Rubinstein Taybi syndrome
Rubinstein-Taybi syndrome also called broad thumb-hallux syndrome, is rare genetic disorder characterized by broad thumbs and toes, short stature, distinctive facial features, and moderate to severe intellectual disability 1. Additional features of Rubinstein-Taybi syndrome can include eye abnormalities, heart and kidney defects, dental problems, and obesity 2. These signs and symptoms vary among affected individuals. People with Rubinstein-Taybi syndrome have an increased risk of developing particular types of noncancerous brain and skin tumors.
Rubinstein-Taybi syndrome is uncommon; it occurs in an estimated 1 in 100,000 to 125,000 newborns 2.
Rubinstein-Taybi syndrome may be caused by a mutation in the CREBBP or EP300 gene, or as the result of a very small loss (microdeletion) of genetic material from the short (p) arm of chromosome 16. In some people with Rubinstein Taybi syndrome, the cause is unknown. While Rubinstein Taybi syndrome can be inherited in an autosomal dominant manner, most cases result from a new (de novo) mutation in the responsible gene and are not inherited from a parent. Rubinstein-Taybi syndrome treatment is symptomatic and supportive 1.
There is no specific treatment for Rubinstein Taybi syndrome. However, the following treatments can be used to manage problems commonly associated with Rubinstein Taybi syndrome.
- Surgery to repair the bones in the thumbs or toes can sometimes improve grasp or relieve discomfort.
- Early intervention programs and special education to address developmental disabilities.
- Referral to behavioral specialists and support groups for family members.
- Medical treatment for heart defects, hearing loss, and eye abnormalities.
- Treatment for constipation and gastroesophageal reflux (GERD).
Figure 1. Rubinstein Taybi syndrome
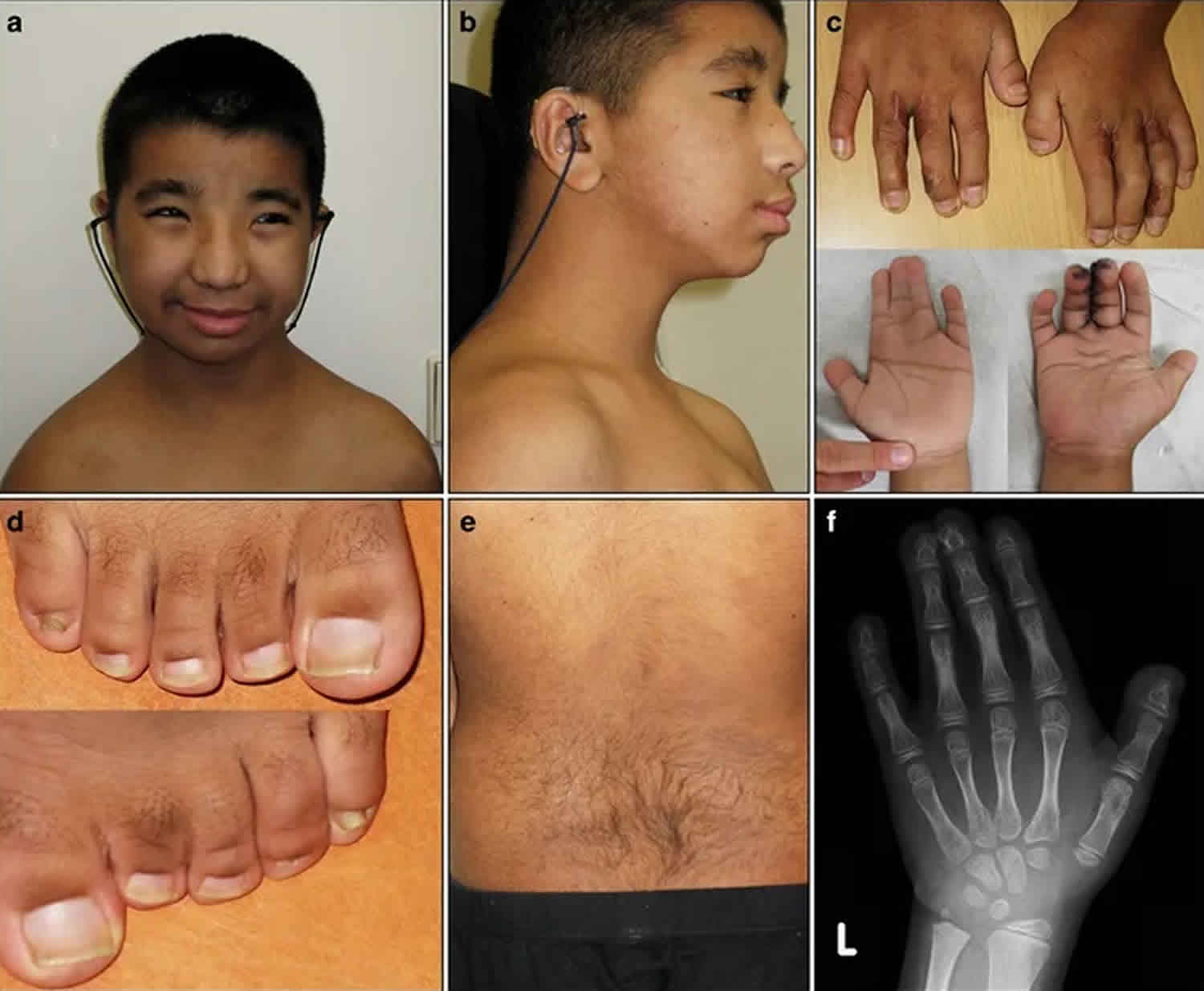
Footnote: (a) Frontal photograph and (b) profile of the proband. Notice the hypertelorism and broad nose. (c) Hands: surgically corrected syndactyly of the third and fourth finger on the right hand, and syndactyly of the second, third, and fourth finger on the left. Note the broad thumbs. (d) Feet: syndactyly of the third and fourth toes. Note the broad first toes. (e) Back: note the hirsutism. (f) An X-ray of the left hand at the age of 10, showing brachydactyly of the distal phalanges and soft tissue syndactyly of the second to fourth finger.
[Source 3 ]Are puberty and menstruation delayed in females with Rubinstein-Taybi syndrome?
A number of studies show that females with Rubinstein-Taybi syndrome start puberty at about 12 years of age (with a range of 11 to 13 years) 4. The average age of menarche (the onset of menstruation) is about 13.6 years (with a range of 11 to 19 years) 5. These ages of puberty and menarche do not differ from those of the general population 6.
How does Rubinstein Taybi syndrome affect eyesight?
