What is Sturge Weber syndrome
Sturge-Weber syndrome is a rare disorder that affects the development of certain blood vessels, causing abnormalities in the brain, skin, and eyes from birth. Sturge-Weber syndrome has three major features: a red or pink birthmark called a port-wine birthmark, a brain abnormality called a leptomeningeal angioma, and increased pressure in the eye (glaucoma). Port-wine birthmarks are caused by enlarged blood vessels right underneath the skin. Leptomeningeal angiomas are clusters of abnormal blood vessels between the layers of tissue that cover the brain and spine. These angiomas can lead to decreased blood flow to the brain, which in turn can cause strokes, seizures, headaches and muscle weakness. These features can vary in severity and not all individuals with Sturge-Weber syndrome have all three features. Other symptoms of Sturge-Weber syndrome may include seizures, muscle weakness, developmental and intellectual disability. Seizures occur in 72% to 80% of Sturge-Weber syndrome patients with unilateral brain lesions and in 93% of patients with bihemispheric involvement 1. Seizures can begin anytime from birth to adulthood, but 75% of those with seizures begin having them during the first year of infancy, 86% by age 2, and 95% before age 5. Glaucoma occurs in 30% to 71% of patients 1. Increased pressure in the eyes (glaucoma) may be diagnosed at birth, during childhood or adulthood.
Sturge-Weber syndrome is estimated to affect 1 in 20,000 to 50,000 individuals.
Sturge-Weber syndrome can be thought of as a spectrum of disease in which individuals may have abnormalities affecting all three of these systems (i.e. brain, skin and eyes), or only two, or only one. Consequently, the specific symptoms and severity of the disorder can vary dramatically from one person to another. Symptoms are usually present at birth (congenital), yet the disorder is not inherited and does not run in families. Some symptoms may not develop until adulthood. Sturge-Weber syndrome is caused by a somatic mutation in the GNAQ gene. The gene mutation is not inherited and the mutation occurs randomly (sporadically) for no known reason 2.
Some publications break down Sturge-Weber syndrome into three main subtypes. Type 1 consists of skin and neurological symptoms. These individuals may or may not have glaucoma. Type 2 consists of skin symptoms and possibly glaucoma, but there is no evidence of neurological involvement. Type 3 consists of neurological involvement, but without skin abnormalities. Glaucoma is usually not present. Type 3 may also be known as the isolated neurological variant.
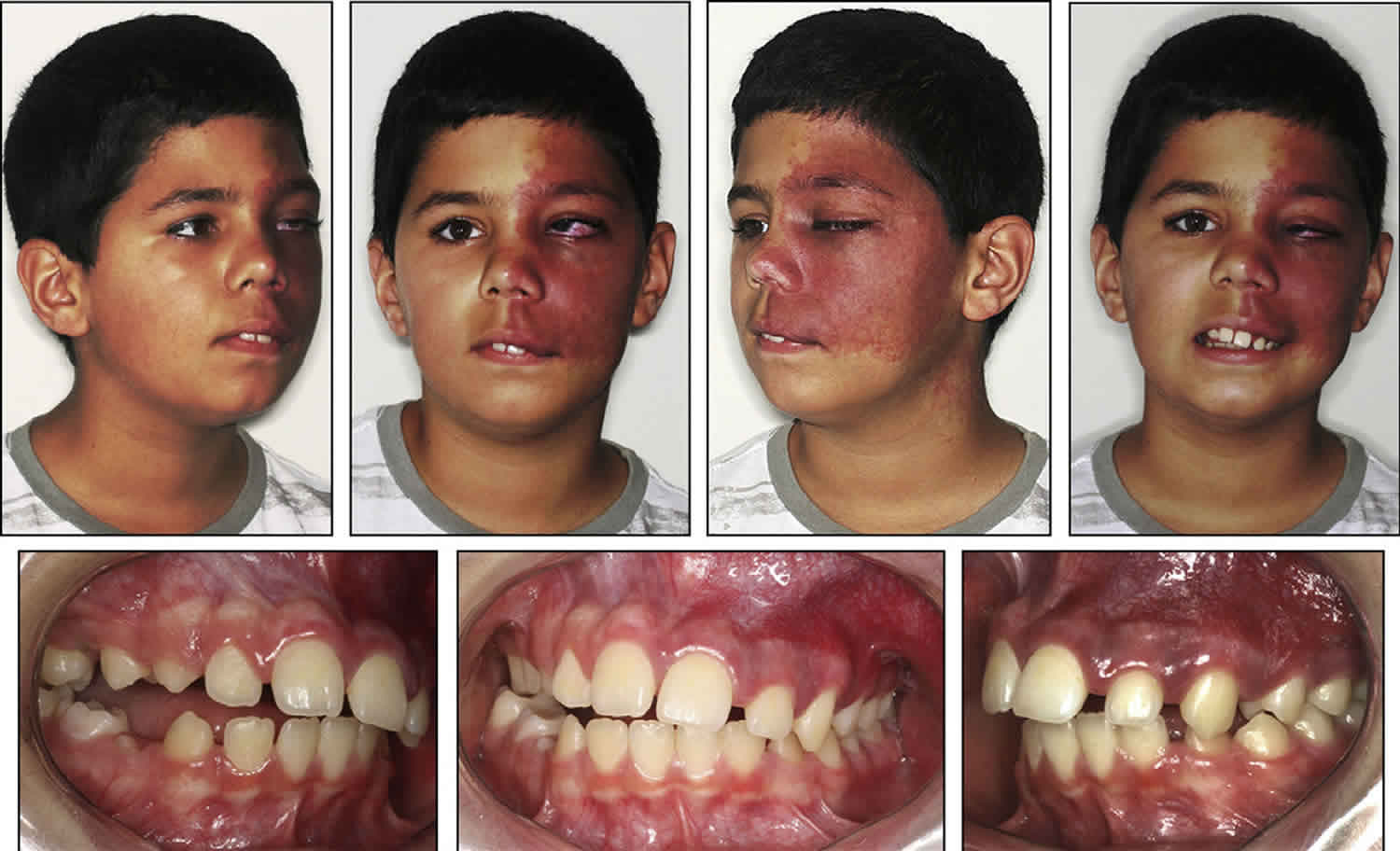
