believed to progress from preexisting peripheral cystoid degeneration in the retina 22. The existing cystic lesions essentially fuse together as neuroretinal and glial supporting elements within each lesion degenerate. This area slowly enlarges over time resulting in separation of the retina into an inner and outer layer. As the retina continues to split, neurons in that area are disrupted, causing irreversible and complete loss of visual function in the affected area 16.
Degenerative retinoschisis symptoms
Most patients are asymptomatic and this is an incidental finding. These patients may be referred to the retinal specialist with the presumptive diagnosis of retinal detachment.
Degenerative retinoschisis complications
The rate of retinoschisis progression to symptomatic retinal detachment is estimated to be between 0.05% and 2.2% 17. However, this progression is responsible for roughly 3% of full retinal detachments overall. It has been shown that those with a family history of retinal detachment have an increased risk of this complication 22. The presence of retinal holes is related to the two types of retinal detachments that are possible from progression of retinoschisis. retinoschisis without retinal breaks in either layer will never lead to a retinal detachment 23. Retinal holes may be seen in the outer or inner retinal layer of retinoschisis lesions, however, outer holes are much more common 19. Outer layer retinal holes may exist with or without a co-exisitng retinal detachment 24. If a retinal detachment is present, it is localized and fairly stable (non-progressive). Outer retinal holes in isolation are more common compared to outer + inner layer holes 23. Outer and inner retinal layer holes are associated with symptomatic, rapidly progressive retinal detachments. This scenario allows for collapse and quick progression into a full thickness rhegmatogenous retinal detachment that requires urgent surgical treatment. These dual layer holes are a rare occurrence in patients with retinoschisis who are initially asymptomatic 23. Treatment with laser demarcation or more invasive surgical intervention would be similar to treatment of conventional retinal tears or detachments 22.
Degenerative retinoschisis diagnosis
Diagnosis is typically based on ophthalmoscopy with scleral depression and contact lens examination, although lately optical coherence tomography (OCT) has been shown to play a significant role in distinguishing acquired retinoschisis from retinal detachment 25. OCT can be used to obtain detailed cross-sectional images of the peripheral retina 26.
- Optical coherence tomography (OCT): The schisis occurs at the junction between the outer plexiform layer and inner nuclear layer of the neurosensory retina in the typical subtype 27.
- Fundoscopy: RS appears as an immobile, dome-shaped elevation of the retina[10]. The height of the schisis cavity will not shallow with scleral indentation 27.
- Visual Field Testing: an absolute visual field defect will be present in the affected area. However, since RS typically occurs anterior to the equator in the periphery of the retina, typically no visual field defect is detected 20.
Acquired retinoschisis diagnostic criteria 26:
- Elevation of the inner layers of the neurosensory retina due to microcystoid degeneration. The lesion appears relatively immobile, transparent, smooth-domed and bullous in appearance.
- Minimal pigment alterations or atrophy of the retinal pigment epithelium layer
Degenerative retinoschisis treatment
Various treatment and management strategies are continually under debate as degenerative retinoschisis has been found to be primarily asymptomatic and non-progressive 19. Most cases of retinoschisis are innocuous and do not affect central vision. Frequency of the follow up visits depends upon the lesion size, its proximity to the macula and the presence or absence of symptoms (6-24 month intervals). Patients should be advised to return urgently if they experience symptoms of retinal detachment. Treatment (laser surgery, incisional surgery) should be considered only in symptomatic cases that threaten the macula and in cases with progressive retinal detachment 19. Although retinal detachment due to underlying retinoschisis can occur, unless the retinal detachment is full thickness, it is possible for patients to maintain good vision without surgical intervention. This is true even with foveal involvement in some cases 28.
Stage of senile retinoschisis
- Without retinal breaks: no treatment (except rarely)
- With outer-layer retinal breaks: no treatment (except rarely)
- With localized ‘schisis-detachment’: no treatment (except rarely)
- With progressive, symptomatic retinal detachment: prompt surgical repair
Retinoschisis symptoms
Retinoschisis symptoms
- Decreased central vision
- Decreased peripheral vision
The symptoms described above may not necessarily mean that you have retinoschisis. However, if you experience one or more of these symptoms, contact your eye doctor for a complete exam.
Retinoschisis diagnosis
The electroretinogram (ERG) is used to assess function of the nerve tissue in the retina. The eye is stimulated with light after either dark or light adaptation. Contact lenses, embedded with an electrode to measure electrical impulses created by the functioning retina, are worn by the patient. The reaction of the eye to various light stimuli is recorded and evaluated. This test documents the type of photoreceptor activity and the overall function of the inner and outer layers of the retina, and is a very important tool in diagnosis.
