Dementia
Dementia is not a disease, but a collection of symptoms that result from damage to the brain or disorders affecting the brain 1. Dementia is not a specific disease 2. Dementia affects thinking, behavior, remembering, reasoning and behavioral abilities to such an extent that it interferes with a person’s daily life and activities 3. In dementia, the brain function is affected enough to interfere with the person’s normal social or working life. People with dementia may not be able to think well enough to do normal activities, such as getting dressed or eating. They may lose their ability to solve problems or control their emotions 2. Their personalities may change 2. They may become agitated or see things (hallucinate) that are not there 2.
Memory loss is a common symptom of dementia 2. However, memory loss by itself does not mean you have dementia 2. People with dementia have serious problems with two or more brain functions, such as memory and language 2. People with advanced dementia may not recognise close family and friends, they may not remember where they live or know where they are. They may find it impossible to understand simple pieces of information, carry out basic tasks or follow instructions.
Although dementia is common in very elderly people, up to half of all people age 85 or older may have some form of dementia. Dementia is not a normal part of aging 2, 3. Many people live into their 90s and beyond without any signs of dementia. One type of dementia, fronto-temporal disorders, is more common in middle-aged than older adults.
Dementia ranges in severity from the mildest stage, when it is just beginning to affect a person’s functioning, to the most severe stage, when the person must depend completely on others for basic activities of living 4. As dementia progresses, memory loss and difficulties with communication often become very severe. It’s common for people with dementia to have increasing difficulty speaking and they may eventually lose the ability to speak altogether. It’s important to keep trying to communicate with them and to recognise and use other, non-verbal means of communication, such as expression, touch and gestures. In the later stages, the person is likely to neglect their own health and require constant care and attention.
Many people with dementia gradually become less able to move about unaided and may appear increasingly clumsy when carrying out everyday tasks. Some people may eventually be unable to walk and may become bedbound. Bladder incontinence is common in the later stages of dementia and some people will also experience bowel incontinence.
Loss of appetite and weight loss are common in the later stages of dementia. It’s important that people with dementia get help at mealtimes to ensure they eat enough.
Many people have trouble eating or swallowing and this can lead to choking, chest infections and other problems.
Signs and symptoms of dementia result when once-healthy neurons (nerve cells) in the brain stop working, lose connections with other brain cells, and die. While everyone loses some neurons as they age, people with dementia experience far greater loss 4.
Memory loss, though common, is not the only sign of dementia. For a person to have dementia, he or she must have 4:
- Two or more core mental functions that are impaired. These functions include memory, language skills, visual perception, and the ability to focus and pay attention. These also include cognitive skills such as the ability to reason and solve problems.
- A loss of brain function severe enough that a person cannot do normal, everyday tasks.
In addition, some people with dementia cannot control their emotions. Their personalities may change. They can have delusions, which are strong beliefs without proof, such as the idea that someone is stealing from them. They also may hallucinate, seeing or otherwise experiencing things that are not real.
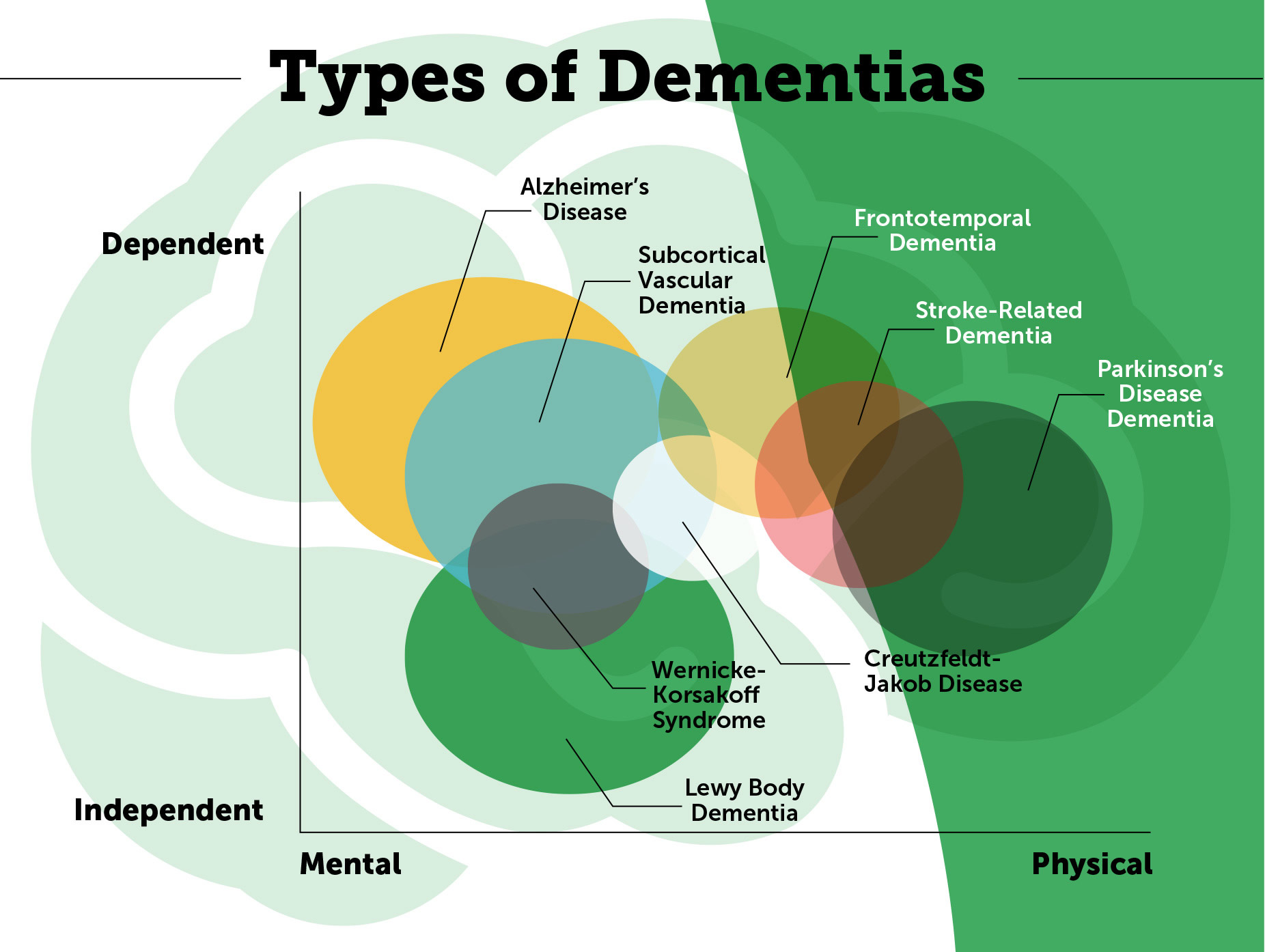
The causes of dementia can vary, depending on the types of brain changes that may be taking place. Many different diseases can cause dementia, including Alzheimer’s disease and stroke 2. Other dementias include Lewy body dementia, frontotemporal disorders, and vascular dementia 4. It is common for people to have mixed dementia—a combination of two or more disorders, at least one of which is dementia 4. For example, some people have both Alzheimer’s disease and vascular dementia. Drugs are available to treat some of these diseases. While these drugs cannot cure dementia or repair brain damage, they may improve symptoms or slow down the disease 2.