The ocular findings in Rubinstein-Taybi syndrome may be physical features affecting the appearance of the eye or functional features affecting vision (eyesight). Purely physical findings may include features like downward-slanted eyes, thick and arched eyebrows, and/or long eyelashes. Features that may affect the function of the eye (vision or eyesight) may include 1:
- Drooping eyelid (ptosis) – if the drooping is severe, it may interfere with vision
- Cataracts – clouding of the lens may make things appear cloudy, fuzzy, foggy or filmy, cause sensitivity to glare, difficulty seeing at night, double vision, loss of color intensity, halos around light, problems seeing shapes against a background and/or differences between shades of colors
- Coloboma – may cause blurred vision, decreased visual acuity, double vision, or ghost images
- Strabismus – when the eyes do not line up, double vision may occur, depth perception may become reduced and vision loss may occur
- Refractive errors – may cause double vision, haziness, glare or halos around light
- Nasolacrimal duct obstruction – a blocked tear duct may cause excessive tearing and/or sticky tears which can affect the eye’s ability to open
- Nystagmus – may affect visual acuity
- Glaucoma – may cause peripheral (side) vision loss, decreased or cloudy vision, halos around light, sensitivity to light, and/or blindness
- Corneal abnormalities – may impair or damage vision
How long can a person with Rubinstein-Taybi syndrome live for?
The long-term outlook (prognosis) for people with Rubinstein Taybi syndrome is generally good, but it may vary due to the range and severity of the health problems that may be present. Most patients have developmental delay and intellectual disability but most of patients older than 6 years of age are able to learn to read 7. The majority of children can learn to read at an elementary level. The majority of children have delayed motor development, but on average, they learn to walk by 2 1/2 years of age. Life expectancy generally does not seem to be affected, except in children with complex cardiac (heart) defects. Cancers and respiratory infections are the most common causes of death 8. Survival rates in general are good and there are many reports of adults with Rubinstein Taybi syndrome 9.
Rubinstein Taybi syndrome cause
Mutations in the CREBBP gene cause about half of cases of Rubinstein-Taybi syndrome. The CREBBP gene provides instructions for making a protein that helps control the activity of many other genes. This protein, called CREB binding protein, plays an important role in regulating cell growth and division and is essential for normal development before birth. Because one copy of the CREBBP gene is deleted or mutated in people with Rubinstein-Taybi syndrome, their cells make only half of the normal amount of CREB binding protein. A reduction in the amount of this protein disrupts normal development before and after birth. Abnormal brain development is thought to underlie intellectual disability in people with Rubinstein-Taybi syndrome. Researchers have not determined how CREBBP gene mutations lead to other signs and symptoms of Rubinstein-Taybi syndrome.
