
What is thalassemia
Thalassemia is a group of inherited blood disorders that can be passed from parents to their children and affect the amount and type of hemoglobin the body produces. “Inherited” means that the disorder is passed from parents to children through genes.
Thalassemias cause the body to make fewer healthy red blood cells and less hemoglobin than normal. Hemoglobin is an iron-rich protein in red blood cells. It carries oxygen to all parts of the body. Hemoglobin also carries carbon dioxide (a waste gas) from the body to the lungs, where it’s exhaled.
People who have thalassemias can have mild or severe anemia. Anemia is caused by a lower than normal number of red blood cells or not enough hemoglobin in the red blood cells.
Hemoglobin (Hb) is a substance present in all red blood cells. Hemoglobin (Hb) is important for proper red blood cell function because it carries the oxygen that red blood cells deliver around the body. One portion of hemoglobin called heme is the molecule with iron at the center. Another portion is made of up four protein chains called globins. Each of the four globin chains holds a heme group containing one iron atom. Depending on their structure, the globin chains are designated as alpha, beta, gamma, or delta.
Not all hemoglobin is the same. Different types of hemoglobin are classified according to the type of globin chains they contain. The type of globin chains present is important in hemoglobin’s ability to transport oxygen.
Normal hemoglobin types include:
- Hemoglobin A – this is the predominant type of Hb in adults (about 95-98%); Hb A contains two alpha (α) protein chains and two beta (ß) protein chains.
- Hb A2 – makes up about 2-3.5% of Hb found in adults; it has two alpha (α) and two delta (δ) protein chains.
- Hb F – makes up to 2% of Hb found in adults; it has two alpha (α) and two gamma (γ) protein chains. Hb F is the primary hemoglobin produced by a developing baby (fetus) during pregnancy. Its production usually falls to a low level within a year after birth.
Normal hemoglobin, also called hemoglobin A, has four protein chains—two alpha globin and two beta globin. The two major types of thalassemia, alpha and beta, are named after defects in these protein chains. People with thalassemia have one or more genetic mutations that they have inherited and that result in a decreased production of normal hemoglobin. When the body doesn’t make enough normal hemoglobin, red blood cells do not function properly and oxygen delivery suffers. This can lead to anemia with signs and symptoms that can range from mild to severe, depending on the type of thalassemia that a person has. Examples of signs and symptoms include weakness, fatigue, and pale skin (pallor).
For hemoglobin, there are four genes (two from each parent) in your DNA that code for the alpha globin chains and two genes (each) for the beta, delta, and gamma globin chains. Since everyone inherits a set of chromosomes from each parent, each person inherits two alpha globulin genes and one beta globulin gene from each parent. A person may inherit mutations in either the alpha or beta globin genes.
With thalassemias, mutations in one or more of the globin genes cause a reduction in the amount of the particular globin chain produced. This can upset the balance of alpha to beta chains, resulting in unusual forms of hemoglobin or an increase in the amount of normally minor hemoglobin, such as Hb A2 or Hb F. The thalassemias are usually classified by the type of globin chain whose synthesis is decreased. For example, the most common alpha chain-related condition is called alpha thalassemia. Alpha thalassemia trait occurs if one or two of the four genes are missing. If more than two genes are missing, moderate to severe anemia occurs. And an increase of minor hemoglobin components, such as Hb A2 or Hb F is called beta thalassemia. The severity of this condition depends on the number of genes affected.
Other types of mutations in the genes coding for the globin chains can result in a globin that is structurally altered, such as hemoglobin S, which causes sickle cell. Together, thalassemia and hemoglobin abnormalities are called hemoglobinopathies.
The most severe form of alpha thalassemia is called alpha thalassemia major or hydrops fetalis. Babies who have this disorder usually die before or shortly after birth.