Who gets dementia?
Most people with dementia are older, but it is important to remember that not all older people get dementia. Dementia is NOT a normal part of ageing.
Dementia can happen to anybody, but it is more common after the age of 65 years. People in their 40s and 50s can also have dementia.
Do memory problems always mean Alzheimer’s disease?
Many people worry about becoming forgetful. They think forgetfulness is the first sign of Alzheimer’s disease. But not all people with memory problems have Alzheimer’s disease 5. Other causes for memory problems can include aging, medical conditions, emotional problems, mild cognitive impairment, or another type of dementia.
Early signs of dementia
There are some common early symptoms that may appear some time before a diagnosis of dementia. These include:
- memory loss
- difficulty concentrating
- finding it hard to carry out familiar daily tasks, such as getting confused over the correct change when shopping
- struggling to follow a conversation or find the right word
- being confused about time and place
- mood changes
These symptoms are often mild and may get worse only very gradually. It’s often termed “mild cognitive impairment” (MCI) as the symptoms are not severe enough to be diagnosed as dementia.
You might not notice these symptoms if you have them, and family and friends may not notice or take them seriously for some time. In some people, these symptoms will remain the same and not worsen. But some people with MCI (mild cognitive impairment) will go on to develop dementia.
Dementia is not a natural part of ageing. This is why it’s important to talk to a doctor sooner rather than later if you’re worried about memory problems or other symptoms.
What is Mild Cognitive Impairment?
Some forgetfulness can be a normal part of aging. However, some people have more memory problems than other people their age. This condition is called mild cognitive impairment (MCI) 6. Mild cognitive impairment is a clinical syndrome in which an individual experiences a mild but noticeable and measurable decline in cognitive abilities, including memory, judgment, language and thinking skills that are greater than normal age-related changes, but the loss doesn’t significantly interfere with your ability to handle everyday activities 7. Mild cognitive impairment is an intermediate stage between normal cognitive changes that may occur with age and more serious symptoms that indicate dementia 8. If you have mild cognitive impairment, you may be aware that your memory or mental function has “slipped.” Your family and close friends also may notice a change. People with mild cognitive impairment can take care of themselves and do their normal activities 9. Mild cognitive impairment is characterized by problems with memory, language, thinking or judgment.
People with mild cognitive impairment (MCI) have a significantly increased risk, but not a certainty, of developing dementia. Overall, about 1% to 3% of older adults develop dementia every year. Numerous international population-based studies have been conducted to document the frequency of mild cognitive impairment, estimating its prevalence to be between 15% and 20% in persons 60 years and older, making it a common condition encountered by clinicians 8. Studies suggest that around 8% to 15% of individuals with mild cognitive impairment (MCI) go on to develop dementia each year 8.
Mild cognitive impairment memory problems may include:
- Losing things often
- Forgetting to go to events and appointments
- Having more trouble coming up with words than other people of the same age
If you have mild cognitive impairment (MCI), you may also experience:
- Depression
- Irritability and aggression
- Anxiety
- Apathy
If you have mild cognitive impairment (MCI), you may be aware that your memory or mental function has “slipped.” Your family and close friends also may notice a change. But these changes aren’t severe enough to significantly interfere with your daily life and usual activities.
Mild cognitive impairment may increase your risk of later developing dementia caused by Alzheimer’s disease or other neurological conditions. But some people with mild cognitive impairment never get worse, and a few eventually get better.
Experts classify mild cognitive impairment based on the thinking skills affected:
- Amnestic mild cognitive impairment: mild cognitive impairment that primarily affects memory. A person may start to forget important information that he or she would previously have recalled easily, such as appointments, conversations or recent events.
- Nonamnestic mild cognitive impairment: mild cognitive impairment that affects thinking skills other than memory, including the ability to make sound decisions, judge the time or sequence of steps needed to complete a complex task, or visual perception.
Researchers have found that more people with mild cognitive impairment than those without it go on to develop Alzheimer’s disease. However, not everyone who has mild cognitive impairment develops Alzheimer’s disease. About 8 of every 10 people who fit the definition of amnestic mild cognitive impairment go on to develop Alzheimer’s disease within 7 years 9. In contrast, 1 to 3 percent of people older than 65 who have normal cognition will develop Alzheimer’s disease in any one year 9.
Research suggests genetic factors may play a role in who will develop mild cognitive impairment, as they do in Alzheimer’s disease 9. Studies are underway to learn why some people with mild cognitive impairment progress to Alzheimer’s disease and others do not.
Symptoms of Mild Cognitive Impairment
Your brain, like the rest of your body, changes as you grow older. Many people notice gradually increasing forgetfulness as they age. It may take longer to think of a word or to recall a person’s name. But consistent or increasing concern about your mental performance may suggest mild cognitive impairment (MCI). People with amnestic mild cognitive impairment have more memory problems than normal for people their age, but their symptoms are not as severe as those of people with Alzheimer’s disease 9. For example, they do not experience the personality changes or other problems that are characteristic of Alzheimer’s disease 9. People with mild cognitive impairment (MCI) are still able to carry out their normal daily activities 9.
Cognitive issues that may indicate possible mild cognitive impairment (MCI) if you experience any or all of the following:
- You forget things more often.
- You forget important events such as appointments or social engagements.
- You lose your train of thought or the thread of conversations, books or movies.
- You feel increasingly overwhelmed by making decisions, planning steps to accomplish a task or understanding instructions.
- You start to have trouble finding your way around familiar environments.
- You become more impulsive or show increasingly poor judgment.
- Your family and friends notice any of these changes.
Signs of Mild Cognitive Impairment
Signs of Mild Cognitive Impairment include 9:
- Losing things often.
- Forgetting to go to events or appointments.
- Having more trouble coming up with words than other people of the same age.
Movement difficulties and problems with the sense of smell have also been linked to mild cognitive impairment.
Mild cognitive impairment causes
There’s no single cause of mild cognitive impairment (MCI), just as there’s no single outcome for the disorder. Symptoms of MCI may remain stable for years, progress to Alzheimer’s disease or another type of dementia, or improve over time.
Current evidence indicates that mild cognitive impairment (MCI) often, but not always, develops from a lesser degree of the same types of brain changes seen in Alzheimer’s disease or other forms of dementia. Some of these changes have been identified in autopsy studies of people with mild cognitive impairment (MCI). These changes include:
- Abnormal clumps of beta-amyloid protein (plaques) and microscopic protein clumps of tau characteristic of Alzheimer’s disease (tangles)
- Lewy bodies, which are microscopic clumps of another protein associated with Parkinson’s disease, dementia with Lewy bodies and some cases of Alzheimer’s disease
- Small strokes or reduced blood flow through brain blood vessels
Brain-imaging studies show that the following changes may be associated with mild cognitive impairment (MCI):
- Shrinkage of the hippocampus, a brain region important for memory
- Enlargement of the brain’s fluid-filled spaces (ventricles)
- Reduced use of glucose, the sugar that’s the primary source of energy for cells, in key brain regions
Risk factors for developing mild cognitive impairment
The strongest risk factors for mild cognitive impairment (MCI) are:
- Increasing age
- Having a specific form of a gene known as APOE e4, also linked to Alzheimer’s disease — though having the gene doesn’t guarantee that you’ll experience cognitive decline
Other medical conditions and lifestyle factors have been linked to an increased risk of cognitive change, including:
- Diabetes
- Smoking
- High blood pressure
- Elevated cholesterol
- Obesity
- Depression
- Lack of physical exercise
- Low education level
- Infrequent participation in mentally or socially stimulating activities
Mild cognitive impairment prevention
Mild cognitive impairment can’t always be prevented. But research has found some environmental factors that may affect the risk of developing the condition. Studies show that these steps may help prevent cognitive impairment:
- Avoid excessive alcohol use.