Mutations in the EP300 gene cause a small percentage of cases of Rubinstein-Taybi syndrome. Like the CREBBP gene, this gene provides instructions for making a protein that helps control the activity of other genes. It also appears to be important for development before and after birth. EP300 gene mutations result in the loss of one functional copy of the gene in each cell, which interferes with normal development and causes the typical features of Rubinstein-Taybi syndrome. The signs and symptoms of this disorder caused by EP300 gene mutations are typically milder than those caused by mutations in the CREBBP gene.
Several cases of severe Rubinstein-Taybi syndrome have resulted from a deletion of genetic material from the short (p) arm of chromosome 16. Multiple genes, including the CREBBP gene, are missing as a result of this deletion. Researchers believe that the loss of multiple genes in this region probably accounts for the serious complications associated with severe Rubinstein-Taybi syndrome. Some researchers suggest that these cases are a separate condition called chromosome 16p13.3 deletion syndrome. However, a few studies indicate that some people with large deletions in the same region of chromosome 16 have characteristic features of Rubinstein-Taybi syndrome rather than a more severe condition.
Nearly 30 to 40 percent of people with Rubinstein-Taybi syndrome do not have an identified mutation in the CREBBP or EP300 gene or a chromosome 16 deletion. The cause of the condition is unknown in these cases. Researchers predict that mutations in other genes can also cause the disorder.
Rubinstein Taybi syndrome inheritance pattern
In most affected children, Rubinstein Taybi syndrome occurs as the result of a new (de novo) genetic mutation that is not present in or carried by the parents. In these cases, the risk of having a second affected child is less than 1%.
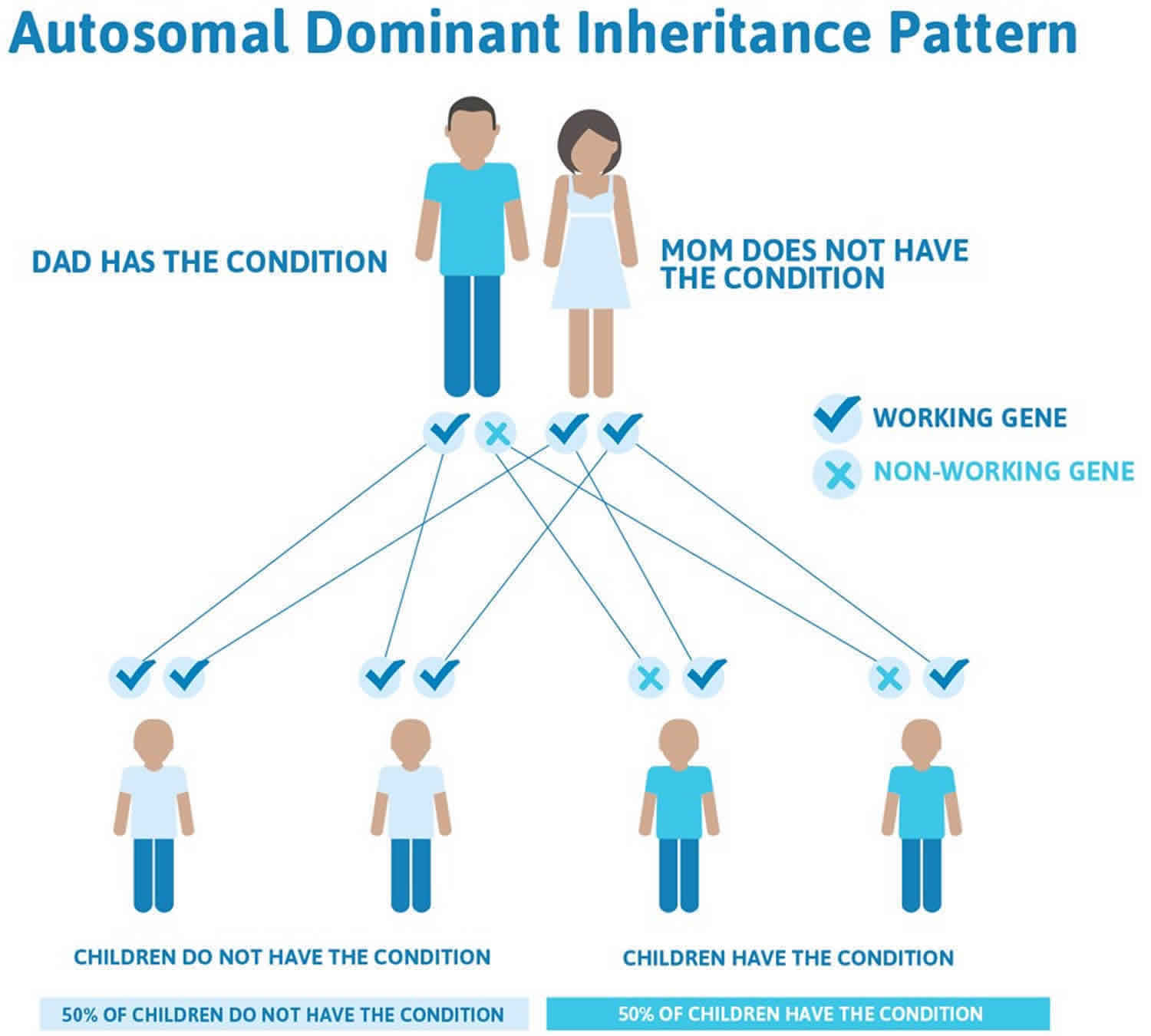
Rubinstein Taybi syndrome may also be inherited in an autosomal dominant pattern, meaning one copy of the altered gene in each cell is sufficient to cause the disorder and that if a person has Rubinstein Taybi syndrome each of his/her children have a 50% chance of having Rubinstein Taybi syndrome.
The most common gene responsible for Rubinstein Taybi syndrome is the CREBBP gene. Pathogenic variants in the CREBBP gene have been identified in 50%-60% of individuals with Rubinstein Taybi syndrome. Mutations in the EP300 gene have been identified in 3%-8% of individuals with Rubinstein Taybi syndrome.
Often autosomal dominant conditions can be seen in multiple generations within the family. If one looks back through their family history they notice their mother, grandfather, aunt/uncle, etc., all had the same condition. In cases where the autosomal dominant condition does run in the family, the chance for an affected person to have a child with the same condition is 50% regardless of whether it is a boy or a girl. These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
- When one parent has the abnormal gene, they will pass on either their normal gene or their abnormal gene to their child. Each of their children therefore has a 50% (1 in 2) chance of inheriting the changed gene and being affected by the condition.