Most people with Sturge-Weber syndrome are born with a port-wine birthmark. This type of birthmark is caused by enlargement (dilatation) of small blood vessels (capillaries) near the surface of the skin. Port-wine birthmarks are typically initially flat and can vary in color from pale pink to deep purple. In people with Sturge-Weber syndrome, the port-wine birthmark is most often on the face, typically on the forehead, temple, or eyelid. The port-wine birthmark is usually only on one side of the face but can be on both sides. Over time, the skin within the port-wine birthmark can darken and thicken.
In Sturge-Weber syndrome, there is usually abnormal formation and growth of blood vessels within the two thin layers of tissue that cover the brain and spinal cord. This abnormality, which is called leptomeningeal angioma, can affect one or both sides of the brain and impair blood flow in the brain and lead to loss of brain tissue (atrophy) and deposits of calcium (calcification) in the brain below the angioma. The decrease in blood flow caused by leptomeningeal angiomas can cause stroke-like episodes in people with Sturge-Weber syndrome. These episodes often involve temporary muscle weakness on one side of the body (hemiparesis), vision abnormalities, seizures, and migraine headaches. In affected individuals, these episodes usually begin by age 2. The seizures usually involve only one side of the brain (focal seizures), during which the port-wine birthmark may darken and individuals may lose consciousness. People with Sturge-Weber syndrome have varying levels of cognitive function, from normal intelligence to intellectual disability. Some individuals have learning disabilities with problems focusing similar to attention deficit hyperactivity disorder (ADHD).
In individuals with Sturge-Weber syndrome, glaucoma typically develops either in infancy or early adulthood and can cause vision impairment. In some affected infants, the pressure can become so great that the eyeballs appear enlarged and bulging (buphthalmos). Individuals with Sturge-Weber syndrome can have tangles of abnormal blood vessels (hemangiomas) in various parts of the eye. When these abnormal blood vessels develop in the network of blood vessels at the back of the eye (choroid), it is called a diffuse choroidal hemangioma and occurs in about one-third of individuals with Sturge-Weber syndrome. A diffuse choroidal hemangioma can cause vision loss. When present, the eye abnormalities typically occur on the same side of the head as the port-wine birthmark.
Sturge-Weber syndrome is diagnosed based on the symptoms. Imaging studies, such as an MRI or CT-scan, are also used to aid in the diagnosis.
