Seckel syndrome
Seckel syndrome also known as bird-headed dwarfism, microcephalic primordial dwarfism or Seckel type dwarfism, is an extremely rare genetic disorder with autosomal recessive inheritance that is characterized by growth delays prior to birth (intrauterine growth retardation) resulting in low birth weight. Growth delays continue after birth (postnatal), resulting in short stature (dwarfism) 1. Other symptoms and physical features associated with Seckel syndrome include an abnormally small head (microcephaly), varying degrees of mental retardation (intellectual disability) and/or unusual characteristic facial features including “beak-like” protrusion of the nose 2. Other facial features may include abnormally large eyes, a narrow face, malformed ears, and/or an unusually small jaw (micrognathia). About less than 25% of the patients also have blood abnormalities 3. Severe sinus bradycardia 3, malignant hypertension 4 and moyamoya-like vasculopathy of the brain 5, superficial femoral artery stenosis 6 are reported. In addition, some affected infants may exhibit permanent fixation of the fifth fingers in a bent position (clinodactyly), malformation (dysplasia) of the hips, dislocation of a bone in the forearm (radial dislocation), and/or other physical abnormalities.
The prevalence of Seckel syndrome is less than 1 per 10,000 live births 7. Although the mode of inheritance is autosomal recessive, cases have been reported in non-consanguineous families 8. This type of inheritance can occur through heterozygous mutations and has more severe phenotypes 9.
Seckel syndrome is a genetically heterogeneous disorder which may be divided in 8 different subtypes according to the specific gene alteration (mutation) in 8 different genes that have been identified to date 9. They include very rare mutations in the ataxia-telangiectasia and Rad3-related (ATR) gene, encoding a phosphatidylinositol 3-kinase-like kinase with distinct functions in DNA damage response, and mutations in ATRIP, CENPJ, RBBP8, NIN, DNA2, CEP63 and CEP152 10. Mutations are most frequently found in CEP152, a centrosomal protein with essential roles during mitosis and in DNA damage response 11.