- There is also a 50% (1 in 2) chance that a child will inherit the normal copy of the gene. If this happens the child will not be affected by the disorder and cannot pass it on to any of his or her children.
Figure 2 illustrates autosomal dominant inheritance. The example below shows what happens when dad has the condition, but the chances of having a child with the condition would be the same if mom had the condition.
Figure 2. Rubinstein Taybi syndrome autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Rubinstein Taybi syndrome symptoms
Rubinstein Taybi syndrome is a rare genetic disorder which may affect many organ systems of the body. Features include distinctively broad and/or angled fingers and toes, developmental delays, growth delays, speech delays, intellectual disability, characteristic abnormalities of the head and face (craniofacial dysmorphism), breathing and feeding difficulties (dysphagia), and urogenital abnormalities. In some people, the skin, heart, and/or respiratory system may also be affected. Symptoms associated with Rubinstein Taybi syndrome vary greatly from person to person.
Most infants with Rubinstein Taybi syndrome have thumbs and/or great toes that are abnormally broad as a result of unusual broadness of the bones in the tips of the thumbs and great toes (terminal phalanges). In addition, the distal bones of the thumbs and great toes may also be angled improperly (misaligned) on a proximal bone that is abnormally shaped (delta phalanx). The fifth fingers may be fixed in a bent position (clinodactyly).
Rubinstein Taybi syndrome symptoms include:
- Broadening of the thumbs and big toes
- Constipation
- Excess hair on body (hirsutism)
- Heart defects, possibly requiring surgery
- Intellectual disability
- Seizures
- Short stature that is noticeable after birth
- Slow development of cognitive skills
- Slow development of motor skills accompanied by low muscle tone
Other signs and symptoms of Rubinstein Taybi syndrome may include:
- Absent or extra kidney, and other problems with kidney or bladder
- An underdeveloped bone in the midface
- Unsteady or stiff walking gait
- Downward-slanted eyes
- Low-set ears or malformed ears
- Drooping eyelid (ptosis)
- Cataracts
- Coloboma (a defect in the iris of the eye)
- Microcephaly (excessively small head)
- Narrow, small, or recessed mouth with crowded teeth
- Prominent or “beaked” nose
- Thick and arched eyebrows with long eyelashes
- Undescended testicle (cryptorchidism), or other testicular problems
Growth and development
While prenatal growth is often normal, in most infants with Rubinstein Taybi syndrome parameters for height, weight, and head circumference fall below the fifth percentile during infancy. Affected infants fail to grow and gain weight at the expected rate (failure to thrive). Although weight gain can be very slow in infancy, children with Rubinstein Taybi syndrome may later show a relative obesity for their height. Feeding difficulties (dysphagia) may occur and many affected individuals are prone to repeated respiratory infections. As infants age, they may continue to experience poor growth and exhibit short stature (most below the third percentile).
