
Sandhoff disease
Sandhoff disease also known as GM2 gangliosidosis type 2 or total hexosaminidase deficiency, is a rare inherited lysosomal storage disease that progressively destroys nerve cells (neurons) in the brain and spinal cord (central nervous system). Sandhoff disease is a severe form of Tay-Sachs disease and it is caused by a deficiency of the enzyme beta-hexosaminidase (a combined beta-hexosaminidase A and hexosaminidase B deficiency), which results in the harmful accumulation of certain fats (lipids) in the brain and other organs of the body. How the lysosomal accumulation of ganglioside might affect the early development of the nervous system is not understood 1. Sandhoff disease features catastrophic neurodegeneration and death in early childhood.
There are 3 forms of Sandhoff disease 2:
- Infantile Sandhoff disease – symptoms appear around 6 months of age.
- Juvenile Sandhoff disease – symptoms appear after the first year of life, typically between ages 2 and 5, but can occur anytime during childhood.
- Late Onset Sandhoff disease – symptoms typically appear in adolescence or early adulthood, but sometimes later.
The most common and severe form of Sandhoff disease becomes apparent in infancy. Infants with Sandhoff disease typically appear normal until the age of 3 to 6 months, when their development slows and muscles used for movement weaken. Affected infants lose motor skills such as turning over, sitting, and crawling. They also develop an exaggerated startle reaction to loud noises. As the disease progresses, children with Sandhoff disease experience seizures, vision and hearing loss, intellectual disability, and paralysis. An eye abnormality called a cherry-red spot, which can be identified with an eye examination, is characteristic of this disorder. Some affected children also have enlarged organs (organomegaly) or bone abnormalities. Children with the severe infantile form of Sandhoff disease usually live only into early childhood.
Other forms of Sandhoff disease are very rare. Signs and symptoms can begin in childhood, adolescence, or adulthood and are usually milder than those seen with the infantile form. Characteristic features include muscle weakness, loss of muscle coordination (ataxia) and other problems with movement, speech problems, and mental illness. These signs and symptoms vary widely among people with late-onset forms of Sandhoff disease.
Sandhoff disease is a rare disorder that is estimated to affect 1 in 1,000,000 individuals 3; its frequency varies among populations. Sandhoff disease appears to be more common in the Creole population of northern Argentina; the Metis Indians in Saskatchewan, Canada; and people from Lebanon; Eastern European and Ashkenazi Jewish descent, but Sandhoff disease is not limited to any ethnic group 4.
Sandhoff disease is caused by mutations in the HEXB gene. The HEXB gene provides instructions for making a protein that is part of two critical enzymes in the nervous system, beta-hexosaminidase A and beta-hexosaminidase B. Sandhoff disease is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. Each parent must carry the defective gene and pass it on to the child. Individuals who carry only one copy of the mutated gene typically do not show signs and symptoms of the disorder. Onset of the disorder usually occurs at 6 months of age. Symptoms may include:
- progressive nervous system deterioration,
- problems initiating and controlling muscles and movement,
- increased startle reaction to sound,
- early blindness,
- seizures,
- spasticity (non-voluntary and awkward movement),
- myoclonus (shock-like contractions of a muscle,
- macrocephaly (an abnormally enlarged head),
- cherry-red spots in the eyes,
- frequent respiratory infections,
- doll-like facial appearance, and
- enlarged liver and spleen.
There is no specific treatment for Sandhoff disease. Supportive treatment includes proper nutrition and hydration and keeping the airway open. Anticonvulsants may initially control seizures 5.
What is lysosome?