Seckel syndrome treatment is supportive 9. Supportive therapy including special education, speech and language therapy, behavioral therapy, occupational therapy, and community services for families. Ritalin® may be helpful in managing hyperkinesia. Seizures are usually responsive to monotherapy with standard antiepileptic drugs 12.
Figure 1. Seckel syndrome
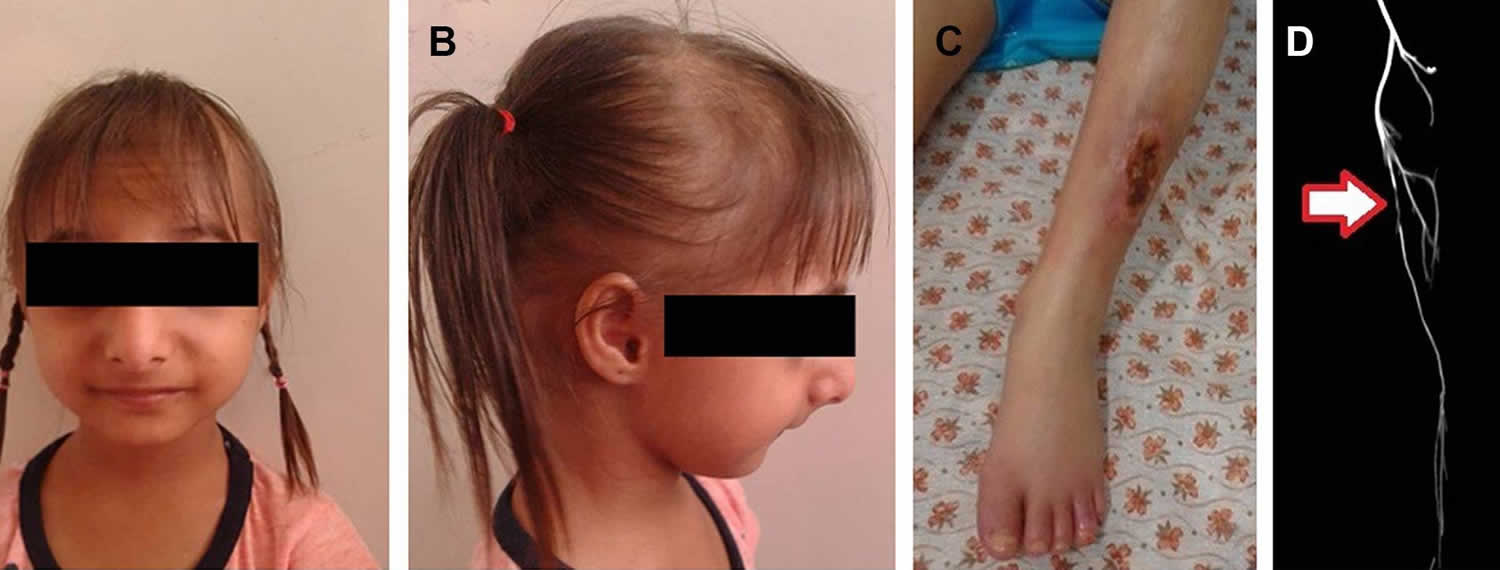
Footnote: Bird-like face, retrognathia, microcephaly and thin hairs were prominent in frontal (A) and lateral (B) views. An erosive ulcer was evident on the left shin (C). The left superficial femoral artery was stenotic, as revealed by CT angiography and shown with a white arrow (D).
[Source 6 ]Seckel syndrome causes
Three variants of Seckel syndrome involve disruptions or changes (mutations) of genes on three different chromosomes. The gene map locations are: Seckel syndrome 1, on chromosome 3 (3q22-q24); Seckel syndrome 2, on chromosome 18 (18p11.31-q11) and Seckel syndrome 3, on chromosome 14 (14q21-q22). The specific gene involved in Seckel syndrome 1 is known as the ataxia-telangiectasia and Rad3-related protein (ATR) gene. The genes involved in Seckel syndrome types 2 and 3 are unknown.
Eight different genes that have been identified to date and they include very rare mutations in the ataxia-telangiectasia and Rad3-related (ATR) gene, encoding a phosphatidylinositol 3-kinase-like kinase with distinct functions in DNA damage response, and mutations in ATRIP, CENPJ, RBBP8, NIN, DNA2, CEP63 and CEP152 10. Mutations are most frequently found in CEP152, a centrosomal protein with essential roles during mitosis and in DNA damage response 11.
Seckel syndrome inheritance pattern