Most infants and children with Rubinstein Taybi syndrome experience varying degrees of intellectual disability (average IQ between 36-51), delays in the acquisition of skills requiring coordination of muscular and mental activities (psychomotor delays), and delayed socialization. Most affected infants and children do not reach certain developmental milestones (e.g., sitting, crawling, standing, walking, etc.) at a time when they would otherwise be expected. Most children with Rubinstein Taybi syndrome experience a significant delay in expressive speech. In addition, there may be diminished muscle tone (hypotonia), abnormally exaggerated reflexes (hyperreflexia), a stiff, unsteady gait, infrequent bowel movements (constipation), and seizures.
The average IQ in one study was 51 and in another study was 36. IQ scores range from 25 to 79. Performance IQ is usually higher than verbal IQ. Some individuals with EP300-Rubinstein Taybi syndrome have normal intellect 10.
In one study of adults with Rubinstein Taybi syndrome, families reported a decline in abilities over time in 32%, including decreased social interaction, more limited speech, and worsening stamina and mobility 11.
Physical features
Infants with Rubinstein Taybi syndrome have several distinctive head and facial (craniofacial) features. Most affected infants have a large, “beak-shaped” or straight nose with a broad nasal bridge. In addition, affected infants may have a characteristic facial appearance and downslanting eyelid openings (palpebral fissures). In some children, the wall (septum) dividing the nostrils may extend below the nostrils (low hanging columella). Children with Rubinstein Taybi syndrome typically have a small head (microcephaly), below the 5th percentile.
Abnormalities of the mouth and jaw may be present including an abnormally small mouth, a short, thin upper lip, a highly arched roof of the mouth (palate), an underdeveloped upper jaw bone (maxilla), and an abnormally small lower jaw (micrognathia) that is displaced farther back than otherwise expected (retrognathia). Many affected infants have irregularly shaped, abnormally crowded teeth, resulting in upper and lower jaws that do not meet properly (malocclusion). Affected individuals may have a boney protuberance on the lingual aspect of the upper front teeth (talon cusps). The soft tissue structure that hangs in the back of the throat may also be divided (bifid uvula). In addition, some affected individuals may appear to be frowning or upset when they smile.
In addition to abnormally broad thumbs and toes, some children with Rubinstein Taybi syndrome may have toes that overlap, unusually shaped bones of the feet (metatarsals), and/or abnormally duplicated bones (proximal or distal phalanges) of the great toes (halluces).
Affected individuals may experience overgrowth of scar tissue at the site of a cut, injury, or surgical incision (keloid formation) or this may occur spontaneously.
Eyes
Affected infants may have abnormalities of the eyes including eyes that appear widely spaced (apparent hypertelorism); crossed eyes (strabismus); upper eyelids that droop (ptosis); and/or extra folds of skin on either side of the nose that may cover the eyes’ inner corners (epicanthal folds).
Skeletal deformities
There may be additional skeletal abnormalities including abnormal side-to-side (scoliosis) or front-to-back (kyphosis) curvature of the spine, abnormal depression of the bone forming the center of the chest (sternum), known as “funnel chest” or pectus excavatum, abnormalities of vertebrae and the pelvis, malformations of ribs, and recurrent dislocation of the knee caps. The lower end of the spinal cord may be abnormally tied down (tethering).
Genitourinary tract
Male infants with Rubinstein Taybi syndrome may have abnormalities of the genitourinary tract including failure of one or both testes to descend into the scrotum (cryptorchidism), an abnormal fold of skin extending around the base of the penis (shawl scrotum), and/or misplacement of the urinary opening, such as on the underside of the penis (hypospadias). In addition, infants with Rubinstein Taybi syndrome may have underdeveloped (hypoplastic) or absent kidney(s), repeated infections of the urinary tract, kidney stones, unusual accumulation of urine in the kidney (hydronephrosis), and/or backflow (reflux) of urine into the tubes (ureters) that normally bring urine to the bladder. In some cases, duplication of the kidneys and/or ureters may also be present.