Two genes (one from each parent) are needed to make enough beta globin protein chains. Beta thalassemia occurs if one or both genes are altered.
The severity of beta thalassemia depends on how much one or both genes are affected. If both genes are affected, the result is moderate to severe anemia. The severe form of beta thalassemia is known as thalassemia major or Cooley’s anemia.
Thalassemias affect males and females. The disorders occur most often among people of Italian, Greek, Middle Eastern, Southern Asian, and African descent. Severe forms usually are diagnosed in early childhood and are lifelong conditions.
Doctors diagnose thalassemias using blood tests. The disorders are treated with blood transfusions, medicines, and other procedures.
Treatments for thalassemias have improved over the years. People who have moderate or severe thalassemias are now living longer and have better quality of life.
However, complications from thalassemias and their treatments are frequent. People who have moderate or severe thalassemias must closely follow their treatment plans. They need to take care of themselves to remain as healthy as possible.
Figure 1. Red blood cell hemoglobin
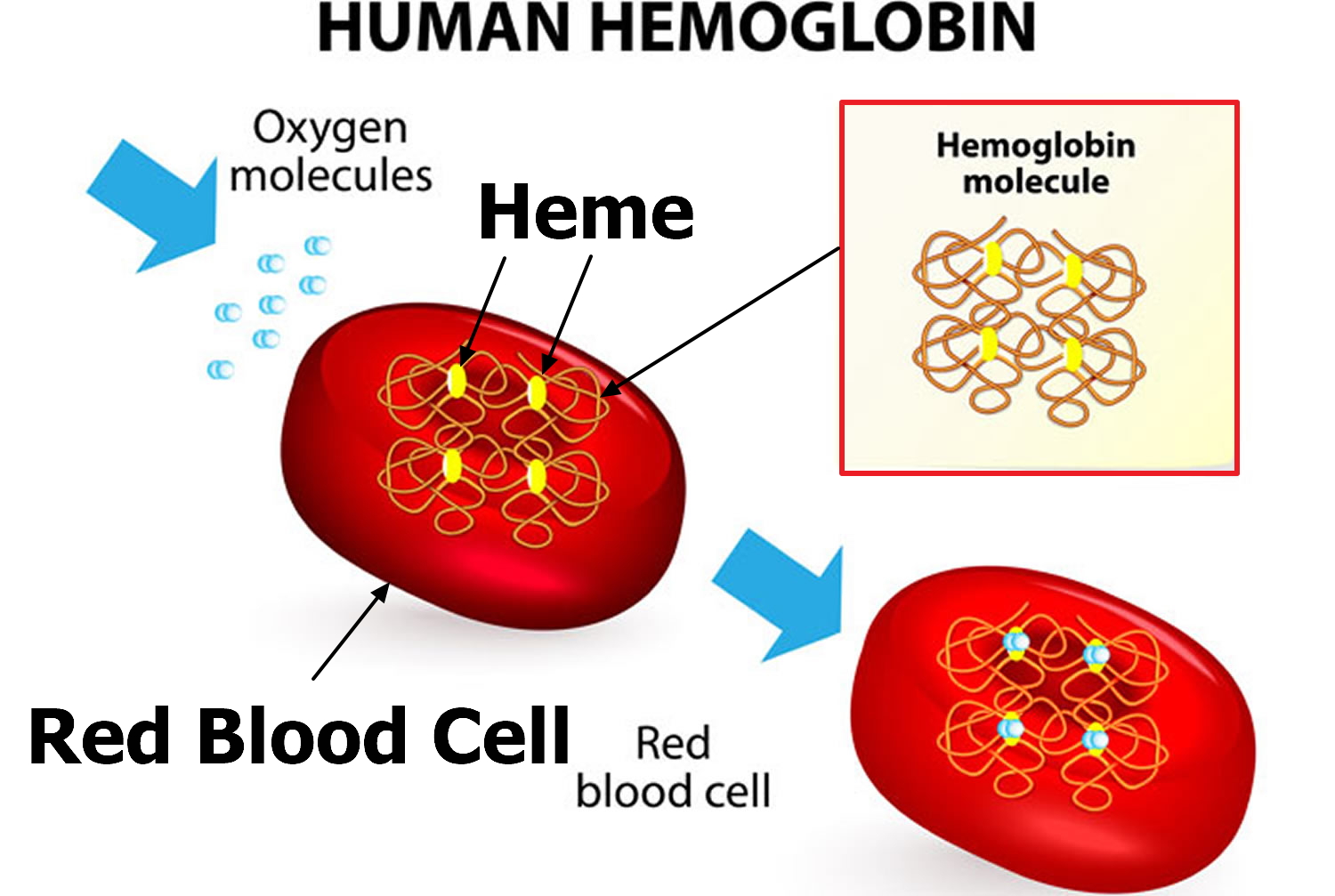 Figure 2. Structure of hemoglobin
Figure 2. Structure of hemoglobin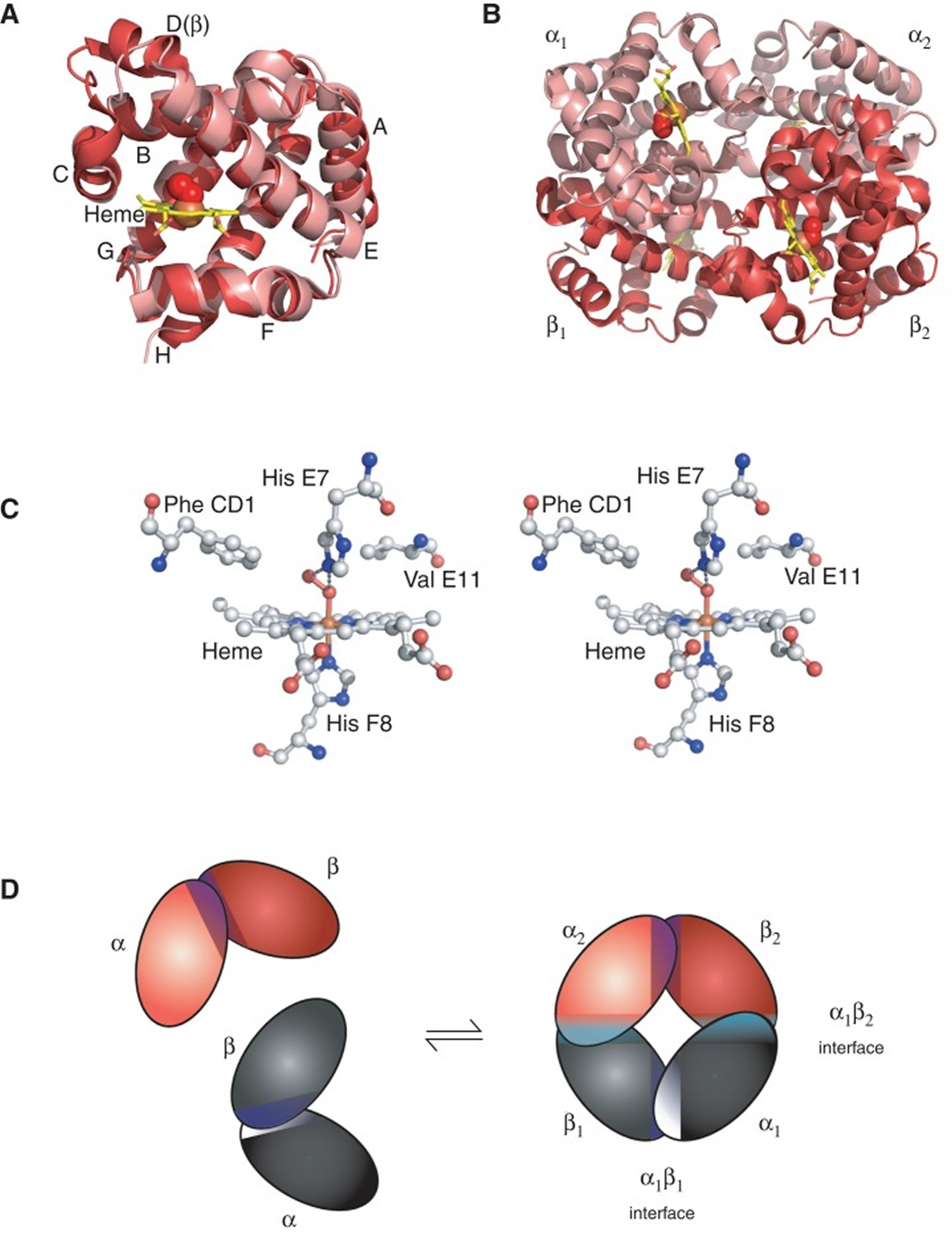 Footnotes:
Footnotes:
The structure of hemoglobin (Hb). (A) The α (pink) and β (red) hemoglobin (Hb) subunits have conserved α-helical folds. Helices are labeled A–H from the amino terminus. The α subunit lacks helix D. (B) The high O2 affinity R state quaternary structure of hemoglobin (Hb) with O2 (red spheres) bound at all four heme sites (protoporphyrin-IX as yellow sticks, with central iron atom as orange sphere). (C) Stereo (wall-eye) diagram of the heme pocket of β showing the proximal (F8) and distal (E7) histidines and selected residues in the distal heme pocket that influence ligand binding and autoxidation. (D) hemoglobin (Hb) tetramer is assembled from two identical αβ dimers (shown in red and gray for clarity). In the tetramer, each subunit makes contact with the unlike chain through a high affinity dimerization α1β1 interface and a lower affinity α1β2 dimer–tetramer interface (cyan).
How do I know if I have thalassemia?
People with moderate and severe forms of thalassemia usually find out about their condition in childhood, since they have symptoms of severe anemia early in life. People with less severe forms of thalassemia may only find out because they are having symptoms of anemia, or maybe because a doctor finds anemia on a routine blood test or a test done for another reason.
Because thalassemias are inherited, the condition sometimes runs in families. Some people find out about their thalassemia because they have relatives with a similar condition.
People who have family members from certain parts of the world have a higher risk for having thalassemia. Traits for thalassemia are more common in people from Mediterranean countries, like Greece and Turkey, and in people from Asia, Africa, and the Middle East. If you have anemia and you also have family members from these areas, your doctor might test your blood further to find out if you have thalassemia.
What should I do if I’m a carrier of thalassemia and I want to get pregnant?
Some severe types of thalassemia can cause babies to die before they are born or soon after. If you or your partner knows you are a carrier for thalassemia, you may want to talk to your doctor or a genetic counselor (find a genetic counselor here: https://www.nsgc.org/page/find-a-genetic-counselor) before getting pregnant. Certain tests may be able to show which type of thalassemia you are carrying. Once you are pregnant, prenatal testing can show whether or not your baby has thalassemia.
How can I prevent thalassemia?
Because thalassemia is passed from parents to children, it is very hard to prevent. However, if you or your partner knows of family members with thalassemia, or if you both have family members from places in the world where thalassemia is common, you can speak to a genetic counselor (go to: https://www.nsgc.org/page/find-a-genetic-counselor) to determine what your risk would be of passing thalassemia to your children.
Thalassemia types
The various types of thalassemia have specific names related to the severity of the disorder.