Seckel syndrome is inherited as an autosomal recessive trait. However, Seckel syndrome cases have been reported in non-consanguineous families 8. This type of inheritance can occur through heterozygous mutations and has more severe phenotypes 9.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
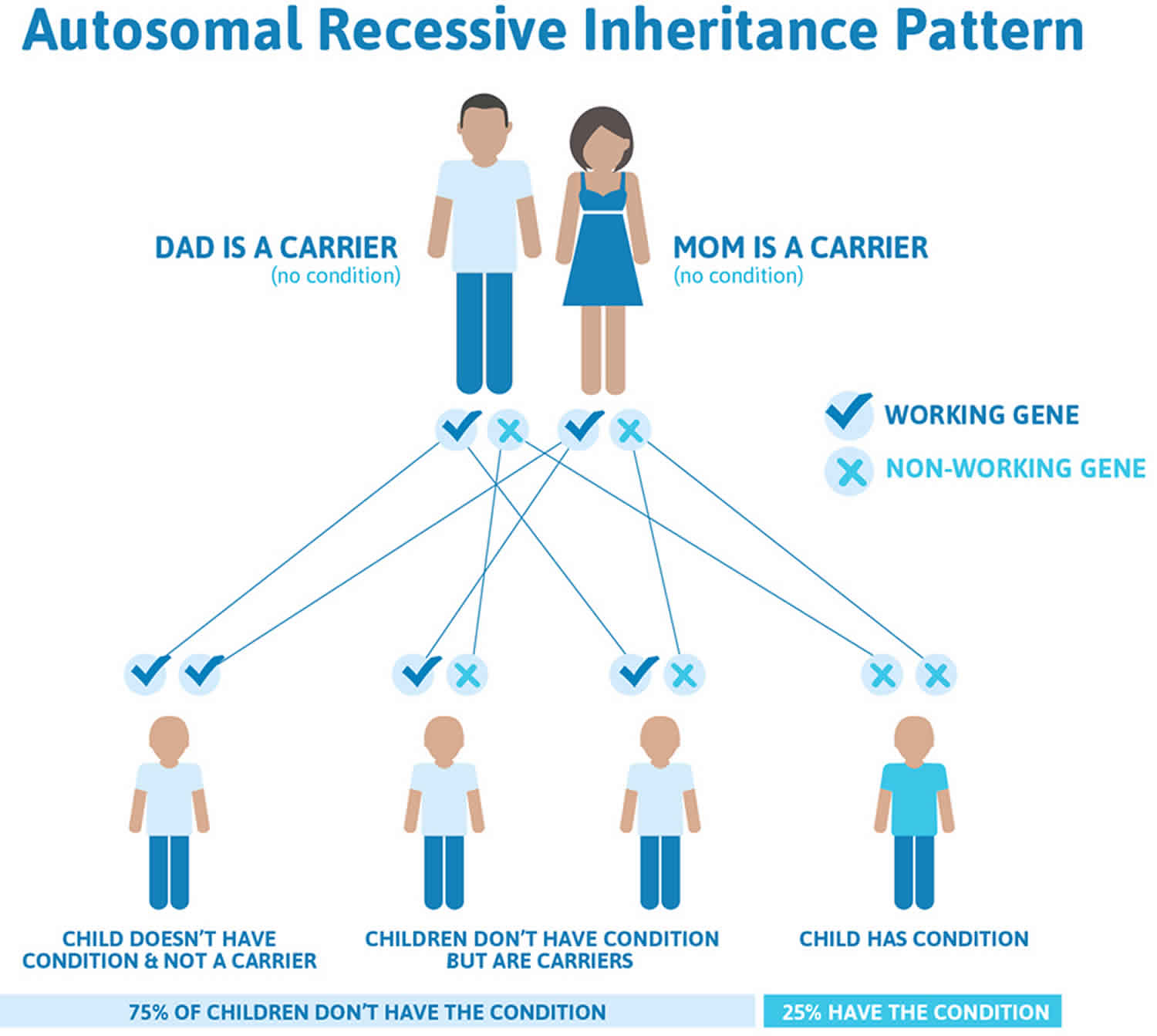
Figure 2 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 2. Seckel syndrome autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Seckel syndrome symptoms
Seckel syndrome is characterized by abnormally slow growth during fetal development (intrauterine growth retardation [IUGR] with average birth weight 1540g), resulting in low birth weight. Unusually slow growth (growth retardation and delayed bone maturation) continues after birth (postnatal) and typically leads to short stature (dwarfism) with proportional development of the arms and legs (as opposed to short stature with abnormally small arms and legs, i.e., short-limbed dwarfism). Moderate to severe mental retardation may also be present at birth (congenital) but may not become apparent until an affected child is older. Half of the patients have an IQ less than 50.
In addition, infants with Seckel syndrome have distinctive abnormalities of the head and facial (craniofacial) area. In most cases, affected infants may have microcephaly, a condition that indicates that the head circumference is smaller than would be expected for an infant’s age and sex; a receding forehead; an unusually small jaw (micrognathia) that is recessed farther back than usual (retrognathia); and/or a curved, triangular “beak-like” nose. Due to such abnormalities the middle portion of the face may appear unusually prominent. In addition, in some cases, certain fibrous joints between the bones of the skull (cranial sutures) may close prematurely (craniosynostosis). As a result, the head may appear abnormally elongated or shortened, depending on which part of the skull is affected.