Cardiac
Approximately one third of infants with Rubinstein Taybi syndrome have an associated heart defect that is present at birth (congenital heart defect). According to the medical literature, patent ductus arteriosus may be the most common congenital heart defect present in infants with Rubinstein Taybi syndrome. Infants with Rubinstein Taybi syndrome may also have extra heart sounds (heart murmurs), abnormal narrowing of the opening between the pulmonary artery and the right ventricle of the heart (pulmonary stenosis), narrowing of the aorta (aortic coarctation), and/or ventricular septal defects (VSDs) and/or atrial septal defects (ASDs). The symptoms associated with a ventricular septal defect or atrial septal defect vary from person to person, depending upon the size and location of the defect. (For more information on these disorders, choose the exact name of the heart defect as your search term in the Rare Disease Database.)
Respiratory
Affected individuals may also have abnormalities of the respiratory system. The lungs may be abnormally divided into small extra sections (lung lobulation) and/or the walls of the voice box (larynx) may be weak and easily collapsible, potentially resulting in swallowing and breathing difficulties (e.g., temporary cessation of normal breathing rhythm during sleep [sleep apnea]).
Individuals with Rubinstein Taybi syndrome can be difficult to intubate because of the easy collapsibility of the laryngeal wall. An anesthesiologist comfortable with managing complex pediatric airway problems should administer general anesthesia when needed.
Neurologic
Occasional craniospinal and posterior fossa abnormalities including Chiari malformation, syringomyelia, os odontoideum, and cervical cord compression have been reported 12. There may also be spinal cord tethering or lipoma. Seizures or abnormal EEG findings can occur.
Behavior
Individuals with Rubinstein Taybi syndrome often exhibit a short attention span, decreased tolerance for noise and crowds, impulsivity, and moodiness. Impulsivity, distractibility, instability of mood, and stereotypies are frequently observed 13. Other abnormal behaviors include attention problems, hyperactivity, self-injurious behaviors, and aggressive behaviors. Approximately 62% of adults with Rubinstein Taybi syndrome were reported to have autistic-like behaviors and one third had unreasonable fears or anxiety 11. There may be an insistence on sameness and repetitive questioning 14. Crawford et al 15 noted higher levels of panic attack, agoraphobia, and obsessive-compulsive disorder.
Tumors
Some persons with Rubinstein Taybi syndrome appear to be more prone to developing certain malignancies (including meningioma, pilomatixoma, rhabdomysarcoma, pheochromocytoma, neuroblastoma, medulloblastona, oligodendroglioma, leioyosarcoma, seminoma, odontoma, choristoma, and leukemia) than the general population. However, this is somewhat controversial as one recent study found only an increased risk for meningiomas and pilomatrixomas, but not for malignancies in general.
Rubinstein Taybi syndrome complications
Rubinstein Taybi syndrome complications depend on what part of the body is affected. Complications may include:
- Feeding problems in infants
- Repeated ear infections and hearing loss
- Problems with the shape of the heart
- Abnormal heartbeat
- Scarring of the skin
Rubinstein Taybi syndrome diagnosis
The diagnosis of Rubinstein Taybi syndrome is primarily based on physical (clinical) features, including a downward to the eyes (downslanted palpebral fissures), a low-hanging nasal septum (columella), a high palate, cusp-like structures (talon cusps) on the front teeth, and/or broad and angulated thumbs and great toes.
The diagnosis may be further supported through x-ray studies revealing malformations of the bones of the hands and feet characteristic to Rubinstein Taybi syndrome. Genetic testing (FISH or sequence analysis) may confirm Rubinstein Taybi syndrome, including pathogenic variants in the CREBBP gene (identified in 50%-60% of affected individuals) or in the EP300 gene (identified in 3%-8% of Rubinstein Taybi syndrome individuals).
Rubinstein Taybi syndrome treatment
To establish the extent of disease and needs in an individual diagnosed with Rubinstein-Taybi syndrome, the evaluations summarized in Table 1 (if not performed as part of the evaluation that led to the diagnosis) are recommended 16.