Alpha Thalassemias
- Alpha thalassemia silent carrier
- Alpha thalassemia minor, also called alpha thalassemia trait
- Hemoglobin H disease
- Alpha thalassemia major, also called hydrops fetalis
Beta Thalassemias
- Beta thalassemia minor, also called beta thalassemia trait
- Beta thalassemia intermedia
- Beta thalassemia major, also called Cooley’s anemia or beta-zero (ß0) thalassemia
- Beta-plus (ß+) thalassemia
- Mediterranean anemia
Alpha thalassemia
Alpha thalassemia is caused by a deletion or mutation in one or more of the four alpha globin gene copies. The mutation causes a decrease in the production of alpha globin. The more genes that are affected, the less alpha globin is produced by the body. The four different types of alpha thalassemia are classified according to the number of genes affected and include:
- Alpha Thalassemia Silent Carrier (1 gene affected). People who have mutation(s) in only one alpha globin gene are silent carriers. They usually have normal hemoglobin levels and red cell indices but can pass on the affected gene to their children. These individuals have no signs or symptoms and are usually identified only after having a child with thalassemia. The only way to identify a silent carrier is by DNA analysis.
- Alpha Thalassemia Minor, also called Alpha Thalassemia Trait (2 genes affected). People who have alpha thalassemia trait have red blood cells that are smaller (microcytic) and paler (hypochromic) than normal, have a decreased MCV (mean corpuscular volume, a measurement of the average size of a single red blood cell), and have a mild chronic anemia. They generally do not have other signs and sometimes may lack symptoms. This form of anemia does not respond to iron supplements. Diagnosis of alpha thalassemia trait is usually done by exclusion of other causes of microcytic anemia. Confirmatory testing by DNA analysis is available but is not routinely done.
- Hemoglobin H Disease (3 genes affected). With this condition, the large decrease in alpha globin chain production causes an excess of beta chains, which then come together into groups of 4 beta chains, known as Hemoglobin H, which is visible inside red blood cells on a specially stained blood smear. Hb H disease can cause moderate to severe anemia and serious health problems such as an enlarged spleen, bone deformities, and fatigue. The signs and symptoms associated with Hb H disease vary widely. Some individuals are asymptomatic while others have severe anemia, requiring regular medical care. Hemoglobin H disease is found most often in individuals of Southeast Asian or Mediterranean descent.
- Alpha Thalassemia Major (also called Hydrops Fetalis, 4 genes affected). This is the most severe form of alpha thalassemia. In this condition, no alpha globin is produced, therefore, no normal hemoglobin is produced. Fetuses affected by alpha thalassemia major become anemic early during the pregnancy. They retain excess fluids (hydropic) and frequently have enlarged hearts and livers. This diagnosis is frequently made in the last months of pregnancy when a fetal ultrasound indicates a hydropic fetus. There are also risks for the pregnant mother. About 80% of the time, the mother will have “toxemia” (protein in the urine, high blood pressure, swollen ankles and feet) and can develop severe postpartum bleeding (hemorrhage). Fetuses with alpha thalassemia major are usually miscarried, stillborn, or die shortly after birth. In very rare cases, children with alpha thalassemia have survived through in utero blood transfusions and extensive medical care.
Alpha thalassemia is a fairly common blood disorder worldwide. Thousands of infants with Hb Bart syndrome and HbH disease are born each year, particularly in Southeast Asia. Alpha thalassemia also occurs frequently in people from Mediterranean countries, Africa, the Middle East, India, and Central Asia.
Alpha thalassemia causes
You need four genes (two from each parent) to make enough alpha globin protein chains. If one or more of the genes is missing, you’ll have alpha thalassemia trait or disease. This means that your body doesn’t make enough alpha globin protein.
- If you’re only missing one gene, you’re a “silent” carrier. This means you won’t have any signs of illness.
- If you’re missing two genes, you have alpha thalassemia trait (also called alpha thalassemia minor). You may have mild anemia.
- If you’re missing three genes, you likely have hemoglobin H disease (which a blood test can detect). This form of thalassemia causes moderate to severe anemia.
- Very rarely, a baby is missing all four genes. This condition is called alpha thalassemia major or hydrops fetalis. Babies who have hydrops fetalis usually die before or shortly after birth.
Alpha thalassemia typically results from deletions involving the HBA1 and HBA2 genes. Both of these genes provide instructions for making a protein called alpha-globin, which is a component (subunit) of hemoglobin.
People have two copies of the HBA1 gene and two copies of the HBA2 gene in each cell. Each copy is called an allele. For each gene, one allele is inherited from a person’s father, and the other is inherited from a person’s mother. As a result, there are four alleles that produce alpha-globin. The different types of alpha thalassemia result from the loss of some or all of these alleles.
Hb Bart syndrome, the most severe form of alpha thalassemia, results from the loss of all four alpha-globin alleles. HbH disease is caused by a loss of three of the four alpha-globin alleles. In these two conditions, a shortage of alpha-globin prevents cells from making normal hemoglobin. Instead, cells produce abnormal forms of hemoglobin called hemoglobin Bart (Hb Bart) or hemoglobin H (HbH). These abnormal hemoglobin molecules cannot effectively carry oxygen to the body’s tissues. The substitution of Hb Bart or HbH for normal hemoglobin causes anemia and the other serious health problems associated with alpha thalassemia.
Two additional variants of alpha thalassemia are related to a reduced amount of alpha-globin. Because cells still produce some normal hemoglobin, these variants tend to cause few or no health problems. A loss of two of the four alpha-globin alleles results in alpha thalassemia trait. People with alpha thalassemia trait may have unusually small, pale red blood cells and mild anemia. A loss of one alpha-globin allele is found in alpha thalassemia silent carriers. These individuals typically have no thalassemia-related signs or symptoms.
Alpha thalassemia inheritance pattern
The inheritance of alpha thalassemia is complex. Each person inherits two alpha-globin alleles from each parent. If both parents are missing at least one alpha-globin allele, their children are at risk of having Hb Bart syndrome, HbH disease, or alpha thalassemia trait. The precise risk depends on how many alleles are missing and which combination of the HBA1 and HBA2 genes is affected.