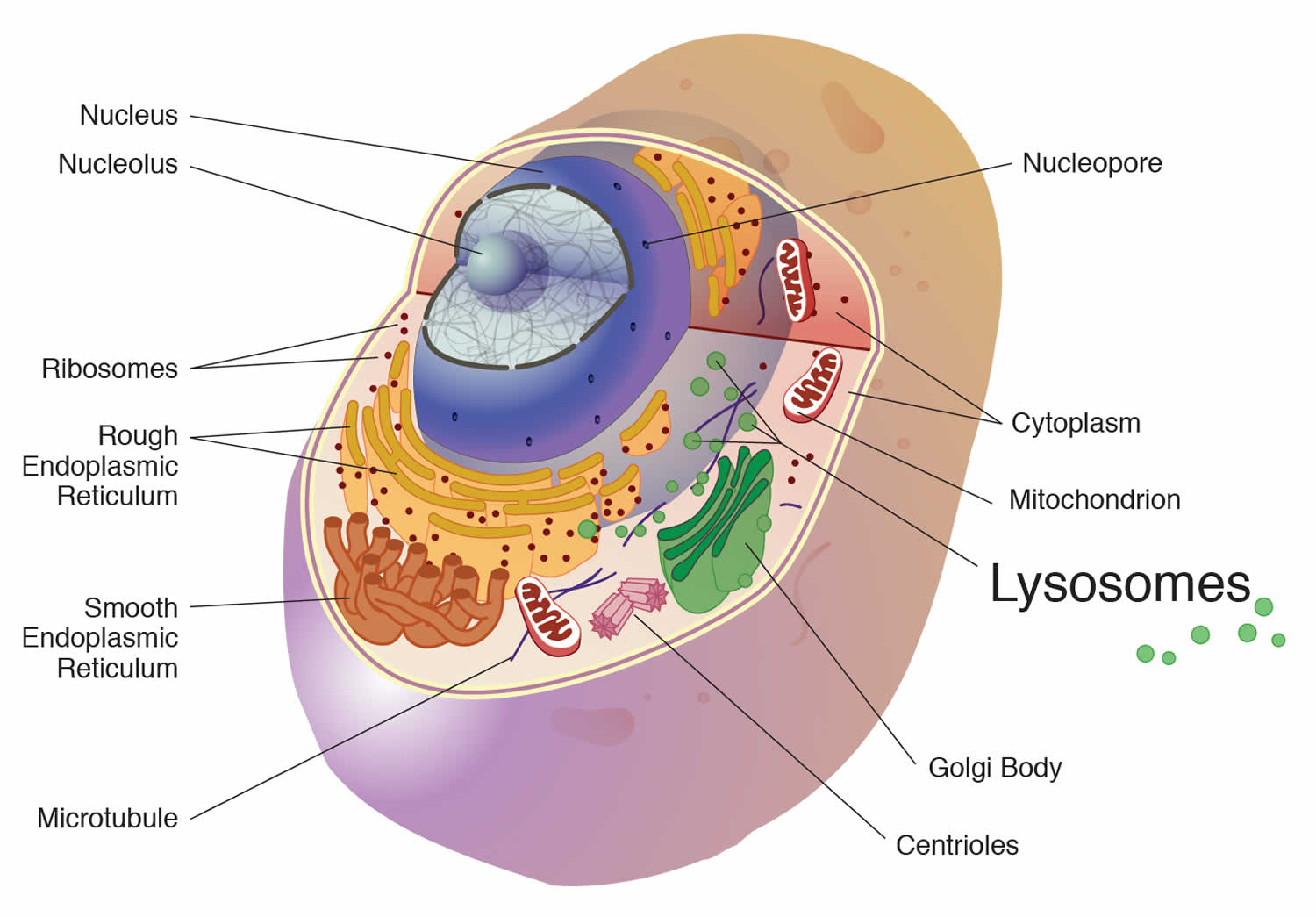
A lysosome is a membrane-bound cell organelle that contains digestive enzymes called hydrolytic enzymes and they break down large molecules into small molecules. Lysosomes digest excess or worn out organelles, food particles, and engulfed viruses or bacteria. The membrane surrounding a lysosome prevents the digestive enzymes inside from destroying the cell. Lysosomes are involved with various cell processes. Lysosomes break down excess or worn-out cell parts. They may be used to destroy invading viruses and bacteria. If the cell is damaged beyond repair, lysosomes can help it to self-destruct in a process called programmed cell death or apoptosis.
Lysosomes also breakdown large proteins into amino acids or large carbohydrates into simple sugars or large lipids into single fatty acids. And when they do that, they provide for the rest of the cell the nutrients that it needs to survive. So, for example, if lysosomes can’t do that, they can’t break down large molecules into small molecules. You’ll have storage of those large molecules, and this is called lysosomal storage diseases. There’s also another type of lysosome storage disease in which the small molecules that are produced from those large molecules can’t get out of the lysosome. They’re stored there because the transporters for moving these small molecules out are missing genetically. And finally, one other function of the lysosome is to ingest bacteria so that the bacteria can be destroyed. So the lysosomes also provide a function against infection, and the cell will often engorge a bacterium and put it into its lysosome for destruction. So here’s an important organelle that has function against infection and function in a way in nutrition to break down large molecules into small molecules so that they can be reutilized.