- Limit exposure to air pollution.
- Reduce your risk of head injury.
- Don’t smoke.
- Manage health conditions such as diabetes, high blood pressure, obesity and depression.
- Practice good sleep hygiene and manage sleep disturbances.
- Eat a nutrient-rich diet that has plenty of fruits and vegetables and is low in saturated fats.
- Engage socially with others.
- Exercise regularly at a moderate to vigorous intensity.
- Wear a hearing aid if you have hearing loss.
- Stimulate your mind with puzzles, games and memory training.
Mild Cognitive Impairment diagnosis
There is no specific test to confirm a diagnosis of mild cognitive impairment (MCI). Your doctor will decide whether mild cognitive impairment is the most likely cause of your symptoms based on the information you provide and results of various tests that can help clarify the diagnosis.
Many doctors diagnose mild cognitive impairment based on the following criteria developed by a panel of international experts:
- You have problems with memory or another mental function. You may have problems with your memory, planning, following instructions or making decisions. Your own impressions should be confirmed by someone close to you.
- You’ve declined over time. A careful medical history reveals that your mental function has declined from a higher level. This change ideally is confirmed by a family member or a close friend.
- Your overall mental function and daily activities aren’t affected. Your medical history shows that overall your daily activities generally aren’t impaired, although specific symptoms may cause worry and inconvenience.
- Mental status testing shows a mild level of impairment for your age and education level. Doctors often assess mental performance with a brief test such as the Short Test of Mental Status, the Montreal Cognitive Assessment (MoCA) or the Mini-Mental State Examination (MMSE). More-detailed neuropsychological testing may help determine the degree of memory impairment, which types of memory are most affected and whether other mental skills also are impaired.
- Your diagnosis isn’t dementia. The problems that you describe and that your doctor documents through corroborating reports, your medical history, and mental status testing aren’t severe enough to be diagnosed as Alzheimer’s disease or another type of dementia.
Neurological exam
As part of your physical exam, your doctor may perform some basic tests that indicate how well your brain and nervous system are working. These tests can help detect neurological signs of Parkinson’s disease, strokes, tumors or other medical conditions that can impair your memory as well as your physical function. The neurological exam may test:
- Reflexes
- Eye movements
- Walking and balance
Lab tests
Blood tests can help rule out physical problems that can affect memory, such as a vitamin B-12 deficiency or an underactive thyroid gland.
Brain imaging
Your doctor may order an MRI or CT scan to check for evidence of a brain tumor, stroke or bleeding.
Mental status testing
Short forms of mental status testing can be done in about 10 minutes. During testing, doctors ask people to conduct several specific tasks and answer several questions, such as naming today’s date or following a written instruction.
Longer forms of neuropsychological testing can provide additional details about your mental function compared with the function of others of a similar age and education level. These tests may also help identify patterns of change that offer clues about the underlying cause of your symptoms.
Mild cognitive impairment treatment
Currently, no mild cognitive impairment (MCI) drugs or other treatments are specifically approved by the Food and Drug Administration (FDA). However, mild cognitive impairment is an active area of research. Clinical studies are underway to shed more light on the disorder and find treatments that may improve symptoms or prevent or delay progression to dementia.
Experts recommend that a person diagnosed with mild cognitive impairment be re-evaluated every six months to determine if symptoms are staying the same, improving or growing worse.
Mild cognitive impairment increases the risk of later developing dementia, but some people with mild cognitive impairment never get worse. Others with mild cognitive impairment later have test results that return to normal for their age and education.
Alzheimer’s drugs
Doctors sometimes prescribe cholinesterase inhibitors – donepezil (Aricept), a type of drug approved for Alzheimer’s disease, for people with mild cognitive impairment whose main symptom is memory loss. However, cholinesterase inhibitors aren’t recommended for routine treatment of mild cognitive impairment. Results of a large, federally funded trial showed that 10 milligrams of donepezil (Aricept) daily reduced the risk of progressing from amnestic mild cognitive impairment to Alzheimer’s disease for about a year, but the benefit disappeared within three years. The study’s principal investigators said the results were not strong enough to clearly recommend donepezil as a treatment for mild cognitive impairment. However, it might be reasonable for patients and their physicians to talk about the possible benefits and risks of such treatment on an individual basis.
Treating other conditions that can affect mental function
Other common conditions besides mild cognitive impairment can make you feel forgetful or less mentally sharp than usual. Treating these conditions can help improve your memory and overall mental function. Conditions that can affect memory include:
- High blood pressure. People with mild cognitive impairment tend to be more likely to have problems with the blood vessels inside their brains. High blood pressure can worsen these problems and cause memory difficulties. Your doctor will monitor your blood pressure and recommend steps to lower it if it’s too high.
- Depression. When you’re depressed, you often feel forgetful and mentally “foggy.” Depression is common in people with mild cognitive impairment. Treating depression may help improve memory, while making it easier to cope with the changes in your life.
- Sleep apnea. In this condition, your breathing repeatedly stops and starts while you’re asleep, making it difficult to get a good night’s rest. Sleep apnea can make you feel excessively tired during the day, forgetful and unable to concentrate. Treatment can improve these symptoms and restore alertness.
Home remedies
Study results have been mixed about whether diet, exercise or other healthy lifestyle choices can prevent or reverse cognitive decline. Regardless, these healthy choices promote good overall health and may play a role in good cognitive health.
- Regular physical exercise has known benefits for heart health and may also help prevent or slow cognitive decline.
- A diet low in fat and rich in fruits and vegetables is another heart-healthy choice that also may help protect cognitive health.
- Omega-3 fatty acids also are good for the heart. Most research showing a possible benefit for cognitive health uses fish consumption as a yardstick for the amount of omega-3 fatty acids eaten.
- Intellectual stimulation may prevent cognitive decline. Studies have shown computer use, playing games, reading books and other intellectual activities may help preserve function and prevent cognitive decline.
- Social engagement may make life more satisfying, and help preserve mental function and slow mental decline.
- Memory training and other thinking (cognitive) training may help improve your function.
There is some evidence of a possible benefit on mild cognitive impairment from non-pharmacological interventions, such as cognitive training and physical exercise, activities that may be neuroprotective or compensatory. A recent review showed how several studies demonstrated the efficacy of cognitive training in mild cognitive impairment measured as improved performances in tests of global cognitive functioning, memory and meta-memory 10. A limitation of these findings is the small sample sizes of the individual studies. Only seven randomized control trials were identified by a systematic review 11, with a total of 296 mild cognitive impairment subjects who were cognitively treated. Most of these studies in fact included samples of fewer than 50 individuals; therefore, replication of the findings in larger randomized control trials is warranted.
