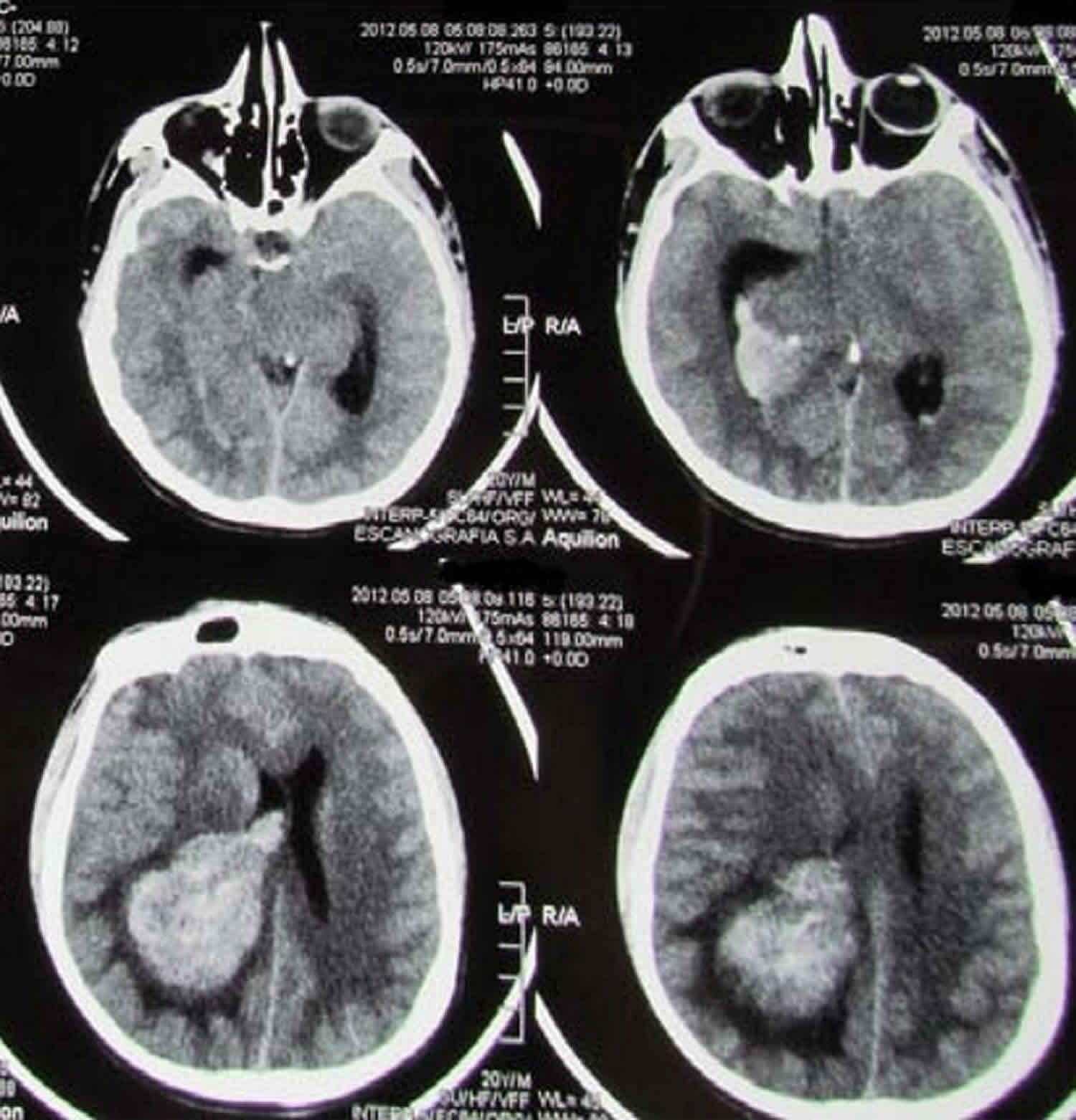
Turcot syndrome
Turcot syndrome is a rare genetic disorder clinically characterized by the association of benign growths (colorectal polyposis or adenomatous polyps) in the mucous lining of the gastrointestinal tract with tumors of the central nervous system (primary brain tumor) 1. Turcot syndrome is commonly seen in association with two other syndromes, namely, hereditary nonpolyposis colorectal cancer (Lynch syndrome or type 1 Turcot syndrome) and familial adenomatous polyposis (type 2 Turcot syndrome). Hereditary nonpolyposis colorectal cancer (Lynch syndrome or type 1 Turcot syndrome) is associated with germ line mutations in deoxyribonucleic acid (DNA) mismatch repair (MMR) genes and familial adenomatous polyposis (type 2 Turcot syndrome) with germ line mutations in adenomatous polyposis coli (APC) gene 2. Turcot syndrome is characterized by an increased risk of colorectal cancer, and an increased risk of brain cancer. Turcot syndrome also has an increased risk for early onset of other tumors including of endometrium, stomach, small intestine, hepatobiliary system, kidney, ureter and ovary.