There is no one treatment for Sturge-Weber syndrome, so management involves treating the specific symptoms that are present. This may include anti-seizure medications, persons with drug-resistant seizures may be treated by surgical removal of epileptic brain tissue. Medications and/or surgery may be performed for glaucoma, and low-dose aspirin to reduce the pressure in the eyes and brain. The port-wine birthmark may be treated with various types of laser treatments. Physical therapy should be considered for infants and children with muscle weakness. Educational therapy is often prescribed for those with impaired cognition or developmental delays. Doctors recommend yearly monitoring for glaucoma.
The long-term outlook for people with Sturge-Weber syndrome is dependent on the severity of symptoms and varies from person to person.
What causes Sturge Weber syndrome?
Sturge-Weber syndrome is caused by a mutation in the GNAQ gene. This gene provides instructions for making a protein called guanine nucleotide-binding protein G(q) subunit alpha (Gαq). The Gαq protein is part of a group of proteins (complex) that regulates signaling pathways to help control the development and function of blood vessels.
The GNAQ gene mutation that causes Sturge-Weber syndrome results in the production of a protein with impaired function. As a result, the altered Gαq protein cannot play its part in regulating signaling pathways, resulting in abnormally increased signaling. The enhanced signaling likely disrupts the regulation of blood vessel development, causing abnormal and excessive formation of vessels before birth in people with Sturge-Weber syndrome.
Sturge-Weber syndrome is not inherited. The GNAQ gene mutation that causes Sturge-Weber syndrome is somatic, which means it occurs after conception. In Sturge-Weber syndrome, the mutation is thought to occur in a cell during early development before birth. As that cell continues to grow and divide, the cells derived from it, specifically certain cells in the brain, eyes, and skin that are involved in blood vessel formation, also have the mutation, while the body’s other cells do not. This situation is called mosaicism. The mosaic nature of the mutations helps to explain why the abnormal blood vessel growth occurs in some parts of the body but not in others.
Sturge Weber syndrome symptoms
Sturge-Weber syndrome is a highly variable disorder. Some individuals may develop characteristic skin abnormalities, but no neurological abnormalities. Less often, individuals develop neurological abnormalities without the characteristic skin issues. Therefore, it is important to note that affected individuals may not have all of the symptoms discussed below and that every individual patient is unique. Parents should talk to their child’s physician and medical team about their specific case, associated symptoms and overall prognosis.
A congenital facial birthmark known as a capillary malformation (port-wine stain or port-wine birthmark or nevus flammeus) is often the most notable initial symptom 3. Port wine birthmarks occur in 3 of 1000 newborns. In a patient with a facial port wine birthmark, the overall risk of having Sturge-Weber syndrome is only about 8% to 15%. The risk of having Sturge-Weber syndrome increases to 25% when half of the face, including the ophthalmic division of the trigeminal nerve is involved and rises to 33% when both sides of the face, including the ophthalmic division of the trigeminal nerve are involved. Port-wine birthmark can range from light pink to reddish to dark purple in color. The size of a port-wine birthmark can vary. Usually, at least one eyelid and/or the forehead of one side of the face are affected, but both sides of the face have been affected less often. In some children, the entire half of one side of the face may be affected. Sometimes, the discoloration may extend slightly onto the other side of the face or both sides of the face may be extensively involved. Rarely, a port-wine birthmark extends all the way to the trunk and/or arms. The port-wine birthmark that characterizes Sturge-Weber syndrome is caused by an overabundance of capillaries just below the surface of the skin in the distribution of the trigeminal nerve. Capillaries are tiny blood vessels that form a fine network throughout the body connecting arteries and veins and are responsible for the exchange of various substances such as oxygen between cells and tissue. If untreated, port-wine birthmarks may deepen in color with age, thicken, and potentially develop blood blisters (blebs) that can burst causing spontaneously bleeding.
The abnormal blood vessels that make up a port-wine birthmark will vary in size, diameter, distribution, and depth from one individual to another and even within the same person in different affected areas. This means that a port-wine birthmark in each individual is unique and can be quite dissimilar from one person to another.
Individuals with Sturge-Weber syndrome may also experience a variety of neurological abnormalities. The extent of neurological involvement can vary dramatically from one person to another. Neurological symptoms are caused by the abnormal growth of blood vessels on the surface of the brain (leptomeningeal angiomas). Seizures, which often begin in infancy or childhood, are a common finding. Seizures usually affect the opposite side of the body as the port-wine birthmark, but sometimes affect both sides of the body. Seizures may vary in frequency and intensity and sometimes may worsen in severity and frequency with age. Affected individuals may also experience muscle weakness or paralysis on one side of the body (hemiparesis), usually on the side opposite the port-wine birthmark. Developmental delays and intellectual disability ranging from mild learning disabilities to severe cognitive deficits may occur in some children; in other children, intelligence and cognition are unaffected. In patients with severe or uncontrolled seizures cognitive impairments are common.