Figure 3. Alpha thalassemia inheritance pattern
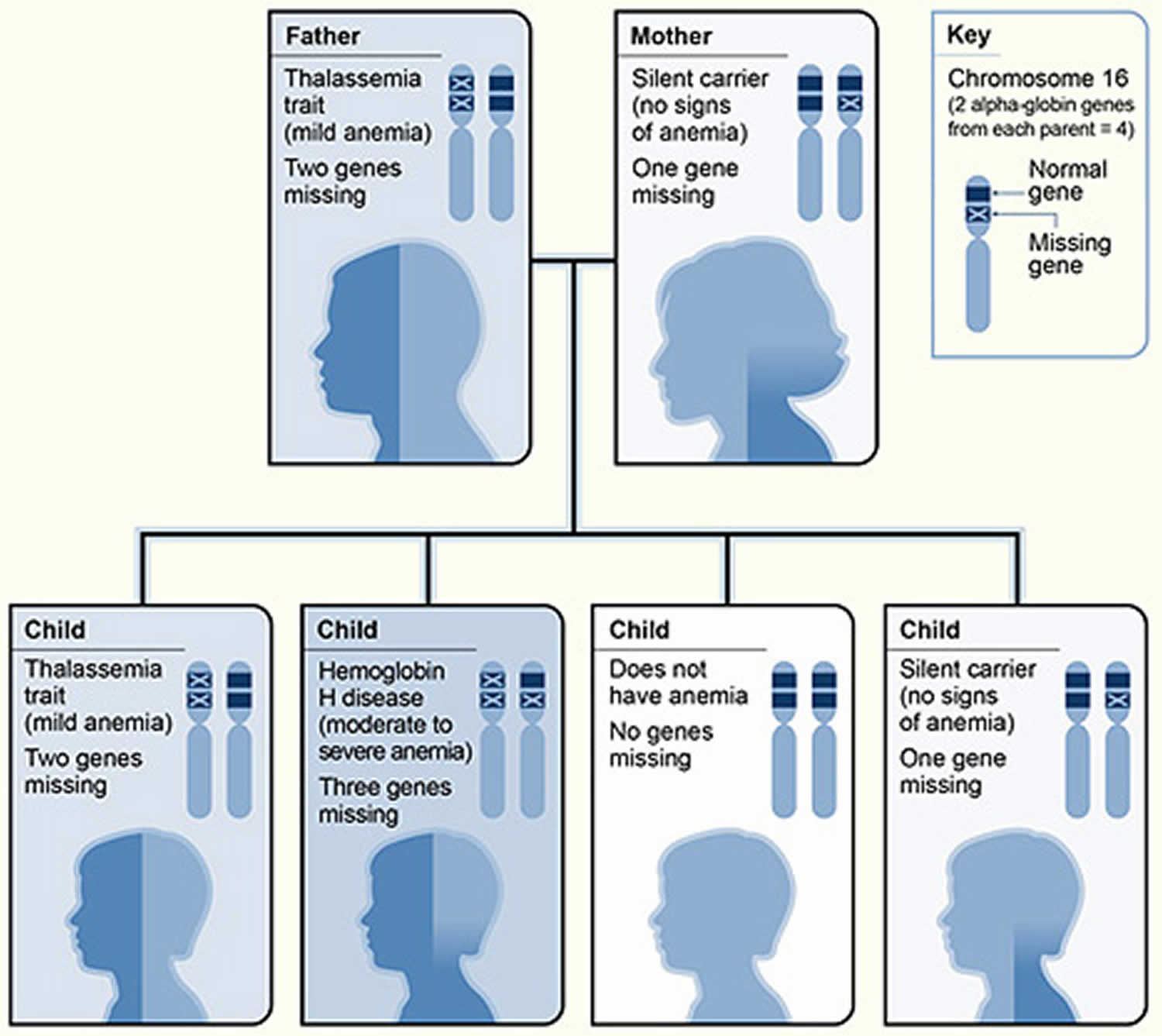
Footnote: The picture shows one example of how alpha thalassemia is inherited. The alpha globin genes are located on chromosome 16. A child inherits four alpha globin genes (two from each parent). In this example, the father is missing two alpha globin genes and the mother is missing one alpha globin gene. Each child has a 25 percent chance of inheriting two missing genes and two normal genes (thalassemia trait), three missing genes and one normal gene (hemoglobin H disease), four normal genes (no anemia), or one missing gene and three normal genes (silent carrier).
[Source 1]Alpha thalassemia symptoms
Alpha thalassemia is a blood disorder that reduces the production of hemoglobin. Hemoglobin is the protein in red blood cells that carries oxygen to cells throughout the body.
In people with the characteristic features of alpha thalassemia, a reduction in the amount of hemoglobin prevents enough oxygen from reaching the body’s tissues. Affected individuals also have a shortage of red blood cells (anemia), which can cause pale skin, weakness, fatigue, and more serious complications.
Two types of alpha thalassemia can cause health problems. The more severe type is known as hemoglobin Bart hydrops fetalis syndrome, which is also called Hb Bart syndrome or alpha thalassemia major. The milder form is called HbH disease.
Hb Bart syndrome is characterized by hydrops fetalis, a condition in which excess fluid builds up in the body before birth. Additional signs and symptoms can include severe anemia, an enlarged liver and spleen (hepatosplenomegaly), heart defects, and abnormalities of the urinary system or genitalia. As a result of these serious health problems, most babies with this condition are stillborn or die soon after birth. Hb Bart syndrome can also cause serious complications for women during pregnancy, including dangerously high blood pressure with swelling (preeclampsia), premature delivery, and abnormal bleeding.
HbH disease causes mild to moderate anemia, hepatosplenomegaly, and yellowing of the eyes and skin (jaundice). Some affected individuals also have bone changes such as overgrowth of the upper jaw and an unusually prominent forehead. The features of HbH disease usually appear in early childhood, and affected individuals typically live into adulthood.
Beta thalassemia
Beta thalassemia is caused by mutations in one or both of the beta globin genes. There have been more than 250 mutations identified, but only about 20 are the most common. The severity of the anemia caused by beta thalassemia depends on which mutations are present and whether there is decreased beta globin production (called beta+ thalassemia) or if production is completely absent (called beta0 thalassemia). The different types of beta thalassemia include:
- Beta Thalassemia Trait or Beta Thalassemia Minor. Individuals with this condition have one normal gene and one with a mutation, causing a mild decrease in beta globin production. They usually have no health problems other than abnormally small red blood cells and a possible mild anemia that will not respond to iron supplements. An individual’s children can inherit this gene.
- Thalassemia Intermedia. In this condition, an affected person has two abnormal genes, causing moderate to severe decrease in beta globin production. These individuals may develop symptoms later than those with thalassemia major (see below) and often with milder symptoms. They rarely require treatment with blood transfusion. The severity of the anemia and health problems experienced depends on the mutation types present. The dividing line between thalassemia intermedia and thalassemia major is the degree of anemia and the number and frequency of blood transfusions required. Those with thalassemia intermedia may need occasional transfusions but do not require them on a regular basis.
- Thalassemia Major or Cooley’s Anemia. This is the most severe form of beta thalassemia. These individuals have two abnormal genes that cause either a severe decrease or complete lack of beta globin production, preventing the production of significant amounts of normal hemoglobin (Hb A). This condition usually appears within the first two years of life and causes life-threatening anemia, poor growth, and skeletal abnormalities during infancy. This anemia requires lifelong regular blood transfusions and considerable ongoing medical care. Over time, these frequent transfusions lead to excessive amounts of iron in the body. Left untreated, this excess iron can deposit in the liver, heart, and other organs and can lead to a premature death from organ failure. Therefore, individuals undergoing transfusion may need chelation therapy to reduce iron overload.
Beta thalassemia is a fairly common blood disorder worldwide. Thousands of infants with beta thalassemia are born each year. Beta thalassemia is found most commonly in populations of Mediterranean, African, and Southeast Asian descent in the U.S. This is likely associated with the incidence of malaria in those regions since thalassemia can increase malaria tolerance. In those regions, thalassemia incidence may be as high as 10%.
Beta thalassemia occurs most frequently in people from Mediterranean countries, North Africa, the Middle East, India, Central Asia, and Southeast Asia.
Other forms of thalassemia occur when a gene for beta thalassemia is inherited in combination with a gene for a hemoglobin variant. The most important of these are:
- Hb E-beta thalassemia. Hb E is one of the most common hemoglobin variants. It is found predominantly in people of Southeast Asian and African descent. If a person inherits one Hb E gene and one beta thalassemia gene, the combination produces Hb E-beta thalassemia, which causes a moderately severe anemia similar to beta thalassemia intermedia.