If a brain tumor arises in a patient with either hereditary nonpolyposis colorectal cancer (Lynch syndrome) or familial adenomatous polyposis (FAP), then the patient is considered to have Turcot syndrome type 1 or Turcot syndrome type 2, respectively, although the association with either of these inherited mutations is not necessary 3.
The type of brain tumor generally depends on whether the Turcot syndrome is more similar to Lynch syndrome (hereditary nonpolyposis colorectal cancer) or more similar to familial adenomatous polyposis (FAP). The 2 most common types of brain tumors in Turcot syndrome are 4:
- Glioblastoma. This type of brain tumor is a very aggressive form of astrocytoma that is commonly found in families who have features of Lynch syndrome.
- Medulloblastoma. This type of brain tumor begins in the cerebellum, the back of the brain. Medulloblastoma most often occurs in children and is commonly found in families who have features of familial adenomatous polyposis.
Turcot syndrome symptoms associated with colorectal polyp formation may include diarrhea, bleeding from the end portion of the large intestine (rectum), fatigue, abdominal pain, and weight loss. Affected individuals may also experience neurological symptoms, depending upon the type, size and location of the associated brain tumor.
Some researchers believe that Turcot syndrome is a variant of familial adenomatous polyposis. Others believe that it is a separate disorder. There have been attempts to reclassify Turcot syndrome as mismatch repair cancer syndrome (MMRCS) or rename it as brain-tumor polyposis syndrome 1 and 2 (BTPS1 and BTPS2) 5. These syndromes suggest a focus on defining the syndrome by the genetic presentation, involving findings in addition to colorectal cancer and primary brain tumors, such as cafe-au-lait spots, hematologic malignancies, among others. However, the original definition of Turcot syndrome is restricted to the phenotypic presentation of solely colorectal cancer plus primary brain tumors. Thus, it is now common to see Turcot syndrome1 and Turcot syndrome type 2, meaning colorectal cancer with primary brain tumor secondary to either MMR gene mutations or APC gene mutations, respectively. Brain-tumor polyposis syndrome is commonly used as a synonym to Turcot syndrome today, though brain-tumor polyposis syndrome is an expanded version which includes additional symptoms mentioned in this paper.
The exact cause of Turcot syndrome is not known 6. Turcot syndrome affects males and females in equal numbers. Approximately 150 cases have been reported in the medical literature 6.
Turcot syndrome causes
Turcot syndrome type 1, sometimes called “true” Turcot syndrome, is inherited as an autosomal recessive trait. Recessive genetic disorders occur when an individual inherits the same abnormal gene for the same trait from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%.
Researchers believe that mutations to two DNA mismatch repair (MMR) genes (i.e., MLH1 and PMS2) may be responsible for development of this form of Turcot syndrome. MLH1 is located on the short arm (p) of chromosome 3 at band number 21.3. PMS2 is located on the short arm (p) of chromosome 7 at band number 22.
Chromosomes, which are present in the nucleus of human cells, carry the genetic information for each individual. Pairs of human chromosomes are numbered from 1 through 22, and an additional 23rd pair of sex chromosomes which include one X and one Y chromosome in males and two X chromosomes in females. Each chromosome has a short arm designated “p” and a long arm designated “q”. Chromosomes are further sub-divided into many bands that are numbered. For example, “chromosome 3p21.3” refers to band 21 on the short arm of chromosome 3. The numbered bands specify the location of the thousands of genes that are present on each chromosome.