A rapidly growing body of evidence suggests that exercise, specifically aerobic exercise, may attenuate cognitive impairment 12. A systematic review of the effect of aerobic exercise on cognitive performance in individuals with neurological disorders found modest improvements in attention and processing speed, executive function and memory 13. Therefore, as for the case of cognitive training, larger randomized control trials specifically in subjects with mild cognitive impairment, are warranted to confirm or refute these preliminary results. In particular, there is a strong need for evidence regarding the combined effect of multiple non-pharmacological interventions on mild cognitive impairment evolution and ongoing multidomain randomized control trials of mild cognitive impairment are particularly relevant 14. A further possibility is to combine pharmacological and non-pharmacological interventions and evaluate whether their joint effect has more therapeutic value than the individual treatments alone. Such studies could also be combined with therapeutic trials.
Alternative medicine
Some supplements — including vitamin E, ginkgo and others — have been purported to help prevent or delay the progression of mild cognitive impairment. However, no supplement has shown any benefit in a clinical trial.
Dementia causes
Dementia symptoms can be caused by a number of conditions and each has its own causes.
The most common types of dementia symptoms are caused by 15 :
- Alzheimer’s disease,
- Vascular dementia and vascular cognitive impairment (to learn more about Vascular Dementia),
- Parkinson’s disease (to learn more about Parkinson’s Disease Dementia),
- Dementia with Lewy bodies (to learn more about Lewy Body Dementia),
- Fronto Temporal Lobar Degeneration (to learn more about Frontotemporal dementia),
- Huntington’s disease,
- Alcohol related dementia (Korsakoff’s syndrome),
- HIV Associated Dementia (AIDS Dementia Complex),
- Creutzfeldt-Jacob disease and
- Mixed dementia, a combination of two or more disorders, at least one of which is dementia 16.
Dementia risk factors
Many factors can eventually contribute to dementia. Some factors, such as age, can’t be changed. Others can be addressed to reduce your risk.
Risk factors that can’t be changed
- Age. The risk rises as you age, especially after age 65. However, dementia isn’t a normal part of aging, and dementia can occur in younger people.
- Family history. Having a family history of dementia puts you at greater risk of developing the condition. However, many people with a family history never develop symptoms, and many people without a family history do. There are tests to determine whether you have certain genetic mutations.
- Down syndrome. By middle age, many people with Down syndrome develop early-onset Alzheimer’s disease.
Risk factors you can change
You might be able to control the following risk factors for dementia.
- Diet and exercise. Research shows that lack of exercise increases the risk of dementia. And while no specific diet is known to reduce dementia risk, research indicates a greater incidence of dementia in people who eat an unhealthy diet compared with those who follow a Mediterranean-style diet rich in produce, whole grains, nuts and seeds.
- Excessive alcohol use. Drinking large amounts of alcohol has long been known to cause brain changes. Several large studies and reviews found that alcohol use disorders were linked to an increased risk of dementia, particularly early-onset dementia.
- Cardiovascular risk factors. These include high blood pressure (hypertension), high cholesterol, buildup of fats in your artery walls (atherosclerosis) and obesity.
- Depression. Although not yet well-understood, late-life depression might indicate the development of dementia.
- Diabetes. Having diabetes may increase your risk of dementia, especially if it’s poorly controlled.
- Smoking. Smoking might increase your risk of developing dementia and blood vessel diseases.
- Air pollution. Studies in animals have indicated that air pollution particulates can speed degeneration of the nervous system. And human studies have found that air pollution exposure — particularly from traffic exhaust and burning wood — is associated with greater dementia risk.
- Head trauma. People who’ve had a severe head trauma have a greater risk of Alzheimer’s disease. Several large studies found that in people age 50 years or older who had a traumatic brain injury (TBI), the risk of dementia and Alzheimer’s disease increased. The risk increases in people with more-severe and multiple TBIs. Some studies indicate that the risk may be greatest within the first six months to two years after the TBI.
- Sleep disturbances. People who have sleep apnea and other sleep disturbances might be at higher risk of developing dementia.
- Vitamin and nutritional deficiencies. Low levels of vitamin D, vitamin B-6, vitamin B-12 and folate can increase your risk of dementia.
- Medications that can worsen memory. Try to avoid over-the-counter sleep aids that contain diphenhydramine (Advil PM, Aleve PM) and medications used to treat urinary urgency such as oxybutynin (Ditropan XL). Also limit sedatives and sleeping tablets and talk to your doctor about whether any of the drugs you take might make your memory worse.
Dementia prevention
There is no certain way to prevent all types of dementia. However, a healthy lifestyle can help lower your risk of developing dementia when you are older. It can also prevent cardiovascular diseases, such as strokes and heart attacks.
To reduce your risk of developing dementia and other serious health conditions, it’s recommended that you:
- Eat a healthy diet. A diet such as the Mediterranean diet — rich in fruits, vegetables, whole grains and omega-3 fatty acids, which are commonly found in certain fish and nuts — might promote health and lower your risk of developing dementia. This type of diet also improves cardiovascular health, which may help lower dementia risk.
- Get enough vitamins. Some research suggests that people with low levels of vitamin D in their blood are more likely to develop Alzheimer’s disease and other forms of dementia. You can get vitamin D through certain foods, supplements and sun exposure. More study is needed before an increase in vitamin D intake is recommended for preventing dementia, but it’s a good idea to make sure you get adequate vitamin D. Taking a daily B-complex vitamin and vitamin C also might help.
- Maintain a healthy weight. Lose weight if you’re overweight.
- Exercise regularly. Physical activity and social interaction might delay the onset of dementia and reduce its symptoms. Aim for 150 minutes of exercise a week.
- Keep your mind active. Mentally stimulating activities, such as reading, solving puzzles and playing word games, and memory training might delay the onset of dementia and decrease its effects.
- Don’t drink too much alcohol
- Stop smoking (if you smoke). Some studies have shown that smoking in middle age and beyond might increase your risk of dementia and blood vessel conditions. Quitting smoking might reduce your risk and will improve your health.
- Make sure to keep your blood pressure at a healthy level. High blood pressure might lead to a higher risk of some types of dementia. More research is needed to determine whether treating high blood pressure may reduce the risk of dementia.
- Treat health conditions. See your doctor for treatment for depression or anxiety. Treat high blood pressure, high cholesterol and diabetes.
- Treat hearing problems. People with hearing loss have a greater chance of developing cognitive decline. Early treatment of hearing loss, such as use of hearing aids, might help decrease the risk.
- Get good-quality sleep. Practice good sleep hygiene, and talk to your doctor if you snore loudly or have periods where you stop breathing or gasp during sleep.
Diet and dementia
A low-fat, high-fiber diet including plenty of fresh fruit and vegetables and wholegrains can help reduce your risk of some kinds of dementia.
Limiting the amount of salt in your diet to no more than 1.5 grams a day can also help. Too much salt will increase your blood pressure, which puts you at risk of developing some types of dementia.
High cholesterol levels may also put you at risk of developing some kinds of dementia, so try to limit the amount of food you eat that is high in saturated fat.