- Hb S-beta thalassemia or sickle cell-beta thalassemia. Hb S is one of the most well known of the hemoglobin variants. Inheritance of one Hb S gene and one beta thalassemia gene results in Hb S-beta thalassemia. The severity of the condition depends on the amount of beta globin produced by the beta gene. If no beta globin is produced, the clinical picture is similar to sickle cell disease but with even worse baseline anemia. The American College of Medical Genetics advises screening all newborns for hemoglobin S/beta-thalassemia as well as sickle cell anemia. It is also required in all 50 states.
Beta thalassemia causes
You need two genes (one from each parent) to make enough beta globin protein chains. If one or both of these genes are altered, you’ll have beta thalassemia. This means that your body won’t make enough beta globin protein.
- If you have one altered gene, you’re a carrier. This condition is called beta thalassemia trait or beta thalassemia minor. It causes mild anemia.
- If both genes are altered, you’ll have beta thalassemia intermedia or beta thalassemia major (also called Cooley’s anemia). The intermedia form of the disorder causes moderate anemia. The major form causes severe anemia.
Mutations in the HBB gene cause beta thalassemia. The HBB gene provides instructions for making a protein called beta-globin. Beta-globin is a component (subunit) of hemoglobin. Hemoglobin consists of four protein subunits, typically two subunits of beta-globin and two subunits of another protein called alpha-globin.
Some mutations in the HBB gene prevent the production of any beta-globin. The absence of beta-globin is referred to as beta-zero (B0) thalassemia. Other HBB gene mutations allow some beta-globin to be produced but in reduced amounts. A reduced amount of beta-globin is called beta-plus (B+) thalassemia. Having either B0 or B+ thalassemia does not necessarily predict disease severity, however; people with both types have been diagnosed with thalassemia major and thalassemia intermedia.
A lack of beta-globin leads to a reduced amount of functional hemoglobin. Without sufficient hemoglobin, red blood cells do not develop normally, causing a shortage of mature red blood cells. The low number of mature red blood cells leads to anemia and other associated health problems in people with beta thalassemia.
Beta thalassemia inheritance pattern
Thalassemia major and thalassemia intermedia are inherited in an autosomal recessive pattern, which means both copies of the HBB gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. Sometimes, however, people with only one HBB gene mutation in each cell develop mild anemia. These mildly affected people are said to have thalassemia minor.
In a small percentage of families, the HBB gene mutation is inherited in an autosomal dominant manner. In these cases, one copy of the altered gene in each cell is sufficient to cause the signs and symptoms of beta thalassemia.
Figure 4. Beta thalassemia inheritance pattern
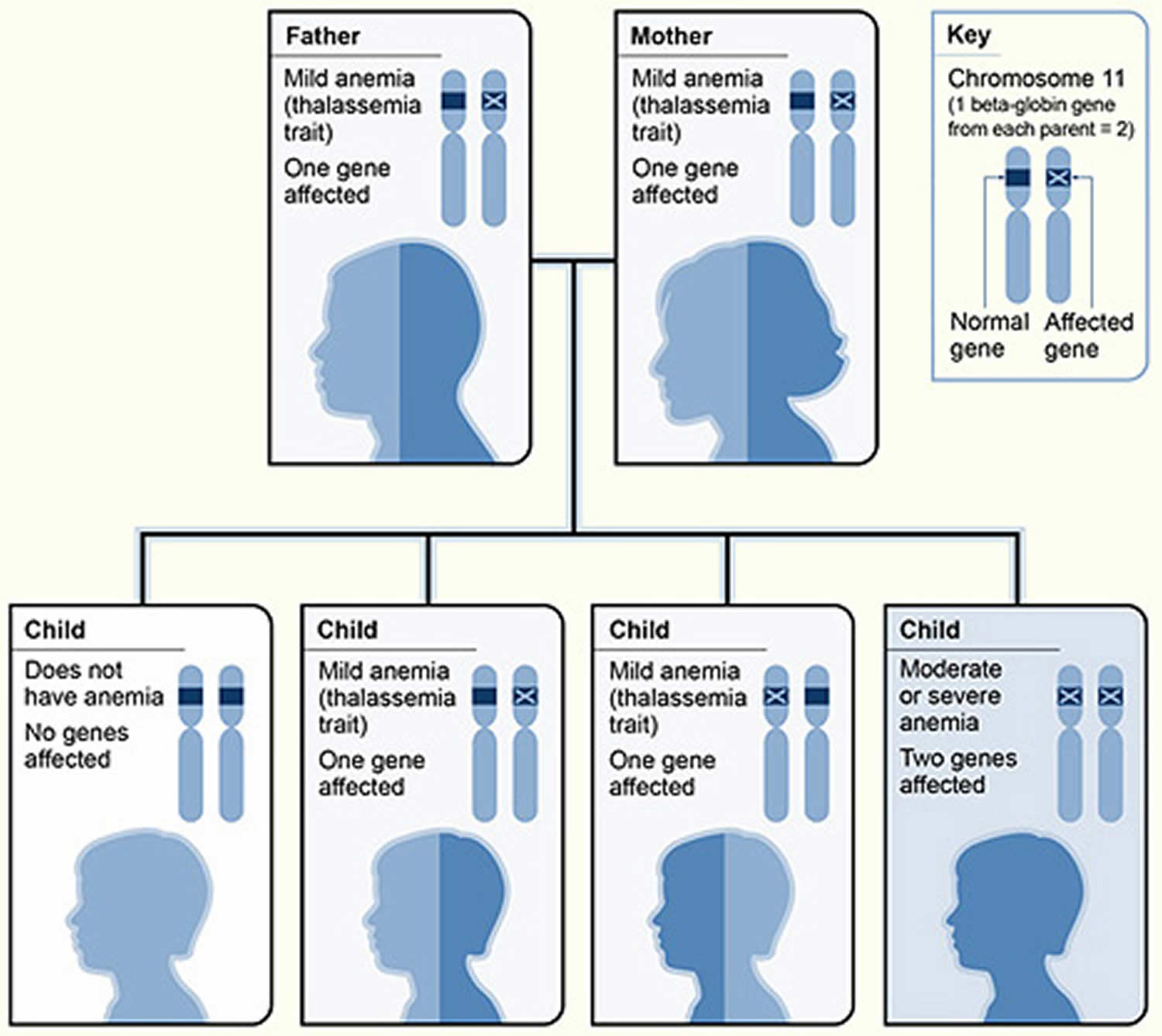
Footnote: The picture shows one example of how beta thalassemia is inherited. The beta globin gene is located on chromosome 11. A child inherits two beta globin genes (one from each parent). In this example, each parent has one altered beta globin gene. Each child has a 25 percent chance of inheriting two normal genes (no anemia), a 50 percent chance of inheriting one altered gene and one normal gene (beta thalassemia trait), or a 25 percent chance of inheriting two altered genes (beta thalassemia major).
[Source 1]Beta thalassemia symptoms
Beta thalassemia is a blood disorder that reduces the production of hemoglobin. Hemoglobin is the iron-containing protein in red blood cells that carries oxygen to cells throughout the body.
In people with beta thalassemia, low levels of hemoglobin lead to a lack of oxygen in many parts of the body. Affected individuals also have a shortage of red blood cells (anemia), which can cause pale skin, weakness, fatigue, and more serious complications. People with beta thalassemia are at an increased risk of developing abnormal blood clots.
Beta thalassemia is classified into two types depending on the severity of symptoms: thalassemia major (also known as Cooley’s anemia) and thalassemia intermedia. Of the two types, thalassemia major is more severe.