Figure 1. Lysosome
Sandhoff disease causes
Mutations in the HEXB (hexosaminidase subunit beta) gene cause Sandhoff disease. The HEXB gene provides instructions for making a protein that is part of two critical enzymes in the nervous system, beta-hexosaminidase A (HexA) and beta-hexosaminidase B (HexB). These enzymes are located in lysosomes, which are structures in cells that break down toxic substances and act as recycling centers. Within lysosomes, these enzymes break down fatty substances, complex sugars, and molecules that are linked to sugars. In particular, beta-hexosaminidase A helps break down a fatty substance called GM2 ganglioside.
Mutations in the HEXB gene disrupt the activity of beta-hexosaminidase A and beta-hexosaminidase B, which prevents these enzymes from breaking down GM2 ganglioside and other molecules. As a result, these compounds can accumulate to toxic levels, particularly in neurons of the brain and spinal cord. A buildup of GM2 ganglioside leads to the progressive destruction of these neurons, which causes many of the signs and symptoms of Sandhoff disease.
Because Sandhoff disease impairs the function of lysosomal enzymes and involves the buildup of GM2 ganglioside, this condition is sometimes referred to as a lysosomal storage disorder or a GM2-gangliosidosis.
Sandhoff disease inheritance pattern
Sandhoff disease is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 2 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 2. Sandhoff disease autosomal recessive inheritance pattern

People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Sandhoff disease symptoms
Infants with the classic form of Sandhoff disease typically appear normal until the age of 3 to 6 months when their development slows and muscles used for movement weaken. Affected infants typically lose motor skills such as turning over, sitting, and crawling. They may also develop an exaggerated startle reaction to loud noises. As the disease progresses, children with Sandhoff disease may experience seizures, vision and hearing loss, intellectual disability, and paralysis. An eye abnormality called a cherry-red spot, which can be identified through an eye examination, is characteristic of this disorder. Some affected children also have an enlarged liver and spleen, frequent respiratory infections, or bone abnormalities 4. Children with the severe infantile form of Sandhoff disease usually live only into early childhood 4.
Forms of Sandhoff disease where the symptoms develop after infancy are very rare. Signs and symptoms can begin in childhood, adolescence, or adulthood and are usually milder than those seen with the infantile form. Characteristic features include muscle weakness, loss of muscle coordination (ataxia) and other problems with movement, speech problems, and mental illness. These signs and symptoms vary widely among people with late-onset forms of Sandhoff disease 4.
Infantile Sandhoff disease
The most common type of Sandhoff disease causes rapidly progressing mental and motor decline in infancy. Within the first six months of life, infants with Sandhoff disease will experience weakness. They lose skills like turning over, sitting, and crawling. They can also have trouble with feeding, overreaction to loud sudden noises, delayed speech, early blindness, seizures, heart murmur, and continuously tight muscles (spasticity). A doctor may notice red spots in the back of the eye (cherry-red spots of the macula) and an abnormal reflex of the foot that indicates damage to the nervous system (the Babinski reflex). Other signs of Sandhoff disease can include a large head (macrocephaly) and unique facial features. Infants with this form of Sandhoff disease usually do not live past 2-5 years.
Juvenile and Adult Sandhoff disease
Sandhoff disease can also happen in older children and adults. These individuals will experience a slower mental and motor decline than in infantile Sandhoff disease. The onset and severity of symptoms can vary. A specific symptom of later-onset Sandhoff disease is muscle weakness affecting the muscles of the arms, legs, and hips. Other symptoms include muscle loss (muscle atrophy), balance problems, uncontrollable muscle contraction (dystonia), damage to nerves controlling involuntary bodily functions (autonomic neuropathy), a loss of intellectual function (cognitive dysfunction), psychiatric illness and dementia.
Sandhoff disease diagnosis
Sandhoff disease is commonly diagnosed by testing the activity of the beta-hexosaminidase A and beta-hexosaminidase B enzymes (enzyme assays). People with Sandoff disease have reduced or absent activity of both enzymes. Genetic testing is used to confirm the diagnosis.