ERGs are easily obtained on adults and children under 24 months of age. However, ERGs performed on children between the ages of two and five can be difficult and often require general anesthesia. Therefore, we encourage the testing be done before age two or after age five. In general, after age five, nearly all children can have an ERG without any difficulties.
Beginning at age 3-4, all children, even without retinoschisis, should have their vision checked yearly and glasses prescribed as necessary. Children who show schisis of the peripheral retina need more frequent examinations. We feel these examinations should be conducted by a subspecialty trained retina surgeon, since surgery would be required if a retinal detachment were to develop. Follow-up intervals are best determined by the doctor who is following your child.
Note: Electroretinogram (ERG) is no longer the primary diagnostic tool used in the diagnosis of X-linked juvenile retinoschisis. While ERG can show selective reduction of the amplitude of the dark-adapted b-wave amplitude with relative preservation of the a-wave amplitude in affected males 29, recent studies have shown that the ERG response is much more variable than previously thought 30. Individuals with X-linked juvenile retinoschisis and an identified RS1 pathogenic variant can have a technically normal ERG in which the b-wave is still present 31. Therefore, this diagnosis cannot be excluded based on a normal ERG.
Retinoschisis treatment
Carbonic anhydrase inhibitors might be beneficial in reducing the cystic spaces observed in X-linked retinoschisis. There is no medical treatment for degenerative retinoschisis; however, vitrectomy surgery is occasionally required for complications related to either type of retinoschisis.
Glasses may improve the overall quality of vision in a patient with retinoschisis who is also near-sighted or farsighted, but will not “repair” the nerve tissue damage from the retinoschisis.
Vitamin A does not appear to help in retinoschisis. Vitamin A may have benefits for other genetic retinal diseases, particularly in cases of retinitis pigmentosa where the retinal nerve cells are slowly dying. However, retinoschisis is quite different from retinitis pigmentosa, since the retinal cells and their connections in retinoschisis are mechanically disrupted but are not thought to be dying.
References- Grayson C, Reid SNM, Ellis JA, Rutherford A, Sowden JC, Yates JRW, et al. Retinoschisin, the X-linked retinoschisis protein, is a secreted photoreceptor protein, and is expressed and released by Weri–Rb1 cells. Hum Mol Genet. 2000 Jul 22;9(12):1873–9.
- Yassur Y, Nissenkorn I, Ben-Sira I, Kaffe S, Goodman RM. Autosomal Dominant Inheritance of Retinoschisis. American Journal of Ophthalmology. 1982 Sep 1;94(3):338–43.
- Sieving PA, MacDonald IM, Chan S. X-Linked Juvenile Retinoschisis. 2003 Oct 24 [Updated 2014 Aug 28]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1222
- The Retinoschisis Consortium. Functional implications of the spectrum of mutations found in 234 cases with X-linked juvenile retinoschisis. Hum Mol Genet. 1998;7:1185–92
- Apushkin MA, Fishman GA, Rajagopalan AS. Fundus findings and longitudinal study of visual acuity loss in patients with X-linked retinoschisis. Retina. 2005;25:612–8
- Bowles K, Cukras C, Turriff A, Sergeev Y, Vitale S, Bush RA, Sieving PA. X-linked retinoschisis: RS1 mutation severity and age affect the ERG phenotype in a cohort of 68 affected male subjects. Invest Ophthalmol Vis Sci. 2011;52:9250–6.
- Sikkink SK, Biswas S, Parry NRA, Stanga PE, Trump D. X-linked retinoschisis: an update. Journal of Medical Genetics. 2007 Apr 1;44(4):225–32.
- Saleheen D, Ali A, Khanum S, Ozair MZ, Zaidi M, Sethi MJ, et al. Molecular analysis of the XLRS1 gene in 4 females affected with X-linked juvenile retinoschisis. Can J Ophthalmol. 2008 Oct;43(5):596–9.
- George NDL, Yates JRW, Moore AT. Clinical Features in Affected Males With X-Linked Retinoschisis. Arch Ophthalmol. 1996 Mar 1;114(3):274–80.