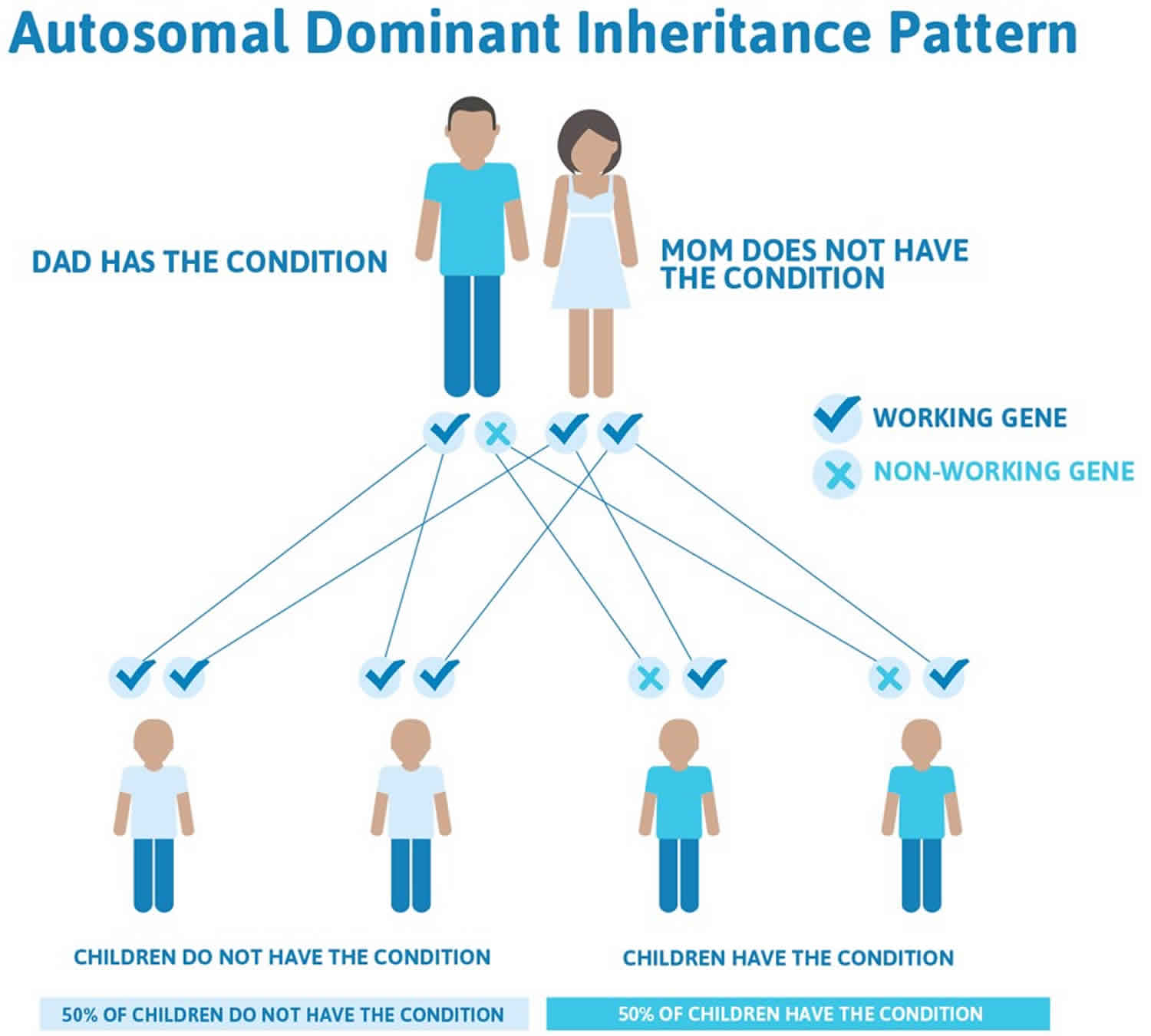
The second type of Turcot syndrome, which is associated with familial adenomatous polyposis, is inherited as an autosomal dominant trait. Dominant genetic disorders occur when only a single copy of an abnormal gene is necessary for the appearance of the disease. The abnormal gene can be inherited from either parent, or can be the result of a new mutation (gene change) in the affected individual. The risk of passing the abnormal gene from affected parent to offspring is 50% for each pregnancy regardless of the sex of the resulting child.