The signs and symptoms of thalassemia major appear within the first 2 years of life. Children develop life-threatening anemia. They do not gain weight and grow at the expected rate (failure to thrive) and may develop yellowing of the skin and whites of the eyes (jaundice). Affected individuals may have an enlarged spleen, liver, and heart, and their bones may be misshapen. Some adolescents with thalassemia major experience delayed puberty. Many people with thalassemia major have such severe symptoms that they need frequent blood transfusions to replenish their red blood cell supply. Over time, an influx of iron-containing hemoglobin from chronic blood transfusions can lead to a buildup of iron in the body, resulting in liver, heart, and hormone problems.
Thalassemia intermedia is milder than thalassemia major. The signs and symptoms of thalassemia intermedia appear in early childhood or later in life. Affected individuals have mild to moderate anemia and may also have slow growth and bone abnormalities.
Thalassemia symptoms
A lack of oxygen in the bloodstream causes the signs and symptoms of thalassemias. The lack of oxygen occurs because the body doesn’t make enough healthy red blood cells and hemoglobin. The severity of symptoms depends on the severity of the disorder.
No Symptoms
Alpha thalassemia silent carriers generally have no signs or symptoms of the disorder. The lack of alpha globin protein is so minor that the body’s hemoglobin works normally.
Mild Anemia
People who have alpha or beta thalassemia trait can have mild anemia. However, many people who have these types of thalassemia have no signs or symptoms.
Mild anemia can make you feel tired. Mild anemia caused by alpha thalassemia trait might be mistaken for iron-deficiency anemia.
Mild to Moderate Anemia and Other Signs and Symptoms
People who have beta thalassemia intermedia have mild to moderate anemia. They also may have other health problems, such as:
- Slowed growth and delayed puberty. Anemia can slow down a child’s growth and development.
- Bone problems. Thalassemia may cause bone marrow to expand. Bone marrow is the spongy substance inside bones that makes blood cells. When bone marrow expands, the bones become wider than normal. They may become brittle and break easily.
- An enlarged spleen. The spleen is an organ that helps your body fight infection and remove unwanted material. When a person has thalassemia, the spleen has to work very hard. As a result, the spleen becomes larger than normal. This makes anemia worse. If the spleen becomes too large, it must be removed.
Severe Anemia and Other Signs and Symptoms
People who have hemoglobin H disease or beta thalassemia major (also called Cooley’s anemia) have severe thalassemia. Signs and symptoms usually occur within the first 2 years of life. They may include severe anemia and other health problems, such as:
- A pale and listless appearance
- Poor appetite
- Dark urine (a sign that red blood cells are breaking down)
- Slowed growth and delayed puberty
- Jaundice (a yellowish color of the skin or whites of the eyes)
- An enlarged spleen, liver, or heart
- Bone problems (especially with bones in the face)
Thalassemias complications
Better treatments now allow people who have moderate and severe thalassemias to live much longer. As a result, these people must cope with complications of these disorders that occur over time.
Heart and Liver Diseases
Regular blood transfusions are a standard treatment for thalassemias. Transfusions can cause iron to build up in the blood (iron overload). This can damage organs and tissues, especially the heart and liver.
Heart disease caused by iron overload is the main cause of death in people who have thalassemias. Heart disease includes heart failure, arrhythmias (irregular heartbeats), and heart attack.
An enlarged spleen
Your spleen helps your body fight infections and filters out damaged blood cells. If you have thalassemia, your spleen may have to work harder than normal, which can cause it to enlarge. If your spleen becomes too large, it may have to be removed.
Infections
Among people who have thalassemias, infections are a key cause of illness and the second most common cause of death. People who have had their spleens removed are at even higher risk because they no longer have this infection-fighting organ.
Osteoporosis
Thalassemia can cause bone deformities in the face and skull. Many people who have thalassemias have bone problems, including osteoporosis. This is a condition in which bones are weak and brittle and break easily.
Too much iron in your blood
This can cause damage to the heart, liver, or endocrine system (glands in the body that make hormones, like the thyroid gland and adrenal glands).
Thalassemia causes
Your body makes three types of blood cells: red blood cells, white blood cells, and platelets. Red blood cells contain hemoglobin, an iron-rich protein that carries oxygen from your lungs to all parts of your body. Hemoglobin also carries carbon dioxide (a waste gas) from your body to your lungs, where it’s exhaled.
Hemoglobin has two kinds of protein chains: alpha globin and beta globin. If your body doesn’t make enough of these protein chains or they’re abnormal, red blood cells won’t form correctly or carry enough oxygen. Your body won’t work well if your red blood cells don’t make enough healthy hemoglobin.
Genes control how the body makes hemoglobin protein chains. When these genes are missing or altered, thalassemias occur.
Thalassemias are inherited disorders—that is, they’re passed from parents to children through genes. People who inherit faulty hemoglobin genes from one parent but normal genes from the other are called carriers. Carriers often have no signs of illness other than mild anemia. However, they can pass the faulty genes on to their children.
People who have moderate to severe forms of thalassemia have inherited faulty genes from both parents.
Alpha Thalassemias
You need four genes (two from each parent) to make enough alpha globin protein chains. If one or more of the genes is missing, you’ll have alpha thalassemia trait or disease. This means that your body doesn’t make enough alpha globin protein.
- If you’re only missing one gene, you’re a “silent” carrier. This means you won’t have any signs of illness.
- If you’re missing two genes, you have alpha thalassemia trait (also called alpha thalassemia minor). You may have mild anemia.
- If you’re missing three genes, you likely have hemoglobin H disease (which a blood test can detect). This form of thalassemia causes moderate to severe anemia.
Very rarely, a baby is missing all four genes. This condition is called alpha thalassemia major or hydrops fetalis. Babies who have hydrops fetalis usually die before or shortly after birth.
Beta Thalassemias
You need two genes (one from each parent) to make enough beta globin protein chains. If one or both of these genes are altered, you’ll have beta thalassemia. This means that your body won’t make enough beta globin protein.
- If you have one altered gene, you’re a carrier. This condition is called beta thalassemia trait or beta thalassemia minor. It causes mild anemia.
- If both genes are altered, you’ll have beta thalassemia intermedia or beta thalassemia major (also called Cooley’s anemia). The intermedia form of the disorder causes moderate anemia. The major form causes severe anemia.
Risk factors for developing thalassemia
Family history and ancestry are the two risk factors for thalassemias.
Family History
- Thalassemias are inherited—that is, the genes for the disorders are passed from parents to their children. If your parents have missing or altered hemoglobin-making genes, you may have thalassemia.
Ancestry
Thalassemias occur most often among people of Italian, Greek, Middle Eastern, Southern Asian, and African descent.
- Alpha thalassemia most often affects people who are of Southeast Asian, Indian, Chinese, or Filipino descent.
- Beta thalassemia most often affects people who are of Mediterranean (Greek, Italian and Middle Eastern), Asian, or African descent.
Thalassemia prevention
You can’t prevent thalassemias because they’re inherited (passed from parents to children through genes). However, prenatal tests can detect these blood disorders before birth.
Family genetic studies may help find out whether people have missing or altered hemoglobin genes that cause thalassemias
If you know of family members who have thalassemias and you’re thinking of having children, consider talking with your doctor and a genetic counselor. They can help determine your risk for passing the disorder to your children.
Thalassemia diagnosis
Doctors diagnose thalassemias using blood tests, including a complete blood count (CBC) and special hemoglobin tests.
- A complete blood count measures the amount of hemoglobin and the different kinds of blood cells, such as red blood cells, in a sample of blood. People who have thalassemias have fewer healthy red blood cells and less hemoglobin than normal in their blood. People who have alpha or beta thalassemia trait may have red blood cells that are smaller than normal.