How weight affects dementia risk
Being overweight can increase your blood pressure, which increases your risk of getting some kinds of dementia. The risk is higher if you are obese. The most scientific way to measure your weight is to calculate your body mass index (BMI).
You can calculate your BMI using the BMI healthy weight calculator. People with a BMI of 25-30 are overweight, and those with a BMI above 30 are obese. People with a BMI of 40 or more are morbidly obese.
Exercise to reduce dementia risk
Exercising regularly will make your heart and blood circulatory system more efficient. It will also help to lower your cholesterol and keep your blood pressure at a healthy level, decreasing your risk of developing some kinds of dementia.
For most people, a minimum of 150 minutes (2 hours and 30 minutes) of moderate-intensity aerobic activity each week, such as cycling or fast walking, is recommended.
Alcohol and dementia
Drinking excessive amounts of alcohol will cause your blood pressure to rise, as well as raising the level of cholesterol in your blood.
Stick to the recommended limits for alcohol consumption to reduce your risk of high blood pressure, cardiovascular disease and dementia.
The recommended daily limit for alcohol consumption is three to four units of alcohol a day for men, and two to three units a day for women. A unit of alcohol is equal to about half a pint of normal-strength lager, a small glass of wine or a pub measure (25ml) of spirits.
Stopping smoking could reduce dementia risk
Smoking can cause your arteries to narrow, which can lead to a rise in your blood pressure. It also increases your risk of developing cardiovascular diseases, cancer and dementia.
Dementia signs and symptoms
Dementia is not a disease itself but it’s a term used to describe a group of symptoms affecting memory, thinking and social abilities severely enough to interfere with your daily life. Dementia is a collection of symptoms that result from damage to the brain caused by different diseases. Although dementia generally involves memory loss, memory loss has different causes. Having memory loss alone doesn’t mean you have dementia, although it’s often one of the early signs of the condition.
Dementia symptoms vary depending on the cause, but common signs and symptoms include:
Cognitive changes
- Memory loss, which is usually noticed by someone else
- Difficulty communicating or finding words
- Difficulty with visual and spatial abilities, such as getting lost while driving
- Difficulty reasoning or problem-solving
- Difficulty handling complex tasks
- Difficulty with planning and organizing
- Difficulty with coordination and motor functions
- Confusion and disorientation
Psychological changes
- Personality changes
- Depression
- Anxiety
- Inappropriate behavior
- Paranoia
- Agitation
- Hallucinations
Dementia can lead to
- Poor nutrition. Many people with dementia eventually reduce or stop eating, affecting their nutrient intake. Ultimately, they may be unable to chew and swallow.
- Pneumonia. Difficulty swallowing increases the risk of choking or aspirating food into the lungs, which can block breathing and cause pneumonia.
Inability to perform self-care tasks. As dementia progresses, it can interfere with bathing, dressing, brushing hair or teeth, using the toilet independently, and taking medications as directed. - Personal safety challenges. Some day-to-day situations can present safety issues for people with dementia, including driving, cooking, and walking and living alone.
- Death. Late-stage dementia results in coma and death, often from infection.
What is the first step to diagnose dementia?
Visiting a family doctor is often the first step for people who are experiencing changes in thinking, movement, or behavior 17. However, neurologists—doctors who specialize in disorders of the brain and nervous system—generally have the expertise needed to diagnose dementia 17. Geriatric psychiatrists, neuropsychologists, and geriatricians may also be skilled in diagnosing the condition 17.
If a specialist cannot be found in your community, ask the neurology department of the nearest medical school for a referral. A hospital affiliated with a medical school may also have a dementia or movement disorders clinic that provides expert evaluation.
Dementia diagnosis
No single test can diagnose dementia, so doctors are likely to run a number of tests that can help pinpoint the problem. To diagnose dementia, doctors first assess whether a person has an underlying treatable condition such as depression, abnormal thyroid function, normal pressure hydrocephalus, or vitamin B12 deficiency 17. Early diagnosis is important, as some causes for symptoms can be treated. In many cases, the specific type of dementia a person has may not be confirmed until after the person has died and the brain is examined.
A medical assessment for dementia generally includes:
- Patient history. Typical questions about a person’s medical and family history might include asking about whether dementia runs in the family, how and when symptoms began, changes in behavior and personality, and if the person is taking certain medications that might cause or worsen symptoms.
- Physical exam. Measuring blood pressure and other vital signs may help physicians detect conditions that might cause or occur with dementia. Such conditions may be treatable.
- Neurological tests. Assessing balance, sensory function, reflexes, vision, eye movements, and other functions helps identify conditions that may affect the diagnosis or are treatable with drugs.
Tests Used to Diagnose Dementia
These tests for dementia are mainly tests of mental abilities, blood tests and brain scans. The following procedures also may be used to diagnose dementia:
Cognitive and neuropsychological tests
These tests measure memory, problem solving, attention, counting, language skills, and other abilities related to mental functioning.
People with symptoms of dementia are often given questionnaires to help test their mental abilities, to see how severe any memory problems may be. One widely used test is called the mini mental state examination (MMSE) 18.
The mini mental state examination (MMSE) assesses a number of different mental abilities, including:
- short- and long-term memory
- attention span
- concentration
- language and communication skills
- ability to plan
- ability to understand instructions
The mini mental state examination (MMSE) is a series of exercises, each carrying a score with a maximum of 30 points. These exercises include:
- memorising a short list of objects and then repeating the list
- writing a short sentence that is grammatically correct, such as “the dog sat on the floor”
- correctly answering time-orientation questions, such as identifying the day of the week, the date or the year
The mini mental state examination (MMSE) is a 30-point test
Advantages
- Relatively quick and easy to perform
- Requires no additional equipment