Headaches, including migraines, and visual field defects such as the loss of vision in half the visual field in one or both eyes (hemaniopsia) may also occur. There is a risk of stroke, stroke-like episodes or mini-strokes (transient ischemic attacks). Stroke-like episodes can be associated with temporary (transient) weakness or paralysis of half of the body and visual field defects. Behavioral problems such as attention deficit disorder, mood disorders, and poorer social skills have also been seen in some children, particularly those with lower cognitive function and a greater frequency of seizures.
Some children are born with glaucoma, a condition marked by increased pressure within the eye. Glaucoma usually affects the eye on the same side of the face as the port-wine birthmark. Glaucoma can potentially damage the optic nerve, the main nerve that transmits signals from the eye to the brain, ultimately resulting in progressive vision loss. The same eye also may become enlarged so that appears to bulge out of or to enlarge its socket (buphthalmos).
Other eye abnormalities can occur including the development of angiomas in the membranes that line the inner surface of the eyelids (conjunctiva), the layer of blood vessels and connective tissue (choroid) between the white of the eye and the retina, and the clear, transparent membrane covering the membrane (cornea). An affected individual’s eyes can be two different colors (e.g. one brown and one blue eye). Additional ocular symptoms can include an abnormal accumulation of fluid inside the eyeball causing enlargement of the eyeball (hydrophthalmos); degeneration of the cranial nerve that transmit lights signals to the brain (optic atrophy); clouding or displacement of the lenses; retinal detachment; streaks resembling blood vessels in the retina (angioid streaks); and/or loss of vision due to an organic lesion in the visual cortex (cortical blindness). Individuals who have neurological abnormalities, but do not have a port-wine birthmark generally do not develop eye problems.
Endocrine disorders have also been reported in some individuals including central hypothyroidism and an increased risk of growth hormone deficiency. Central hypothyroidism is characterized by underactivity of the thyroid gland due to insufficient stimulation of thyroid stimulating hormone in an otherwise healthy thyroid. Central hypothyroidism in Sturge-Weber syndrome may be due to anti-seizure medication.
Additional symptoms may occur including an abnormally large head (macrocephaly), overgrowth (hypertrophy) of the certain soft tissues underlying the port-wine birthmark, and lymphatic malformations, which are non-malignant masses consisting of fluid-filled channels or spaces thought to be caused by abnormal development of the lymphatic system. These symptoms are consistent with a related rare disorder known as Klippel-Trenaunay syndrome and most children with these findings are classified as having Klippel-Trenaunay syndrome. Klippel-Trenaunay syndrome (congenital dysplastic angiopathy) is a congenital vascular disorder of unknown cause. Klippel-Trenaunay syndrome is characterized by a triad of symptoms: Port Wine Birthmark (capillary malformation) covering one or more limbs, congenital vascular anomalies, usually venous varicosities, absence or duplication of a venous structure, malformation and hypertrophy (enlargement of the limb) or atrophy (withering or smaller limb). Klippel-Trenaunay syndrome involves the lower limbs in approximately 90% of the cases. Researchers are not sure whether Sturge-Weber syndrome and Klippel-Trenaunay syndrome are related disorders that overlap or whether they are similar, yet distinct, rare disorders.
Sturge Weber syndrome diagnosis
A diagnosis of Sturge-Weber syndrome is based upon identification of characteristic symptoms (e.g. port-wine birthmark), a detailed patient history, a thorough clinical evaluation and a variety of specialized tests. All newborn babies with a port wine birthmark affecting the eyelids and/or brain involvement should see an ophthalmologist in the first few weeks of life. Children with port wine mark involving lids may be at life-long risk of glaucoma and require periodic eye examination, often under anesthesia or sedation in the first years of life until able to be awake for testing. A diagnosis may be straightforward in an infant with a port-wine birthmark, glaucoma, evidence of cerebral involvement and neuroimaging findings consistent with a diagnosis of Sturge-Weber syndrome. Diagnosis can be more difficult in infants who have a port-wine birthmark, but no neurological symptoms.