This form of Turcot syndrome results from mutations to the APC gene (for “adenomatous polyposis coli”), which has been mapped to the long arm (q) of chromosome 5 (5q21-q22). Evidence suggests that the APC gene functions as a tumor suppressor gene. Mutations to the APC gene are associated with familial adenomatous polyposis and Gardner syndrome.
Turcot syndrome inheritance
Recent research indicates that one type of Turcot syndrome is inherited as an autosomal recessive trait and type 2 Turcot syndrome as an autosomal dominant trait.
Figure 1. Turcot syndrome autosomal recessive inheritance pattern
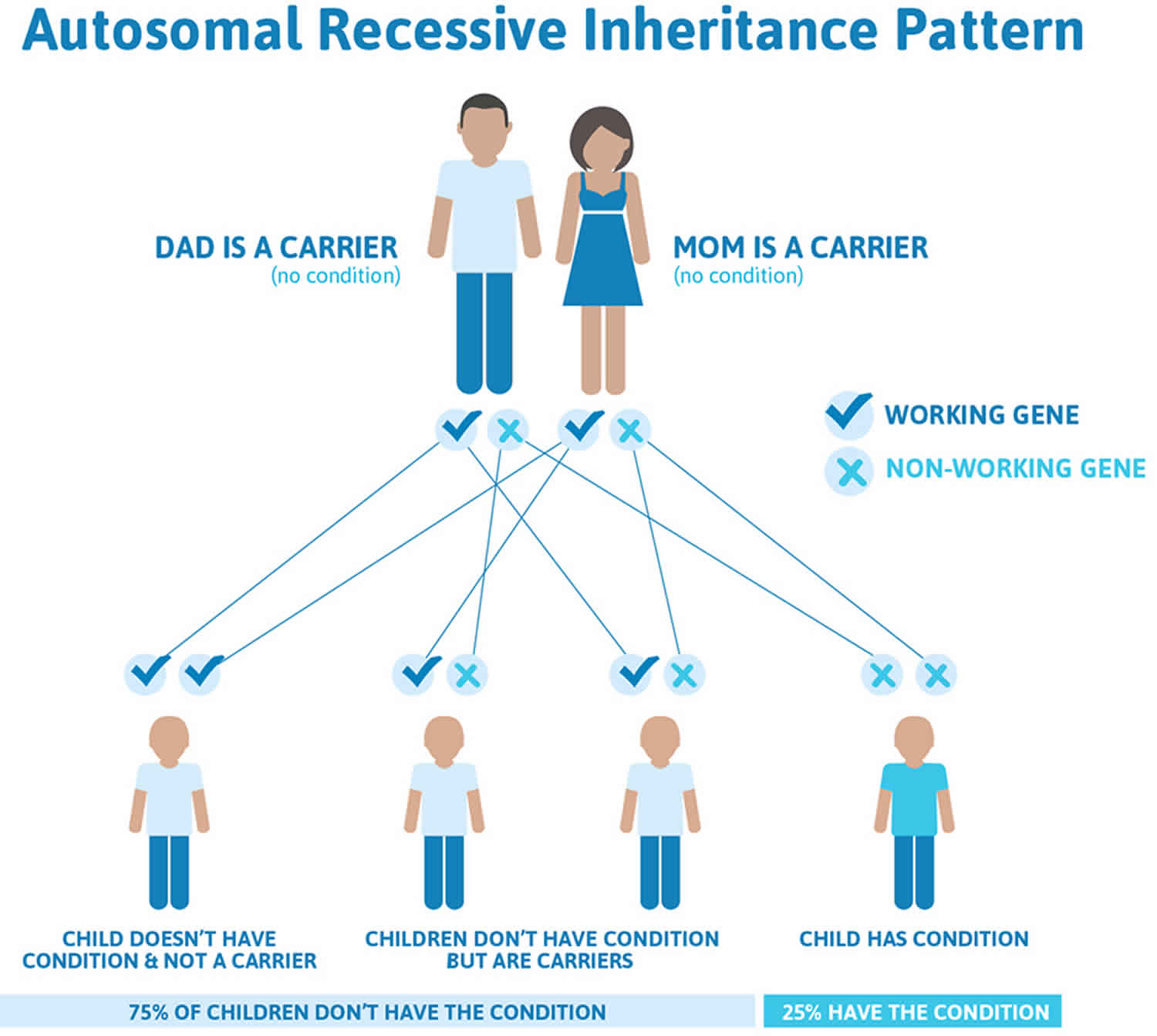
Figure 2. Turcot syndrome autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Turcot syndrome pathophysiology
Turcot syndrome can arise from mutations in the MMR genes (Turcot syndrome type 1, associated with hereditary nonpolyposis colorectal cancer) or the APC genes (Turcot syndrome type 2, associated with familial adenomatous polyposis). Mutations of either gene can result in colorectal cancer and brain tumors; most commonly, glioblastomas and medulloblastomas 7. Glioblastomas are associated with MMR gene mutations, specifically, in the hMLH1 DNA mismatch repair gene. Medulloblastomas are associated with APC gene mutations 7. The mutations that result in the development of Turcot syndrome have generally been shown to be biallelic mutations, compared to the single allele, or heterozygous mutation manifestations in hereditary nonpolyposis colorectal cancer and familial adenomatous polyposis 8.
A general summary of the pathological process is that tumor suppressor gene deletion leads normal colonic mucosa to transform into invasive carcinoma. Normal mucosa develops into polyps, then the polyps continue to develop into invasive cancer. There are several different types of polyps including non-neoplastic hamartomas, hyperplastic proliferation of the mucosa, and adenomatous polyps. Colorectal cancers generally arise from adenomatous polyps. It is adenomatous polyps that develop in type 1 and type 2 of Turcot syndrome. Homozygous MMR or APC gene mutations further the progression of the disease. These mutations manifest as tumors existing beyond the colon, namely brain tumors in Turcot syndrome.