In some infants with Seckel syndrome, other craniofacial abnormalities may be present including unusually large eyes with downwardly slanting eyelid folds (palpebral fissures); crossed eyes (strabismus); low-set, malformed (dysplastic) ears with absent ear lobes; and/or a highly-arched roof of the mouth (highly-arched palate) that may be incompletely formed (cleft palate). In addition, in some cases, one side of the face may appear larger than the other (facial asymmetry). Some affected infants and children may have dental abnormalities including underdevelopment (enamel hypoplasia) of tooth enamel and/or crowding and/or improper positioning of the teeth.
In addition, some children with Seckel syndrome may have various skeletal abnormalities including dislocation of the head of the forearm bone on the thumb side of the hand (radial dislocation), dislocation of the elbows, dislocation and/or malformation (dysplasia) of the hips, and/or an inability to fully extend the knees. In some cases, affected children may develop abnormal front-to-back and/or side-to-side curvature of the spine (kyphoscoliosis). Additional skeletal abnormalities may include permanent fixation of the fifth fingers in a bent position (clinodactyly), malformation of the foot in a twisted position (clubfoot), and/or absence of one pair of ribs (i.e., exhibit 11 rather than 12 pairs of ribs).
In some cases, males with Seckel syndrome may exhibit failure of the testes to descend normally into the scrotum (cryptorchidism) and/or affected females may have an abnormally enlarged clitoris (clitoromegaly). In addition, affected children may have excessive body hair (hirsutism), and/or a single, deep crease across the palms of the hands (simian crease).
In some cases, individuals with Seckel syndrome may also have associated blood (hematological) disorders including deficiency of all bone marrow elements including red blood cells, white blood cells, and platelets (pancytopenia). A low level of circulating red blood cells is known as anemia.
Seckel syndrome diagnosis
With the advent of technically superior ultasonography, Seckel syndrome may be diagnosed before birth (prenatally). In fetal ultrasonography, reflected sound waves are used to create an image of the developing fetus. Intrauterine ultrasound features, including abnormal head appearance, extreme microcephaly, cystic hygroma, encephalocele, posterior fossa cysts, cortical dysplasia and corpus callosum agenesis, can also be exploited for prenatal diagnosis 13. In addition, fetal MRI can show brain migration disorders and 3D sonography helps in the diagnosis 14. After birth, Seckel syndrome may be suspected based upon a thorough clinical evaluation, a detailed patient history, and a variety of specialized tests 15. Although distinctive craniofacial, skeletal, and/or other physical abnormalities associated with Seckel syndrome may be present at birth (congenital), a diagnosis of Seckel syndrome may not be confirmed, in some cases, until an affected child ages and the full syndrome develops (e.g., when short stature and/or mental retardation becomes apparent).
Short stature associated with Seckel syndrome involves proportional growth of the arms and legs, which allows for differential diagnosis from syndromes that involve short stature and abnormally small arms and legs (short-limbed dwarfism).
Seckel syndrome treatment
Seckel syndrome treatment is supportive 9. Supportive therapy including special education, speech and language therapy, behavioral therapy, occupational therapy, and community services for families. Ritalin may be helpful in managing hyperkinesia. Seizures are usually responsive to monotherapy with standard antiepileptic drugs 12. Hematological abnormalities have been reported in a few cases with Seckel syndrome, including anemia, pancytopenia, and acute myeloid leukemia, and medical treatment is proposed depending on the symptoms 16. In addition, severe retinal detachment probably due to chorioretinal degeneration has been reported in Seckel syndrome, so the risk of sudden-onset loss of vision should be considered in the care of these patients 17. Dental care is also very important in Seckel syndrome because gingival hyperplasia, significant dental crowding, enamel hypoplasia and early loss of permanent teeth have been described 18.
Seckel syndrome life expectancy
Data on life expectancy in Seckel syndrome have not been reported; however, anecdotal reports document survival without obvious complications in persons over age 50 years 12. The risk for hematologic complication has not been assessed; no data link those with hematologic problems to a specific genotype.
References- Seckel HP. Bird-headed dwarfs: studies in developmental anthropology including human proportions. In: Thomas CC, editor. IL: Springfield; 1960. (pub.).