The management of Rubinstein Taybi syndrome is directed toward the specific symptoms of each individual. Management may require the coordinated efforts of a team of specialists, including pediatricians, physicians who diagnose and treat heart abnormalities (cardiologists), skeletal abnormalities (orthopedists), hearing problems (audiologists), urinary tract abnormalities (urologists), kidney dysfunction(nephrologists), as well as dental specialists, physical therapists, speech pathologists, dietitians, and/or other health care professionals. Growth parameters should be regularly plotted on an Rubinstein Taybi syndrome-specific growth chart. There should be yearly eye and hearing evaluations and routine monitoring for cardiac, dental, and renal abnormalities.
Orthopedic surgery, physical therapy, and/or other supportive techniques may help treat certain skeletal abnormalities potentially associated with Rubinstein Taybi syndrome, such as scoliosis. In some cases, surgery may be performed on the hands and/or feet, particularly when there are extra (supernumerary) fingers and/or toes, or when the fingers are severely misaligned.
Affected individuals may require early intervention to prevent and/or monitor respiratory and feeding difficulties. Special education programs, vocational training, speech, and/or behavioral therapy may also be recommended.
Genetic counseling is recommended for affected individuals and their family members.
Table 1. Recommended evaluations following initial diagnosis in individuals with Rubinstein-Taybi syndrome
| System/Concern | Evaluation | Comment |
|---|---|---|
| Neurologic | Ultrasound of spinal canal in neonatal period to screen for tethered cord | MRI of spinal canal should be performed in older children if symptomatic. |
| Constitutional | Measurement of growth | Plot parameters on Rubinstein Taybi syndrome growth charts. |
| Neurodevelopmental | Multidisciplinary developmental &/or neuropsychological evaluation | Assess: gross & fine motor, speech/language, cognitive, & vocational skills; behavior. |
| Ophthalmologic | Ophthalmologic examination | Evaluate for strabismus, refractory errors, ptosis, nasolacrimal duct obstruction, cataracts, coloboma, nystagmus, glaucoma, & corneal abnormalities. |
| Audiologic | Hearing evaluation | Recommended: auditory brain stem evoked response testing |
| Pulmonary | Evaluation for obstructive sleep apnea by polysomnography | If indicated by snoring, particular sleeping posture, night wakefulness, & excessive daytime sleepiness |
| Cardiac | Echocardiogram | Evaluation by cardiologist for structural heart defects |
| Genitourinary |
| Refer to urologist for undescended testes by age 6-12 mos. |
| Gastrointestinal |
| Upper GI study if symptoms of malrotation |
| Orthopedic | Assess thumbs & halluces, joints, & spine. | |
| Dental/Orthodontic | Dental & orthodontic evaluations | |
| Other | Consultation w/clinical geneticist &/or genetic counselor |
Table 2. Treatment of manifestations in individuals with Rubinstein-Taybi syndrome
| Manifestation/Concern | Treatment | Considerations/ Other |
|---|---|---|
| Developmental & behavioral concerns |
| Refer family to support groups & other resources. |
| Ocular manifestations | Standard treatment as per ophthalmologist | |
| Hearing loss | Standard treatment as per audiologist | |
| Obstructive sleep apnea | Treatment as per pulmonologist | |
| Cardiac anomalies | Standard treatment as per cardiologist | |
| Renal anomalies | Standard treatment as per nephrologist &/or urologist | |
| Cryptorchidism | Standard treatment as per urologist | |
| Gastroesophageal reflux &/or constipation |
| |
| Significantly angulated thumbs or duplicated halluces | Surgical repair as per orthopedist | |
| Dental anomalies | Standard treatment as per dentist &/or orthodontist |
Prevention of secondary complications
Individuals with Rubinstein Taybi syndrome can be difficult to intubate because of the easy collapsibility of the laryngeal wall. An anesthesiologist comfortable with managing complex pediatric airway problems should therefore administer general anesthesia when needed. Individuals with Rubinstein Taybi syndrome may require earlier intubation and later extubation than other individuals undergoing similar procedures.
Surveillance
Table 3. Recommended surveillance for individuals with Rubinstein-Taybi syndrome
| System/Concern | Evaluation | Frequency |
|---|---|---|
| Growth | Monitor weight & linear growth w/RSTS growth charts. | Frequently during 1st yr of life & at regular checkups |
| Ocular manifestations | Ophthalmologic evaluation | Annually or as necessary |
| Hearing loss | Audiologic evaluation |
|
| Dental anomalies | Dental & orthodontic evaluation | Beginning at age 1 yr; continue every 6 mos or as per dentist/orthodontist |
Rubinstein Taybi syndrome prognosis
The long-term outlook (prognosis) for people with Rubinstein Taybi syndrome is generally good, but it may vary due to the range and severity of the health problems that may be present. Most patients have developmental delay and intellectual disability but most of patients older than 6 years of age are able to learn to read 7. The majority of children can learn to read at an elementary level. The majority of children have delayed motor development, but on average, they learn to walk by 2 1/2 years of age. Life expectancy generally does not seem to be affected, except in children with complex cardiac (heart) defects. Cancers and respiratory infections are the most common causes of death 8. Survival rates in general are good and there are many reports of adults with Rubinstein Taybi syndrome 9.