In contrast to the single allelic loss of the APC gene which results in familial adenomatous polyposis, Turcot syndrome which results in colorectal cancer and brain tumors seems to require a biallelic loss of the APC gene 3 and in the MMR genes. Cancer develops either by the loss of tumor suppressor genes, the activation of oncogenes, or both. The APC gene is a tumor suppressor gene. When it is missing, it contributes to a lack of regulation in cell division. Hence, in familial adenomatous polyposis, there is an uncontrolled development of adenomatous colonic polyps that eventually turn invasive. These polyps found in Turcot syndrome type 2 can turn invasive by 20, and by 40 years of age, most of these patients will have developed cancer if the colon is not removed 9. Turcot syndrome patients that develop deadly brain tumors do not tend to live that long 10.
The APC gene mutation starts via KRAS protooncogene point mutations. This leads to hypomethylation of DNA which leads to proto-oncogene activation. Then there is a loss of the APC alleles, which is a tumor suppressor gene. The APC gene is found on the long arm of chromosome 5. There has also been mention of loss of the DCC gene on chromosome 18q and 17p. The specific MMR genes which undergo mutation are the MLH1 and MSH2 genes, and occasionally hPMS2 8. These genes are also involved in the pathogenesis of hereditary nonpolyposis colorectal cancer, but there they are found as heterozygous mutations as opposed to homozygous mutations that lead to brain tumor development that would qualify the disease as Turcot syndrome.
The medulloblastomas associated with Turcot syndrome2 come from neural stem cell precursors containing the homozygous APC mutation in the cerebellum 11. Glioblastomas are generally found in Turcot syndrome1 associated with the homozygous MMR gene mutations 12.
Turcot syndrome symptoms
Turcot syndrome is characterized by the formation of multiple benign growths (polyps) in the colon that occur in association with a primary brain tumor. These growths are associated with bleeding from the rectum, diarrhea, constipation, abdominal pain, and/or weight loss. The number and size of these polyps may vary greatly from case to case, ranging from fewer than 10 to more than 100.