- Can provide a method of monitoring deterioration over time
Disadvantages
- Biased against people with poor education due to elements of language and mathematical testing
- Bias against visually impaired
- Limited examination of visuospatial cognitive ability
- Poor sensitivity at detected mild/early dementia
The Mini Mental State Examination (MMSE)
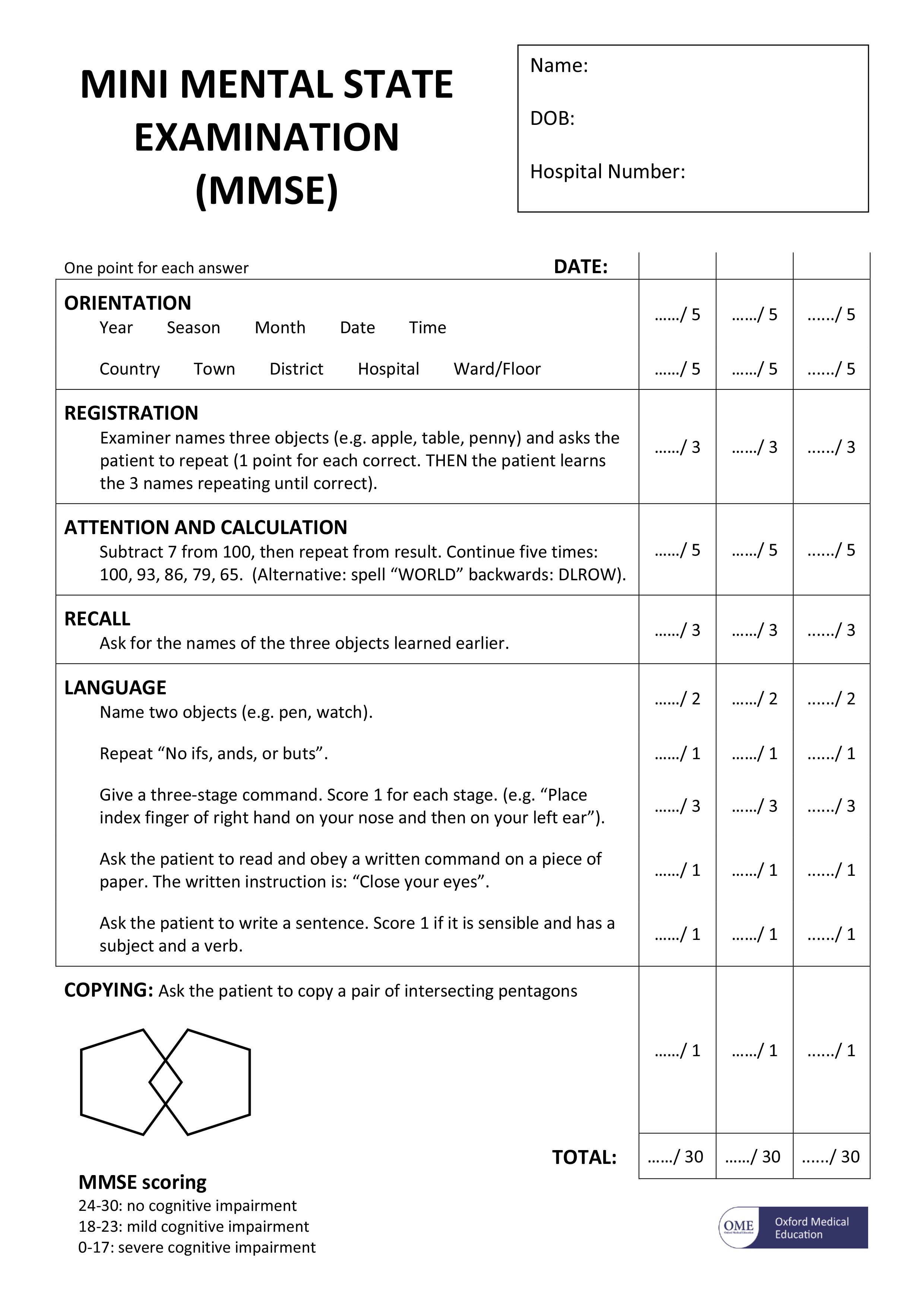
Footnote: The mini mental state examination (MMSE) is not a test to diagnose dementia. However, it is useful for assessing the level of mental impairment that a person with dementia may have. Test scores may be influenced by a person’s level of education. For example, someone who cannot read or write very well may have a lower score, but they may not have dementia. Similarly, someone with a higher level of education may achieve a higher score, but still have dementia.
[Source 19 ]Laboratory tests
Blood and urine tests can help rule out possible causes of symptoms.
Blood tests (in roughly descending order of importance)
- Thyroid function tests: Rule out hypothyroidism as a cause of a dementia-like presentation
- Vitamin B12: Low levels can cause memory impairment and mood changes
- Blood glucose: Independent risk factor for dementia. Unclear if high blood sugars are direct cause. Iatrogenic hypoglycaemic episodes can also cause worsening dementia. Indeed, tightly controlled blood sugars in the elderly has a negative impact on mortality.
- Urea and electrolytes: Severe disturbances can cause cognitive impairment (e.g. renal failure and hyperuraemia). Sodium and calcium are particularly important.
- Liver function tests: Metabolic causes in liver dysfunction (e.g. hyperammonaemia in cirrhosis). Rarer causes (e.g. Wilson’s disease).
- Infective screen: FBC, ESR, CRP to look for superimposed infection as cause of confusion. HIV-associated or neurosyphilis dementia.
- Autoimmune screen: Should be considered in rapidly progressive dementia.
- Further tests: More specific tests are rarely needed. However, if there is diagnostic uncertainly they may include: serum and urinary copper and caeruloplasmin (Wilsons disease); ammonia (liver disease and inherited metabolic abnormalities); HIV; syphilis serology.
Brain scans
Brain scans are often used for diagnosing dementia once other simpler tests have ruled out other problems. They are needed to check for evidence of other possible problems that could explain a person’s symptoms, such as a major stroke or a brain tumor and other problems that can cause dementia. Scans also identify changes in the brain’s structure and function. The most common scans are:
- Computed tomography (CT) scan, which uses X-rays to produce images of the brain and other organs. A computerised tomography (CT) scan can be used to check for signs of stroke or a brain tumor. However, unlike an MRI scan, a CT scan cannot provide detailed information about the structure of the brain. The National Institute for Health recommends using a magnetic resonance imaging (MRI) scan to help confirm a diagnosis of dementia 18.CT scans are quick, painless and generally safe. However, there’s a small risk you could have an allergic reaction to the contrast dye used and you will be exposed to X-ray radiation. The amount of radiation you’re exposed to during a CT scan varies, depending on how much of your body is scanned. CT scanners are designed to make sure you’re not exposed to unnecessarily high levels. Generally, the amount of radiation you’re exposed to during each scan is the equivalent to between a few months and a few years of exposure to natural radiation from the environment.
- Magnetic resonance imaging (MRI), which uses magnetic fields and radio waves to produce detailed images of body structures, including tissues, organs, bones, and nerves. An MRI scan can provide detailed information about the blood vessel damage that occurs in vascular dementia, plus any shrinking of the brain. In frontotemporal dementia, the frontal and temporal lobes are mainly affected by shrinking 18. MRI scans don’t involve exposing the body to X-ray radiation. This means people who may be particularly vulnerable to the effects of radiation, such as pregnant women and babies, can use them if necessary. An MRI scan is a painless and safe procedure. You may find it uncomfortable if you have claustrophobia, but most people find this manageable with support from the radiographer. Extensive research has been carried out into whether the magnetic fields and radio waves used during MRI scans could pose a risk to the human body. No evidence has been found to suggest there’s a risk, which means MRI scans are one of the safest medical procedures currently available. However, not everyone can have an MRI scan. For example, they’re not always possible for people who have certain types of implants fitted, such as a pacemaker (a battery-operated device that helps to control an irregular heartbeat).
- Positron emission tomography (PET) scan or a single photon-emission computed tomography (SPECT) scan, which uses radiation to provide pictures of brain activity may be recommended if the result of your CT or MRI scan is uncertain. These scans look at how the brain functions and can pick up abnormalities with the blood flow in the brain. They’re particularly helpful for investigating confirmed cases of cancer, to determine how far the cancer has spread and how well it’s responding to treatment. They can also help diagnose some conditions that affect the normal workings of the brain, such as dementia. PET scanners work by detecting the radiation given off by a substance called a radiotracer as it collects in different parts of your body.In most PET scans a radiotracer called fluorodeoxyglucose (FDG) is used, which is similar to naturally occurring glucose (a type of sugar) so your body treats it in a similar way. Before the scan, the radiotracer is injected into a vein in your arm or hand. You will need to wait quietly for about an hour, to give it time to be absorbed by the cells in your body. By analysing the areas where the radiotracer does and doesn’t build up, it’s possible to work out how well certain body functions are working and identify any abnormalities. For example, a concentration of fluorodeoxyglucose (FDG) in the body’s tissues can help identify cancerous cells because cancer cells use glucose at a much faster rate than normal cells.In a standard PET scan the amount of radiation you’re exposed to is small – about the same as the amount you get from natural sources, such as the sun, over three years.The radiotracer becomes quickly less radioactive over time and will usually be passed out of your body naturally within a few hours. Drinking plenty of fluid after the scan can help flush it from your body.As a precaution, you may be advised to avoid prolonged close contact with pregnant women, babies or young children for a few hours after a PET scan, as you will be slightly radioactive during this time.