Clinical Testing and Workup
Various imaging techniques can be used to identify and assess neurological complications including x-rays of the skull (skull radiography) or magnetic resonance imaging (MRI) with gadolinium. A head computed tomography (CT) scan can show intracranial calcification in certain areas of the brain. An MRI uses a magnetic field and radio waves to produce cross-sectional images of particular organs and bodily tissues. Gadolinium is a contrast agent that is used to enhance the scanning results and supply a more detailed picture of tissues such as the brain or blood vessels.
Newer neuroimaging techniques such as susceptibility-weighted imaging (SWI) have proven useful in evaluating individuals for brain abnormalities potentially associated with Sturge-Weber syndrome. SWI uses a different type of contrast to enhance traditional MRIs and may allow physicians to diagnose brain abnormalities earlier. SWI is particularly effective at evaluating venous structures in the brain.
Computerized tomography (CT) scanning may also be used to aid in diagnosing Sturge-Weber syndrome. During CT scanning, a computer and x-rays are used to create a film showing cross-sectional images of certain tissue structures. A single-photon emission computed tomography scan (SPECT), which is a specialized CT scan, can reveal areas of involvement in the brain that may not show in MRI or traditional CT scans. SPECT scanning may be used in conjunction with other scanning techniques to evaluate the brain of individuals suspected of having Sturge-Weber syndrome.
Traditional angiography designed to evaluate the health and function of blood vessels) are not usually recommended for individuals suspected of having Sturge-Weber syndrome, but occasionally may be required to exclude a high flow lesion such as an arterial venous malformation or arterial venous fistula. An electroencephalogram (EEG) can be used to evaluate and localize seizure activity.
A complete ophthalmological exam can reveal glaucoma and other eye abnormalities potentially associated with Sturge-Weber syndrome. Because of the high risk of glaucoma, complete eye examination should be performed regularly, especially in infants and young children. Follow-up examination should continue into adulthood even if results are normal through childhood.
Sturge Weber syndrome treatment
The treatment of Sturge-Weber syndrome is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, neurologists, neurosurgeons, dermatologists, ophthalmologists, and other healthcare professionals may need to systematically and comprehensively plan an affect child’s treatment. Psychosocial support for the entire family is essential as well.
Laser treatment can lighten or remove the port-wine birthmark in affected individuals, even infants as young as one month old. However, port-wine birthmarks tend to return or darken again, necessitating multiple laser therapy sessions. Pulse dye laser therapy is the most common technique for treating individuals with Sturge-Weber syndrome 4. However, because each port-wine birthmark is dissimilar (e.g., they vary in size, diameter, distribution and depth), the most effective therapy for one person will not be the same for another person and no one form of laser therapy is effective for all affected individuals. In fact, different laser therapies may be required for different affected areas of the same individual. Topical sirolimus is now being used by many providers in combination with laser treatments to prevent regrowth of abnormal vessels.
Seizures are treated with anti-seizure (anti-convulsant) medications. The effectiveness of these medications in treating people with Sturge-Weber syndrome is highly variable. Some individuals do not respond to anti-seizure medications (refractory seizures) despite an aggressive treatment regimen. Refractory cases may ultimately require surgery. Surgical techniques that have been used to control seizures in Sturge-Weber syndrome include hemispherectomy, focal cortical resection, and vagal nerve stimulation.
Hemispherectomy involves the surgical removal or disabling of half of the brain, specifically the half of the brain which is repeatedly damaged by chronic seizure activity. This surgical procedure can be associated with significant adverse effects including weakness on one side of the body possibly affecting walking (hemiparetic gait), little use of the affected hand, or hemianopsia. In some cases, such abnormalities may already be present before the surgery as a consequence of Sturge-Weber syndrome. In certain cases, hemispherectomy may be recommended for individuals with repeated stroke-like episodes and progressive neurological deficits.
Focal cortical resection is used when seizure activity arises from one specific area of the brain. This area of the brain can be isolated through brain mapping, a scientific method of studying brainwave activity. A neurosurgeon will remove the affected piece of the brain (focal resection). This procedure requires removing a small piece of the skull in order to gain access to the brain. A focal resection is less likely to produce neurologic deficits but is also less likely to result in full seizure control.
Vagus nerve stimulation is a procedure in which a device called a pulse generator is inserted into the chest and a wire is run underneath the skin to the vagus nerve in the neck. The pulse generator is similar to a pacemaker and transmits mild, electrical impulses to the brain via the vagus nerve. These impulses prevent seizures from occurring. This intensity and timing of the nerve impulses are determined based upon each individual’s needs.