Some researchers have separated Turcot syndrome into two forms. type 1 is characterized by the presence of fewer than 100 colonic polyps. These polyps are large in size and more likely to become malignant (cancerous). Type 2 is characterized by smaller, more numerous colonic polyps. This type of Turcot syndrome closely resembles familial adenomatous polyposis.
Individuals with Turcot syndrome often have neurological abnormalities that vary, depending upon the type, size, and location of the associated brain tumor. In cases of Turcot syndrome, the brain tumor is often a glioma. Additional brain tumors that have been associated with Turcot syndrome include medulloblastomas, glioblastomas, ependymomas, and astrocytomas. Medulloblastomas occur with greater frequency in the type 2 form of Turcot syndrome.
Individuals with Turcot syndrome have a much greater risk than the general population of developing colon cancer later in life. Affected individuals also have a predisposition to develop malignant (cancerous) tumors in areas outside the colon, including thyroid, adrenal, and/or abdominal tumors.
Additional symptoms associated with Turcot syndrome include small, coffee-colored spots on the skin (cafe-au-lait spots), the formation of multiple, benign fatty tumors (lipomas), and/or the development of a type of skin cancer known as basal cell carcinoma. Basal cell carcinoma is characterized by the formation of small, shiny, firm masses of tissues (nodules); flat, scar-like lesions (plaques); or red patches covered by thick, dry, silvery scales on the skin.
Turcot syndrome diagnosis
A diagnosis of Turcot syndrome is made based upon a detailed patient history, a thorough clinical evaluation, and a variety of specialized tests. Because children of an affected parent have a genetic risk of developing Turcot syndrome, regular screening via sigmoidoscopy is required until approximately age 35 to 40 to help ensure early detection and prompt, appropriate treatment. During sigmoidoscopy, a viewing instrument is used to examine the rectum and the last part of the large intestine (sigmoid colon). In addition, in some cases, DNA testing may be available to help detect family members who have inherited certain changes (mutations) of the APC gene or DNA mismatch repair genes, potentially diagnosing the disorder before polyp development. In addition, x-rays of the large intestine may reveal the presence of polyps. X-rays of the brain may reveal the presence of a central nervous system tumor.
Diagnostic testing for Turcot syndrome also includes direct visual examination of the intestines by the insertion of a flexible, tube-like instrument (colonoscope) into the rectum (colonoscopy) or the removal and microscopic examination of small samples of rectal tissue (biopsy).
Turcot syndrome treatment
The treatment of Turcot syndrome is directed toward the specific symptoms that are apparent in each individual. Surgical removal of the large intestine and the rectum (proctocolectomy) may prevent the risk of such malignancies. However, if procedures are performed to remove the large intestine and surgically join the rectum and small intestine (ileoproctostomy), rectal polyps may regress. Therefore, some physicians may recommend such procedures as an alternative to proctocolectomy. In such cases, the remaining rectal region must be regularly examined through sigmoidoscopy to ensure prompt detection and surgical removal or destruction of any new polyps.
In some affected individuals, rapid development of new polyps may necessitate additional surgical treatment, such as removal of the rectum and surgical creation of a connection between the small intestine and the abdominal wall (ileostomy). In other cases, physicians may initially recommend other surgical procedures, such as a technique in which the large intestine is removed (colectomy) and the small intestine and the anus are surgically joined (ileoanal anastomosis).
Affected individuals should also receive periodic neurological screenings to test for the presence of a brain tumor. Treatment for brain tumors depends upon the type, size, and location of the tumor. It may include surgery to remove as much of the tumor as is possible without causing damage to the surrounding tissue. Surgery is often followed or accompanied by radiation and/or chemotherapy treatments.
Genetic counseling may be of benefit for affected individuals and their families. Other treatment is symptomatic and supportive.
Turcot syndrome prognosis
Much of the information about the prognosis of Turcot syndrome has been gained from analyses of reported cases 5. The overall survival for children with medulloblastoma alone with treatment is 52% at 10 years 13. In patients with APC mutations or Turcot syndrome type 2, 7 of 8 patients who died in a familial analysis done by Hamilton et al. died because of a brain tumor 3.