- In some cases, an electroencephalogram (EEG) may be taken to record the brain’s electrical signals (brain activity).
- Psychiatric evaluation. This evaluation will help determine if depression or another mental health condition is causing or contributing to a person’s symptoms.
- Genetic tests. Some dementias are caused by a known gene defect. In these cases, a genetic test can help people know if they are at risk for dementia. People should talk with family members, a primary care doctor, and a genetic counselor before getting tested.
Dementia treatment
Most types of dementia can’t be cured, but there are ways to manage your symptoms.
Dementia medications
The following are used to temporarily improve dementia symptoms:
- Cholinesterase inhibitors. These medications — including donepezil (Aricept), rivastigmine (Exelon) and galantamine (Razadyne) — work by boosting levels of a chemical messenger involved in memory and judgment. Although primarily used to treat Alzheimer’s disease, these medications might also be prescribed for other dementias, including vascular dementia, Parkinson’s disease dementia and Lewy body dementia. Side effects can include nausea, vomiting and diarrhea. Other possible side effects include slowed heart rate, fainting and sleep disturbances.
- Memantine. Memantine (Namenda) works by regulating the activity of glutamate, another chemical messenger involved in brain functions, such as learning and memory. In some cases, memantine is prescribed with a cholinesterase inhibitor. A common side effect of memantine is dizziness.
- Other medications. Your doctor might prescribe medications to treat other symptoms or conditions, such as depression, sleep disturbances, hallucinations, parkinsonism or agitation.
Dementia therapies
Several dementia symptoms and behavior problems might be treated initially using nondrug approaches, such as:
- Occupational therapy. An occupational therapist can show you how to make your home safer and teach coping behaviors. The purpose is to prevent accidents, such as falls; manage behavior and prepare you for the dementia progression.
- Modifying the environment. Reducing clutter and noise can make it easier for someone with dementia to focus and function. You might need to hide objects that can threaten safety, such as knives and car keys. Monitoring systems can alert you if the person with dementia wanders.
- Simplifying tasks. Break tasks into easier steps and focus on success, not failure. Structure and routine also help reduce confusion in people with dementia.
The following techniques may help reduce agitation and promote relaxation in people with dementia.
- Music therapy, which involves listening to soothing music
- Light exercise
- Watching videos of family members
- Pet therapy, which involves use of animals, such as visits from dogs, to promote improved moods and behaviors in people with dementia
- Aromatherapy, which uses fragrant plant oils
- Massage therapy
- Art therapy, which involves creating art, focusing on the process rather than the outcome
Alternative medicine
Several dietary supplements, herbal remedies and therapies have been studied for people with dementia. But there’s no convincing evidence for any of these. Use caution when considering taking dietary supplements, vitamins or herbal remedies, especially if you’re taking other medications. These remedies aren’t regulated, and claims about their benefits aren’t always based on scientific research.
While some studies suggest that vitamin E supplements may be helpful for Alzheimer’s disease, the results have been mixed. Also, high doses of vitamin E can pose risks. Taking vitamin E supplements is generally not recommended, but including foods high in vitamin E, such as nuts, in your diet, is.
Dementia home remedies
Dementia symptoms and behavior problems will progress over time. Caregivers and care partners might try the following suggestions:
- Enhance communication. When talking with your loved one, maintain eye contact. Speak slowly in simple sentences, and don’t rush the response. Present one idea or instruction at a time. Use gestures and cues, such as pointing to objects.
- Encourage exercise. The main benefits of exercise in people with dementia include improved strength, balance and cardiovascular health. Exercise might also help with symptoms such as restlessness. There is growing evidence that exercise also protects the brain from dementia, especially when combined with a healthy diet and treatment for risk factors for cardiovascular disease. Some research also shows that physical activity might slow the progression of impaired thinking in people with Alzheimer’s disease, and it can lessen symptoms of depression.
- Engage in activity. Plan activities the person with dementia enjoys and can do. Dancing, painting, gardening, cooking, singing and other activities can be fun, can help you connect with your loved one, and can help your loved one focus on what he or she can still do.
- Establish a nighttime ritual. Behavior is often worse at night. Try to establish going-to-bed rituals that are calming and away from the noise of television, meal cleanup and active family members. Leave night lights on in the bedroom, hall and bathroom to prevent disorientation. Limiting caffeine, discouraging napping and offering opportunities for exercise during the day might ease nighttime restlessness.
- Keep a calendar. A calendar might help your loved one remember upcoming events, daily activities and medication schedules. Consider sharing a calendar with your loved one.
- Plan for the future. Develop a plan with your loved one while he or she is able to participate that identifies goals for future care. Support groups, legal advisers, family members and others might be able to help. You’ll need to consider financial and legal issues, safety and daily living concerns, and long-term care options. See Dementia Care Plans below.
Alzheimer’s disease
Alzheimer’s disease is an irreversible, progressive brain disorder that slowly destroys memory and thinking skills and eventually, the ability to carry out the simplest tasks 20. In most people with the disease—those with the late-onset type—symptoms first appear in their mid-60s. Early-onset Alzheimer’s occurs between a person’s 30s and mid-60s and is very rare. Alzheimer’s disease is the most common cause of dementia among older adults 21.
Alzheimer’s disease begins slowly and it usually begins after age 60 21. The risk goes up as you get older. Your risk is also higher if a family member has had the disease 21. It first involves the parts of the brain that control thought, memory and language. People with Alzheimer’s disease may have trouble remembering things that happened recently or names of people they know. A related problem, mild cognitive impairment, causes more memory problems than normal for people of the same age. Many, but not all, people with mild cognitive impairment will develop Alzheimer’s Disease 21.
In Alzheimer’s disease, over time, symptoms get worse. People may not recognize family members. They may have trouble speaking, reading or writing. They may forget how to brush their teeth or comb their hair. Later on, they may become anxious or aggressive, or wander away from home. Eventually, they need total care. This can cause great stress for family members who must care for them.
No treatment can stop the disease. However, some drugs may help keep symptoms from getting worse for a limited time.