- Shanske A, Caride DG, Menasse-Palmer L, Bogdanow A, Marion RW. Central nervous system anomalies in Seckel syndrome: report of a new family and review of the literature. Am J Med Genet. 1997;70(2):155–158. doi:10.1002/(SICI)1096-8628(19970516)70:2<155::AID-AJMG10>3.0.CO;2-I
- Ramasamy C, Satheesh S, Selvaraj R. Seckel syndrome with severe sinus bradycardia. Indian J Pediatr. 2015;82(3):292–293. doi:10.1007/s12098-014-1568-3
- Di Bartolomeo R, Polidori G, Piastra M, Viola L, Zampino G, Chiaretti A. Malignant hypertension and cerebral haemorrhage in Seckel syndrome. Eur J Pediatr. 2003;162(12):860–862. doi:10.1007/s00431-003-1310-z
- Rahme R, Crevier L, Dubois J, Mercier C. Moyamoya-like vasculopathy and Seckel syndrome: just a coincidence? Childs Nerv Syst. 2010;26(7):983–986. doi:10.1007/s00381-010-1142-x
- Saeidi M, Shahbandari M. A Child with Seckel Syndrome and Arterial Stenosis: Case Report and Literature Review. Int Med Case Rep J. 2020;13:159-163. Published 2020 May 14. doi:10.2147/IMCRJ.S241601 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7234957
- Can E, Bulbul A, Uslu S, et al. A case of Seckel syndrome with tetralogy of fallot. Genet Couns. 2010;21(1):49–51.
- Faivre L, Le Merrer M, Lyonnet S, et al. Clinical and genetic heterogeneity of Seckel syndrome. Am J Med Genet. 2002;112(4):379–383. doi:10.1002/ajmg.10677
- Yigit G, Brown KE, Kayserili H, et al. Mutations in CDK5RAP2 cause Seckel syndrome. Mol Genet Genomic Med. 2015;3(5):467-480. doi:10.1002/mgg3.158 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4585455
- Shaheen R, Faqeih E, Ansari S, Abdel-Salam G, Al-Hassnan ZN, Al-Shidi T, et al. Genomic analysis of primordial dwarfism reveals novel disease genes. Genome Res. 2014;24:291–299.
- Kalay E, Yigit G, Aslan Y, Brown KE, Pohl E, Bicknell LS, et al. CEP152 is a genome maintenance protein disrupted in Seckel syndrome. Nat. Genet. 2011;43:23–26.
- Verloes A, Drunat S, Gressens P, et al. Primary Autosomal Recessive Microcephalies and Seckel Syndrome Spectrum Disorders – RETIRED CHAPTER, FOR HISTORICAL REFERENCE ONLY. 2009 Sep 1 [Updated 2013 Oct 31]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK9587
- Akkurt MO, Pakay K, Akkurt I, Temur M, Korkmazer E. Prenatal diagnosis of Seckel syndrome at 21 weeks’ gestation and review of the literature. J Matern Fetal Neonatal Med. 2019;32(11):1905–1908. doi:10.1080/14767058.2017.1419467
- Gupta A, Fazal TS, Arora R. Antenatal diagnosis of seckel syndrome. J Obstet Gynaecol India. 2014;64(Suppl 1):6–8. doi:10.1007/s13224-012-0339-1
- Sisodia R, Raj RK, Goel V. Seckel syndrome: a rare case report. J Indian Soc Pedod Prev Dent. 2014;32(2):160–163. doi:10.4103/0970-4388.130983
- Faivre L. Cormier-Daire V. Seckel syndrome. Orphanet encyclopedia, April 2005. http://www.orpha.net/data/patho/GB/uk-Seckel(05).pdf
- Krzyzanowska-Berkowska P, Szumny D, Mlynczak T, Kisza K, Oficjalska J. Bilateral retinal detachment in Seckel syndrome. Can J Ophthalmol. 2014;49(5):e130–1. doi:10.1016/j.jcjo.2014.07.013
- Ramalingam K, Kaliyamurthy SD, Govindarajan M, Swathi S. Seckel syndrome: a report of a case. J Indian Soc Pedod Prev Dent. 2012;30(3):258–261. doi:10.4103/0970-4388.105021





{kind=link}