Since there are several deadly comorbidities in Turcot syndrome, the prognosis depends on the disease presentation. One case report describes a patient with glioblastoma multiforme early in life that was resected and radiated who subsequently developed adenocarcinoma of the colon. The patient survived into his 60s 14.
Patients who develop any malignancy that metastasizes have a poor prognosis. Inherited mismatch repair or APC mutations cause colorectal cancer 70% to 100% of the time if no treatment or prevention is applied.
Prognosis may be worse in those patients that present with both central nervous system (brain and spinal cord) tumor, specifically, glioma, and colorectal cancer. The development of glioblastoma multiforme seems to render Turcot syndrome patients the worst prognosis, with an average survival being 27 months 15.
Also of note, familial adenomatous polyposis patients who undergo a prophylactic colectomy most frequently end up dying because of duodenal cancer, usually located in the peri-ampullary region of the duodenum 16.
References- Sarma, Y.S., Bhaskararao, G., Sriharibabu, M., & Chakravarthy, D.K. (2015). Case Report : A rare case of Turcot syndrome. http://svimstpt.ap.nic.in/jcsr/Jul-Sep15_files/1cr15.pdf
- Ward RL, Hicks S, Hawkins NJ. Population-based molecular screening for Lynch syndrome : Implications for personalized medicine. J Clin Oncol 2013;31:2554-62.
- Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, Krush AJ, Berk T, Cohen Z, Tetu B. The molecular basis of Turcot’s syndrome. N. Engl. J. Med. 1995 Mar 30;332(13):839-47.
- Familial Adenomatous Polyposis. https://www.cancer.net/cancer-types/familial-adenomatous-polyposis
- Khattab A, Monga DK. Turcot Syndrome. [Updated 2020 Jun 29]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK534782
- Turcot Syndrome. https://rarediseases.org/rare-diseases/turcot-syndrome
- Paraf F, Jothy S, Van Meir EG. Brain tumor-polyposis syndrome: two genetic diseases? J. Clin. Oncol. 1997 Jul;15(7):2744-58.
- Ramachandra C, Challa VR, Shetty R. Constitutional mismatch repair deficiency syndrome: Do we know it? Indian J Hum Genet. 2014 Apr;20(2):192-4.
- Gorovoy IR, de Alba Campomanes A. A potential life-saving diagnosis–recognizing Turcot syndrome. J AAPOS. 2014 Apr;18(2):186-8.
- Fritch Lilla SA, Yi JS, Hall BA, Moertel CL. A novel APC gene mutation associated with a severe phenotype in a patient with Turcot syndrome. J. Pediatr. Hematol. Oncol. 2014 Apr;36(3):e177-9.
- Crawford JR, MacDonald TJ, Packer RJ. Medulloblastoma in childhood: new biological advances. Lancet Neurol. 2007 Dec;6(12):1073-85.
- Lusis EA, Travers S, Jost SC, Perry A. Glioblastomas with giant cell and sarcomatous features in patients with Turcot syndrome type 1: a clinicopathological study of 3 cases. Neurosurgery. 2010 Sep;67(3):811-7; discussion 817.
- Stavrou T, Bromley CM, Nicholson HS, Byrne J, Packer RJ, Goldstein AM, Reaman GH. Prognostic factors and secondary malignancies in childhood medulloblastoma. J. Pediatr. Hematol. Oncol. 2001 Oct;23(7):431-6.
- Rutz HP, de Tribolet N, Calmes JM, Chapuis G. Long-time survival of a patient with glioblastoma and Turcot’s syndrome. Case report. J. Neurosurg. 1991 May;74(5):813-5.
- Dipro S, Al-Otaibi F, Alzahrani A, Ulhaq A, Al Shail E. Turcot syndrome: a synchronous clinical presentation of glioblastoma multiforme and adenocarcinoma of the colon. Case Rep Oncol Med. 2012;2012:720273
- Trimbath JD, Griffin C, Romans K, Giardiello FM. Attenuated familial adenomatous polyposis presenting as ampullary adenocarcinoma. Gut. 2003 Jun;52(6):903-4.





{kind=link}