- Hemoglobin tests measure the types of hemoglobin in a blood sample. People who have thalassemias have problems with the alpha or beta globin protein chains of hemoglobin.
Moderate and severe thalassemias usually are diagnosed in early childhood. This is because signs and symptoms, including severe anemia, often occur within the first 2 years of life.
People who have milder forms of thalassemia might be diagnosed after a routine blood test shows they have anemia. Doctors might suspect thalassemia if a person has anemia and is a member of an ethnic group that’s at increased risk for thalassemias.
Doctors also test the amount of iron in the blood to find out whether the anemia is due to iron deficiency or thalassemia. Iron-deficiency anemia occurs if the body doesn’t have enough iron to make hemoglobin. The anemia in thalassemia occurs because of a problem with either the alpha globin or beta globin chains of hemoglobin, not because of a lack of iron.
Because thalassemias are passed from parents to children through genes, family genetic studies also can help diagnose the disorder. These studies involve taking a family medical history and doing blood tests on family members. The tests will show whether any family members have missing or altered hemoglobin genes.
If you know of family members who have thalassemias and you’re thinking of having children, consider talking with your doctor and a genetic counselor. They can help determine your risk for passing the disorder to your children.
If you’re expecting a baby and you and your partner are thalassemia carriers, you may want to consider prenatal testing.
Prenatal testing involves taking a sample of amniotic fluid or tissue from the placenta. (Amniotic fluid is the fluid in the sac surrounding a growing embryo. The placenta is the organ that attaches the umbilical cord to the mother’s womb.) Tests done on the fluid or tissue can show whether your baby has thalassemia and how severe it might be.
Thalassemia test
Several laboratory tests may be used to help detect and diagnose thalassemia:
Complete blood count (CBC). The complete blood count is an evaluation of the cells in the blood. Among other things, the CBC determines the number of red blood cells present and how much hemoglobin is in them. It evaluates the size and shape of the red blood cells present, reported as the red cell indices. These include the mean corpuscular volume (MCV), a measurement of the size of the red blood cells. A low MCV (mean corpuscular volume) is often the first indication of thalassemia. If the MCV is low and iron deficiency has been ruled out as a cause, thalassemia should be considered.
Blood smear (also called peripheral smear and manual differential). In this test, a trained laboratory professional examines a thin layer of blood that is treated with a special stain, on a slide, under a microscope. The number and type of white blood cells, red blood cells, and platelets are evaluated to see if they are normal and mature. With thalassemia, the red blood cells often appear smaller than normal (microcytic, low MCV). Red cells may also:
- Be paler than normal (hypochromic)
- Vary in size and shape (anisocytosis and poikilocytosis)
- Be nucleated (normal, mature red blood cells do not have a nucleus)
- Have uneven hemoglobin distribution (producing “target cells” that look like a bull’s-eye under the microscope)
The greater the percentage of abnormal-looking red blood cells, the greater the likelihood of an underlying disorder and decreased ability of the red blood cells to carry oxygen.
Iron studies. These may include: iron, ferritin, unsaturated iron binding capacity (UIBC), total iron binding capacity (TIBC), and percent saturation of transferrin. These tests measure different aspects of the body’s iron storage and usage. The tests are ordered to help determine whether an iron deficiency is the cause of a person’s anemia. One or more of them may also be ordered to help monitor the degree of iron overload in an individual with thalassemia.
- Alpha thalassemia is sometimes confused with iron deficiency anemia because both disorders have smaller than usual (microcytic) red blood cells. If someone has thalassemia, his or her iron levels are not expected to be low. Iron therapy will not help people with alpha thalassemia and may lead to iron overload, which can cause organ damage over time.
- Erythrocyte porphyrin tests may be used to distinguish an unclear beta thalassemia minor diagnosis from iron deficiency or lead poisoning. Individuals with beta thalassemia will have normal porphyrin levels, but those with the latter conditions will have elevated porphyrin.
Hemoglobinopathy (Hb) evaluation (hemoglobin electrophoresis). This test assess the type and relative amounts of hemoglobin present in red blood cells. Hemoglobin A (Hb A), composed of both alpha and beta globin, is the type of hemoglobin that normally makes up 95% to 98% of hemoglobin in adults. Hemoglobin A2 (HbA2) is usually 2% to 3% of hemoglobin in adults, while hemoglobin F usually makes up less than 2%.
Beta thalassemia upsets the balance of beta and alpha hemoglobin chain formation and causes an increase in those minor hemoglobin components. So individuals with the beta thalassemia major usually have larger percentages of Hb F. Those with beta thalassemia minor usually have elevated fraction of Hb A2. Hb H is a less common form of hemoglobin that may be seen in some cases of alpha thalassemia. Hb S is the hemoglobin more common in people with sickle cell disease.
Hemoglobinopathy (Hb) evaluations are used for state-mandated newborn hemoglobin screening and prenatal screening when parents are at high risk for hemoglobin abnormalities.
DNA analysis. These tests are used to help confirm mutations in the alpha and beta globin-producing genes. DNA testing is not routinely done but can be used to help diagnose thalassemia and to determine carrier status, if indicated.
- For beta thalassemia, the hemoglobin beta gene, HBB, may be analyzed or sequenced to confirm the presence of thalassemia-causing mutations. Genetic tests may also be given for other HBB mutations such as Hb S mutation, which is associated with sickle cell disease. More than 250 mutations have been associated with beta thalassemia, though some cause no signs or symptoms. However, others decrease the amount of beta globin production and some prevent it completely. The presence of one of those mutations confirms a diagnosis of beta thalassemia.
- The primary molecular test available for alpha thalassemia detects common mutations (e.g., deletions) in the two alpha genes HBA1 and HBA2. Each person has two copies of each of these genes, called alleles, in their cells, one from their mother and one from their father. These alleles govern alpha globin production and if mutations lead to functional loss of one or more of alpha genes, alpha thalassemia occurs.
Since having relatives who carry mutations for thalassemia increases a person’s risk of carrying the same mutant gene, family studies may be done to evaluate carrier status and the types of mutations present in other family members if deemed necessary by a healthcare practitioner.
Genetic testing of amniotic fluid is used in the rare instances a fetus is at increased risk for thalassemia. This is especially important if both parents likely carry a mutation because that increases the risk that their child may inherit a combination of abnormal genes, causing a more severe form of thalassemia.
Thalassemia treatment
Treatments for thalassemias depend on the type and severity of the disorder. People who are carriers or who have alpha or beta thalassemia trait have mild or no symptoms. They’ll likely need little or no treatment.
Most individuals with mild thalassemia traits require no treatment. They may want to consider genetic counseling, however, because they may pass the mutant gene on to their children.
People with hemoglobin H disease or beta thalassemia intermedia will experience variable amounts of anemia throughout their life. They can live relatively normal lives but will require regular monitoring and may occasionally need blood transfusion. Folic acid supplementation is often given, but iron supplementation is not recommended.
Those with beta thalassemia major will usually require regular blood transfusions, as frequently as every few weeks, and chelation therapy to remove iron throughout their life. These transfusions help maintain hemoglobin at a high enough level to provide oxygen to the body and prevent growth abnormalities and organ damage. Frequent transfusions, however, can raise body iron to toxic levels, resulting in deposits of iron in the liver, heart, and other organs. Regular iron chelation therapy is used to help decrease iron in the body.
Bone marrow transplant known as hematopoietic stem cell transplantation can also be used for treatment of beta thalassemia major.