The decision to undergo surgery to treat refractory seizures in children with Sturge-Weber syndrome is difficult because of the varied pattern, frequency and severity of seizures in each child. Some children experience clusters of seizures that occur close together only to be followed by a seizure-free period that can last for many months or years. Some physicians advocate earlier surgery for seizures in order to protect against refractory seizures, developmental delays, cognitive dysfunction, and hemiparesis.
Low-dose aspirin has been used to treat individuals with Sturge-Weber syndrome. Low-dose aspirin has led to a reduction in the frequency of seizures and stroke-like episodes. In some children, the decrease in seizure activity is significant. Complications have included increased bruising and gum or nose bleeding. Most reports in the medical literature suggest low-dose aspirin can safely be used in individuals with Sturge-Weber syndrome and provides benefit. However, studies are needed to determine the long-term safety and effectiveness of low dose aspirin and whether this treatment improves long-term cognitive function and overall quality of life. Increasingly, low dose aspirin is being used by many centers in the treatment of Sturge-Weber syndrome.
The Atkins version of the ketogenic diet has been reported to be successful in reducing the frequency of seizures in five children with Sturge-Weber syndrome. These children had seizures that failed to respond to medical treatment and occurred at least monthly. However, the Atkins diet is difficult for many to maintain over long periods of time.
Recently a small open label trial was published on the use of Epidiolex (cannabidiol) for the treatment of refractory seizures in Sturge-Weber syndrome. This early data suggests that Epidiolex is safe and effective however more data is needed from a multi-centered study. Furthermore, currently a small open label study is underway of sirolimus for cognitive impairments in Sturge-Weber syndrome.
Decisions concerning the use of particular drug regimens, surgery, and/or other treatments should be made by physicians and other members of the health care team in careful consultation with parents or a patient based upon the specifics of an individual case; a thorough discussion of the potential benefits and risks, including possible side effects and long-term effects; patient preference; and other appropriate factors.
Preventive (prophylactic) treatment of migraines and headaches may be recommended and may include medications such as propranolol or verapamil. Some anti-seizure medications such as gabapentin, topiramate, and valproic acid may also help to treat migraines or headaches.
Affected infants and children should receive regular ophthalmological exams in order to promptly detect and treat glaucoma and any increase in intraocular pressure. Certain medications usually delivered as eye drops or orally may be used to treat glaucoma. Ultimately, glaucoma often requires surgery with medications used as a follow up (adjunct) therapy. There are several different surgical techniques used to treat glaucoma in individuals with Sturge-Weber syndrome depending upon the individual case.
Additional therapy includes physical therapy for muscle weakness, special education for children with developmental delays or intellectual disability as well as other medical, social or vocational services.
Sturge Weber syndrome life expectancy
The symptoms of Sturge-Weber syndrome tend to get worse with age 4. However, most people with Sturge-Weber syndrome have mild symptoms which are not life-threatening. The long-term outlook varies depending on the severity of symptoms, and how well seizures and glaucoma can be controlled or prevented. More severe seizures at an early age are associated with an increased chance for developmental and intellectual disability 4. Adults with Sturge-Weber syndrome may have psychological issues that require additional intervention 4.
Although it is possible for the birthmark and atrophy in the cerebral cortex to be present without symptoms, most infants will develop convulsive seizures during their first year of life. There is a greater likelihood of intellectual impairment when seizures start before the age of 2 and are resistant to treatment. Prognosis is worst in the minority of children who have both sides of the brain affected by the blood vessel abnormalities.
References- Understanding Sturge-Weber. https://sturge-weber.org/new-to-swf/
- Comi A. Sturge-Weber syndrome. Handbk of Clin Neuro. 2015; 132:157-168. https://ncbi.nlm.nih.pubmed/26564078
- Sturge-Weber Syndrome Information Page. https://www.ninds.nih.gov/Disorders/All-Disorders/Sturge-Weber-Syndrome-Information-Page
- De la Torre AJ, Luat AF, Juhász C, Ho ML, Argersinger DP et al. A Multidisciplinary Consensus for Clinical Care and Research Needs for Sturge-Weber Syndrome. Pediatr Neurol. Jul 2018; 84:11-20. https://www.ncbi.nlm.nih.gov/pubmed/29803545





{kind=link}