Carrier testing
Carrier testing is performed to identify individuals who have a gene mutation and may be at risk for having a child or other family members with the same mutation. Carriers usually do not have symptoms related to the gene mutation. Carrier testing is typically offered to individuals who have family members with a genetic condition, family members of an identified carrier, and individuals in ethnic or racial groups known to have a higher carrier rate for a particular condition.
There two types of carrier screening tests: DNA and biochemical. Sandhoff carrier screening is available through DNA or biochemical testing. Most molecular screening only looks for several DNA mutations (changes in genetic code) that lead to decreased production of enzyme. This type of screening often focuses on the mutations seen in one ethnic group and misses those seen in people of other backgrounds. For instance, for Tay-Sachs disease, most molecular screening detects >90% of carriers of Jewish background but only 60% of the mutations found in people of other backgrounds.
The second type of molecular screening is called sequencing. This type of genetic carrier screening is accomplished by reading across the DNA code of a specific gene to determine if there are any known mutations (changes in the DNA code). A negative result significantly reduces the chance that you are a carrier, but it does not eliminate the chance, as it is possible that you carry a mutation that has not been discovered yet or that our current technology is unable to detect. It is important to talk with a genetics professional about how your screening results may impact your family’s health.
DNA carrier screening detects specific known mutations that are ‘looked’ for in the test. A negative DNA carrier result does not eliminate an individual’s chances of being a carrier because of the possibility of carrying an unknown mutation or one not ‘looked’ for in the test. Biochemical testing, also called an enzyme assay, detects the level of the enzyme(s) in question in the blood. Enzyme assays can be done using serum or leukocytes (white blood cells). Serum is typically the standard test, but leukocyte testing is recommended when the person being tested is pregnant, on birth control pills or taking any medications that affect hormones because all of these situations can potentially interfere with the accuracy of the serum test. DNA carrier screening may be recommended if the results of the biochemical test are uncertain 6.
Sandhoff disease is associated with deficiencies of both hexosaminidase A (hex A) and hexosaminidase B (hex B) enzyme activity. Carriers of Sandhoff disease have reduced (but adequate) amounts of both hex A and hex B. While most hex A assays are performed to identify Tay-Sachs carriers, the test also can also identify individuals that are carriers of Sandhoff disease. Looking at the total hexosaminidase activity in combination with the percent of hex A activity present can aid in determining whether an individual is a carrier of Sandhoff disease. Typically, a decreased amount of total hexosaminidase activity along with an increase in the proportion of hex A activity in leukocytes is suggestive of a Sandhoff carrier 7. In contrast, Tay-Sachs carriers have a decrease in the amount of hex A activity. When the hex A enzyme result indicates that an individual is a possible Sandhoff carrier, the next step is typically to offer carrier testing to the individual’s partner. If the partner is negative, the risk for the couple to have a child affected with the disorder is very significantly decreased. If the partner is also a possible carrier, more comprehensive testing may be offered.
Sandhoff disease treatment
There is no specific treatment or cure for Sandhoff disease 5. Management is symptomatic and supportive. Supportive treatment includes proper nutrition and hydration and keeping the airway open. Anticonvulsants may be used to control seizures 5. There are current research efforts underway, including experimental gene therapy, substrate reduction therapy, bone marrow transplants, and stem cell therapy 2.
Genetic counseling is recommended for affected individuals and their families.
Sandhoff disease prognosis
The prognosis for individuals with Sandhoff disease is poor. Death usually occurs by age 3 or 4 and is often caused by respiratory infections 5.
References- Allende ML, Cook EK, Larman BC, et al. Cerebral organoids derived from Sandhoff disease-induced pluripotent stem cells exhibit impaired neurodifferentiation. J Lipid Res. 2018;59(3):550-563. doi:10.1194/jlr.M081323 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5832932
- Sandhoff Disease. https://www.ntsad.org/index.php/the-diseases/sandhoff
- Sandhoff disease. https://rarediseases.org/rare-diseases/sandhoff-disease/
- Sandhoff disease. https://ghr.nlm.nih.gov/condition/sandhoff-disease
- Sandhoff Disease Information Page. https://www.ninds.nih.gov/Disorders/All-Disorders/Sandhoff-Disease-Information-Page
- Types of Screening. https://www.ntsad.org/index.php/carrier-screening/types-of-screening
- Hexosaminidase A and Total Hexosaminidase, Leukocytes. https://www.mayocliniclabs.com/test-catalog/Overview/8775





{kind=link}