- Eksandh LC, Ponjavic V, Ayyagari R, Bingham EL, Hiriyanna KT, Andreasson S, Ehinger B, Sieving PA. Phenotypic expression of juvenile X-linked retinoschisis in Swedish families with different mutations in the XLRS1 gene. Arch Ophthalmol. 2000;118:1098–104
- Molday RS, Kellner U, Weber BH. X-linked juvenile retinoschisis: clinical diagnosis, genetic analysis, and molecular mechanisms. Prog Retin Eye Res. 2012;31:195–212
- Kim LS, Seiple W, Fishman GA, Szlyk JP. Multifocal ERG findings in carriers of X-linked retinoschisis. Doc Ophthalmol. 2007;114:21–6
- Lamey T, Laurin S, Chelva E, De Roach J. Genotypic analysis of X-linked retinoschisis in Western Australia. Adv Exp Med Biol. 2010;664:283–91
- Gupta K, Das D, Bhattacharjee H, Deka H, Magdalene D, Deshmukh S. Juvenile X-Linked retinoschisis: Response to topical dorzolamide therapy. TNOA J Ophthalmic Sci Res 2018;56:35-7 http://www.tnoajosr.com/temp/TNOAJOphthalmicSciRes56135-8787715_022627.pdf
- Rishi E, Gopal L, Rishi P, Deshmukh H. Congenital x-linked retinoschisis: a novel approach for management of a large schitic cavity overhanging the macula. Retin Cases Brief Rep. 2009;3(1):105–7.
- Lewis H. Peripheral retinal degenerations and the risk of retinal detachment. Am J Ophthalmol 2003;136(1):155-160.
- Buch H, Vinding T, Nielsen NV. Prevalence and long-term natural course of retinoschisis among elderly individuals: the Copenhagen City Eye Study. Ophthalmology 2007;114(4):751-755.
- Byer NE. Clinical study of senile retinoschisis. Arch Ophthalmol 1968;79(1):36-44.
- Byer NE. Long-term natural history study of senile retinoschisis with implications for management. Ophthalmology 1986;93(9):1127-1137
- Shea M, Schepens CL, Von Pirquet SR. Retionoschisis. I. Senile type: a clinical report of one hundred seven cases. Arch Ophthalmol 1960;63:1-9.
- Straatsma BR, Foss RY. Typical and reticular degenerative retinoschisis. Am J Ophthalmol 1973;75(4):551-575
- Fletcher EC, al. e. Retina: Retinoschisis. In: Cunningham ET, Riordan-Eva P, editors. Vaughan & Asbury General Ophthalmology. 18 ed. New York: McGraw-Hill; 2011; p. Ch. 10.
- Lewis H. Peripheral retinal degenerations and the risk of retinal detachment. Am J Ophthalmol 2003;136(1):155-160
- Zimmerman LE, Spencer WH. The pathologic anatomy of retinoschisis with a report of two cases diagnosed clinically as malignant melanoma. Arch Ophthalmol 1960;63:10-19.
- Yeoh J, Rahman W, Chen FK, da Cruz L. Use of spectral-domain optical coherence tomography to differentiate acquired retinoschisis from retinal detachment in difficult cases. Retina 2012;32(8):1574-1580.
- Stehouwer M, Tan SH, van Leeuwen TG, Verbraak FD. Senile retinoschisis versus retinal detachment, the additional value of peripheral retinal OCT scans (SL SCAN-1, Topcon). Acta Ophthalmol 2014;92(3):221-227
- Yeoh J, Rahman W, Chen FK, da Cruz L. Use of spectral-domain optical coherence tomography to differentiate acquired retinoschisis from retinal detachment in difficult cases. Retina 2012;32(8):1574
- Zaidi A, Lujan B. Combined retinoschisis-detachment involving the fovea managed with observation. Retin Cases Brief Rep 2014;8(4):254-256.
- Nakamura M, Ito S, Terasaki H, Miyake Y. Japanese X-linked juvenile retinoschisis: conflict of phenotype and genotype with novel mutations in the XLRS1 gene. Arch Ophthalmol. 2001;119:1553–4
- Molday RS, Kellner U, Weber BH. X-linked juvenile retinoschisis: clinical diagnosis, genetic analysis, and molecular mechanisms. Prog Retin Eye Res. 2012;31:195–212.
- Renner AB, Kellner U, Fiebig B, Cropp E, Foerster MH, Weber BH. ERG variability in X-linked congenital retinoschisis patients with mutations in the RS1 gene and the diagnostic importance of fundus autofluorescence and OCT. Doc Ophthalmol. 2008;116:97–109





{kind=link}