Fetuses with alpha thalassemia major are usually miscarried, stillborn, or die shortly after birth. Experimental treatments, such as fetal blood transfusions and even fetal marrow transplant, have been successful in a very few cases in bringing a baby to term.
Standard Treatments
Doctors use three standard treatments for moderate and severe forms of thalassemia. These treatments include blood transfusions, iron chelation therapy, and folic acid supplements. Other treatments have been developed or are being tested, but they’re used much less often.
Blood Transfusions
Transfusions of red blood cells are the main treatment for people who have moderate or severe thalassemias. This treatment gives you healthy red blood cells with normal hemoglobin.
Blood transfusion is the mainstay of care for individuals with thalassemia major and many with intermedia. The purpose of blood transfusion is twofold: to improve the anemia and to suppress the ineffective erythropoiesis. Chronic transfusions prevent most of the serious growth, skeletal, and neurological complications of thalassemia major. However, once started, the transfusion-related complications become a major source of morbidity. Standards must be developed and maintained to ensure a safe and rational approach to the use of blood transfusions in the management of these rare disorders.
During a blood transfusion, a needle is used to insert an intravenous (IV) line into one of your blood vessels. Through this line, you receive healthy blood. The procedure usually takes 1 to 4 hours.
Red blood cells live only for about 120 days. So, you may need repeated transfusions to maintain a healthy supply of red blood cells.
If you have hemoglobin H disease or beta thalassemia intermedia, you may need blood transfusions on occasion. For example, you may have transfusions when you have an infection or other illness, or when your anemia is severe enough to cause tiredness.
If you have beta thalassemia major (Cooley’s anemia), you’ll likely need regular blood transfusions (often every 2 to 4 weeks). These transfusions will help you maintain normal hemoglobin and red blood cell levels.
Blood transfusions allow you to feel better, enjoy normal activities, and live into adulthood. This treatment is lifesaving, but it’s expensive and carries a risk of transmitting infections and viruses (for example, hepatitis). However, the risk is very low in the United States because of careful blood screening.
Iron Chelation Therapy
The hemoglobin in red blood cells is an iron-rich protein. Thus, regular blood transfusions can lead to a buildup of iron in the blood. This condition is called iron overload. It damages the liver, heart, and other parts of the body.
To prevent this damage, doctors use iron chelation therapy to remove excess iron from the body. Two medicines are used for iron chelation therapy.
- Deferoxamine is a liquid medicine that’s given slowly under the skin, usually with a small portable pump used overnight. This therapy takes time and can be mildly painful. Side effects include problems with vision and hearing.
- Deferasirox is a pill taken once daily. Side effects include headache, nausea (feeling sick to the stomach), vomiting, diarrhea, joint pain, and tiredness.
Folic Acid Supplements
Folic acid is a B vitamin that helps build healthy red blood cells. Your doctor may recommend folic acid supplements in addition to treatment with blood transfusions and/or iron chelation therapy.
Other Treatments
Other treatments for thalassemias have been developed or are being tested, but they’re used much less often.
Blood and Marrow Stem Cell Transplant
A blood and marrow stem cell transplant replaces faulty stem cells with healthy ones from another person (a donor). Stem cells are the cells inside bone marrow that make red blood cells and other types of blood cells.
A stem cell transplant is the only treatment that can cure thalassemia. But only a small number of people who have severe thalassemias are able to find a good donor match and have the risky procedure.
Possible Future Treatments
Researchers are working to find new treatments for thalassemias. For example, it might be possible someday to insert a normal hemoglobin gene into stem cells in bone marrow. This will allow people who have thalassemias to make their own healthy red blood cells and hemoglobin.
Researchers also are studying ways to trigger a person’s ability to make fetal hemoglobin after birth. This type of hemoglobin is found in fetuses and newborns. After birth, the body switches to making adult hemoglobin. Making more fetal hemoglobin might make up for the lack of healthy adult hemoglobin.
Treating complications
Better treatments now allow people who have moderate and severe thalassemias to live longer. As a result, these people must cope with complications that occur over time.
An important part of managing thalassemias is treating complications. Treatment might be needed for heart or liver diseases, infections, osteoporosis, and other health problems.
Living with thalassemia
Survival and quality of life have improved for people who have moderate or severe thalassemias. This is because:
- More people are able to get blood transfusions now.
- Blood screening has reduced the number of infections from blood transfusions. Also, treatments for other kinds of infections have improved.
- Iron chelation treatments are available that are easier for some people to take.
- Some people have been cured through blood and marrow stem cell transplants.
Living with thalassemia can be challenging, but several approaches can help you cope.
Follow your treatment plan
Following the treatment plan your doctor gives you is important. For example, get blood transfusions as your doctor recommends, and take your iron chelation medicine as prescribed.
Iron chelation treatment can take time and be mildly painful. However, don’t stop taking your medicine. The leading cause of death among people who have thalassemias is heart disease caused by iron overload. Iron buildup can damage your heart, liver, and other organs.
Several chelation treatments are now available, including injections and pills. Your doctor will talk with you about which treatment is best for you.
Take folic acid supplements if your doctor prescribes them. Folic acid is a B vitamin that helps build healthy red blood cells. Also, talk with your doctor about whether you need other vitamin or mineral supplements, such as vitamins A, C, or D or selenium.
Get ongoing medical care
Keep your scheduled medical appointments, and get any tests that your doctor recommends.
These tests may include:
- Monthly complete blood counts and tests for blood iron levels every 3 months
- Yearly tests for heart function, liver function, and viral infections (for example, hepatitis B and C and HIV)
- Yearly tests to check for iron buildup in your liver
- Yearly vision and hearing tests
- Regular checkups to make sure blood transfusions are working
- Other tests as needed (such as lung function tests, genetic tests, and tests to match your tissues with a possible donor if a stem cell transplant is being considered)
Children who have thalassemias should receive yearly checkups to monitor their growth and development. The checkups include a physical exam, including a height and weight check, and any necessary tests.
Take steps to stay healthy
Take steps to stay as healthy as possible. Follow a healthy eating plan and your doctor’s instructions for taking iron supplements.
Get vaccinations as needed, especially if you’ve had your spleen removed. You may need vaccines for the flu, pneumonia, hepatitis B, and meningitis. Your doctor will advise you about which vaccines you need.
Watch for signs of infection (such as a fever) and take steps to lower your risk for infection (especially if you’ve had your spleen removed). For example:
- Wash your hands often.
- Avoid crowds during cold and flu season.
- Keep the skin around the site where you get blood transfusions as clean as possible.
- Call your doctor if a fever develops.
Emotional issues and support
If you or your child has thalassemia, you may have fear, anxiety, depression, or stress. Talk about how you feel with your health care team. Talking to a professional counselor also can help. If you’re very depressed, your doctor may recommend medicines or other treatments that can improve your quality of life.
Joining a patient support group may help you adjust to living with thalassemia. You can see how other people who have the same symptoms have coped with them. Talk with your doctor about local support groups or check with an area medical center.
Support from family and friends also can help relieve stress and anxiety. Let your loved ones know how you feel and what they can do to help you.
Some teens and young adults who have thalassemias may have a hard time moving from pediatric care to adult care. Doctors and other health professionals who care for these children might not be familiar with adult issues related to the disorder, such as certain complications.
Also, it might be hard for adults who have thalassemias to find doctors who specialize in treating the disorder. Ask your child’s doctor to help you find a doctor who can care for your child when the time comes to make the switch. Planning and good communication can help this move go smoothly.
References




{kind=link}