References- Stevens CA. Rubinstein-Taybi Syndrome. 2002 Aug 30 [Updated 2019 Aug 22]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1526
- Rubinstein-Taybi syndrome. https://ghr.nlm.nih.gov/condition/rubinstein-taybi-syndrome
- de Vries, T., R Monroe, G., van Belzen, M. et al. Mosaic CREBBP mutation causes overlapping clinical features of Rubinstein–Taybi and Filippi syndromes. Eur J Hum Genet 24, 1363–1366 (2016). https://doi.org/10.1038/ejhg.2016.14
- Wiley S, Swayne S, Rubinstein JH, Lanphear NE, Stevens CA. Rubinstein-Taybi syndrome medical guidelines. Am J Med Genet A. 2003 Jun 1; 119A(2):101-10. http://www.ncbi.nlm.nih.gov/pubmed/12749047
- Stevens CA, Carey JC, Blackburn BL. Rubinstein-Taybi syndrome: a natural history study. Am J Med Genet Suppl. 1990; 6:30-7. http://www.ncbi.nlm.nih.gov/pubmed/2118775
- Stevens C, Carey J. Rubinstein-Taybi syndrome: Book for Families. http://rubinstein-taybi.com/medical-7/book-for-families
- Rubinstein Taybi syndrome. https://rarediseases.org/rare-diseases/rubinstein-taybi-syndrome
- Rubinstein-Taybi syndrome. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=783
- Genetics of Rubinstein-Taybi Syndrome. https://emedicine.medscape.com/article/948453-overview
- Fergelot P, van Belzen M, van Gils J, Afenjar A, Armour CM, Arveiler B, Beets L, Burglen L, Busa T, Collet M, Deforges J, de Vries BBA, Dominguez Garrido E, Dorison N, Dupont J, Francannet C, Garcia-Minaur S, Gabau Vila E, Gebre-Medhin S, Gener Querol B, Genevieve D, Gerard M, Gervanisi CG, Goldenberg A, Josifova D, Lachlan K, Mas S, Maranda B, Moilanen JS, Nordgren A, Parent P, Rankin J, Reardon W, Rio M, Roume J, Shaw A, Smigiel R, Sojo A, Solomon B, Stembalska A, Stumpel C, Suarez F, Terhal P, Thomas S, Touraine R, Verloes A, Vincent-Delorme C, Wincent J, Peters DJM, Bartsch O, Larizza L, Lacombe D, Hennekam RC. Phenotype and genotype in 52 patients with Rubinstein-Taybi syndrome caused by EP300 mutations. Am J Med Genet A. 2016;170:3069–82
- Stevens CA, Pouncey J, Knowles D. Adults with Rubinstein-Taybi syndrome. Am J Med Genet. 2011;155A:1680–4.
- Marzuillo P, Grandone A, Coppola R, Cozzolino D, Festa A, Messa F, Luongo C, Del Giudice EM, Perrone L. Novel cAMP binding protein-BP (CREBBP) mutation in a girl with Rubinstein-Taybi syndrome, GH deficiency, Arnold Chiari malformation and pituitary hypoplasia. BMC Med Genet. 2013;14:28.
- Verhoeven WMA, Tuinier S, Kuijpers HJH, Egger JIM. Psychiatric profile in Rubinstein-Taybi syndrome. Psychopathology. 2010;43:63–8.
- Waite J, Moss J, Beck SR, Richards C, Nelson L, Arron K, Burbidge C, Berg K, Oliver C. Repetitive behavior in Rubinstein-Taybi syndrome: parallels with autism spectrum phenomenology. J Autism Dev Disord. 2015;2015;45:1238–53.
- Crawford H, Waite J, Oliver C. Diverse profiles of anxiety related disorders in fragile X, Cornelia de Lange and Rubinstein-Taybi syndromes. J Autism Dev Disord. 2017;47:3728–40.
- Wiley S, Swayne S, Rubinstein JH, Lanphear NE, Stevens CA. Rubinstein-Taybi syndrome medical guidelines. Am J Med Genet A. 2003;119A:101–10.





{kind=link}