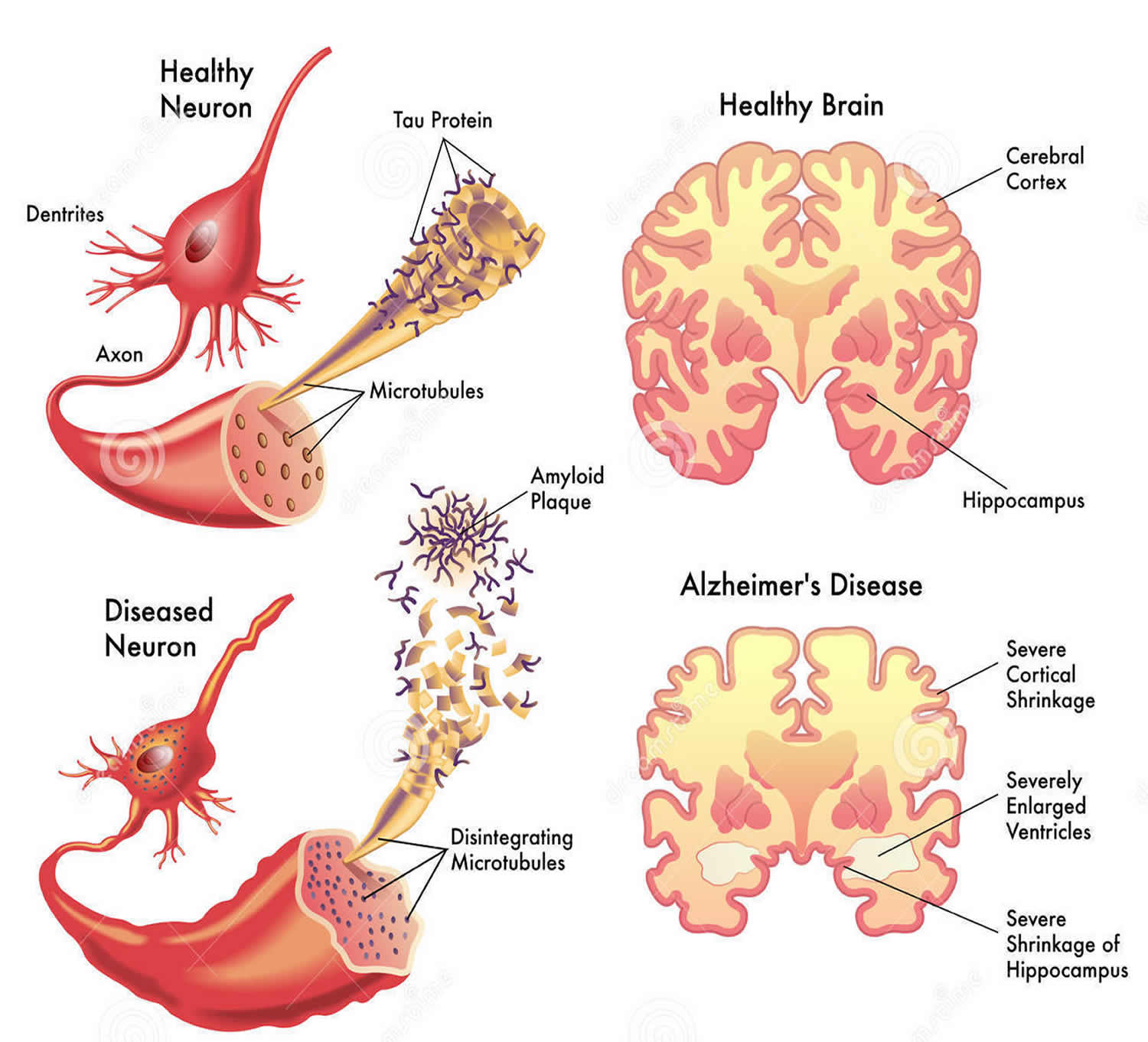
Alzheimer’s disease is named after Dr. Alois Alzheimer. In 1906, Dr. Alzheimer noticed changes in the brain tissue of a woman who had died of an unusual mental illness 20. Her symptoms included memory loss, language problems, and unpredictable behavior. After she died, he examined her brain and found many abnormal clumps (now called amyloid plaques) and tangled bundles of fibers (now called neurofibrillary, or tau, tangles).
These plaques and tangles in the brain are still considered some of the main features of Alzheimer’s disease 20. Another feature is the loss of connections between nerve cells (neurons) in the brain. Neurons transmit messages between different parts of the brain and from the brain to muscles and organs in the body. Many other complex brain changes are thought to play a role in Alzheimer’s disease, too.
This damage initially appears to take place in the hippocampus, the part of the brain essential in forming memories. As neurons die, additional parts of the brain are affected. By the final stage of Alzheimer’s disease, damage is widespread, and brain tissue has shrunk significantly.
What Happens to the Brain in Alzheimer’s Disease?
The healthy human brain contains tens of billions of neurons—specialized cells that process and transmit information via electrical and chemical signals. They send messages between different parts of the brain, and from the brain to the muscles and organs of the body. Alzheimer’s disease disrupts this communication among neurons, resulting in loss of function and cell death.
Key Biological Processes in the Brain
Most neurons have three basic parts: a cell body, multiple dendrites, and an axon (see Figure 2).
- The cell body contains the nucleus, which houses the genetic blueprint that directs and regulates the cell’s activities.
- Dendrites are branch-like structures that extend from the cell body and collect information from other neurons.
- The axon is a cable-like structure at the end of the cell body opposite the dendrites and transmits messages to other neurons.
The function and survival of neurons depend on several key biological processes:
- Communication. Neurons are constantly in touch with neighboring brain cells. When a neuron receives signals from other neurons, it generates an electrical charge that travels down the length of its axon and releases neurotransmitter chemicals across a tiny gap, called a synapse. Like a key fitting into a lock, each neurotransmitter molecule then binds to specific receptor sites on a dendrite of a nearby neuron. This process triggers chemical or electrical signals that either stimulate or inhibit activity in the neuron receiving the signal. Communication often occurs across networks of brain cells. In fact, scientists estimate that in the brain’s communications network, one neuron may have as many as 7,000 synaptic connections with other neurons.
- Metabolism. Metabolism—the breaking down of chemicals and nutrients within a cell—is critical to healthy cell function and survival. To perform this function, cells require energy in the form of oxygen and glucose, which are supplied by blood circulating through the brain. The brain has one of the richest blood supplies of any organ and consumes up to 20 percent of the energy used by the human body—more than any other organ.
- Repair, remodeling, and regeneration. Unlike many cells in the body, which are relatively short-lived, neurons have evolved to live a long time—more than 100 years in humans. As a result, neurons must constantly maintain and repair themselves. Neurons also continuously adjust, or “remodel,” their synaptic connections depending on how much stimulation they receive from other neurons. For example, they may strengthen or weaken synaptic connections, or even break down connections with one group of neurons and build new connections with a different group. Adult brains may even generate new neurons—a process called neurogenesis. Remodeling of synaptic connections and neurogenesis are important for learning, memory, and possibly brain repair.
Neurons are a major player in the central nervous system, but other cell types are also key to healthy brain function. In fact, glial cells are by far the most numerous cells in the brain, outnumbering neurons by about 10 to 1 (see Figure 3). These cells, which come in various forms—such as microglia, astrocytes, and oligodendrocytes—surround and support the function and healthy of neurons. For example, microglia protect neurons from physical and chemical damage and are responsible for clearing foreign substances and cellular debris from the brain. To carry out these functions, glial cells often collaborate with blood vessels in the brain. Together, glial and blood vessel cells regulate the delicate balance within the brain to ensure that it functions at its best.
Figure 1. Alzheimer’s disease brain
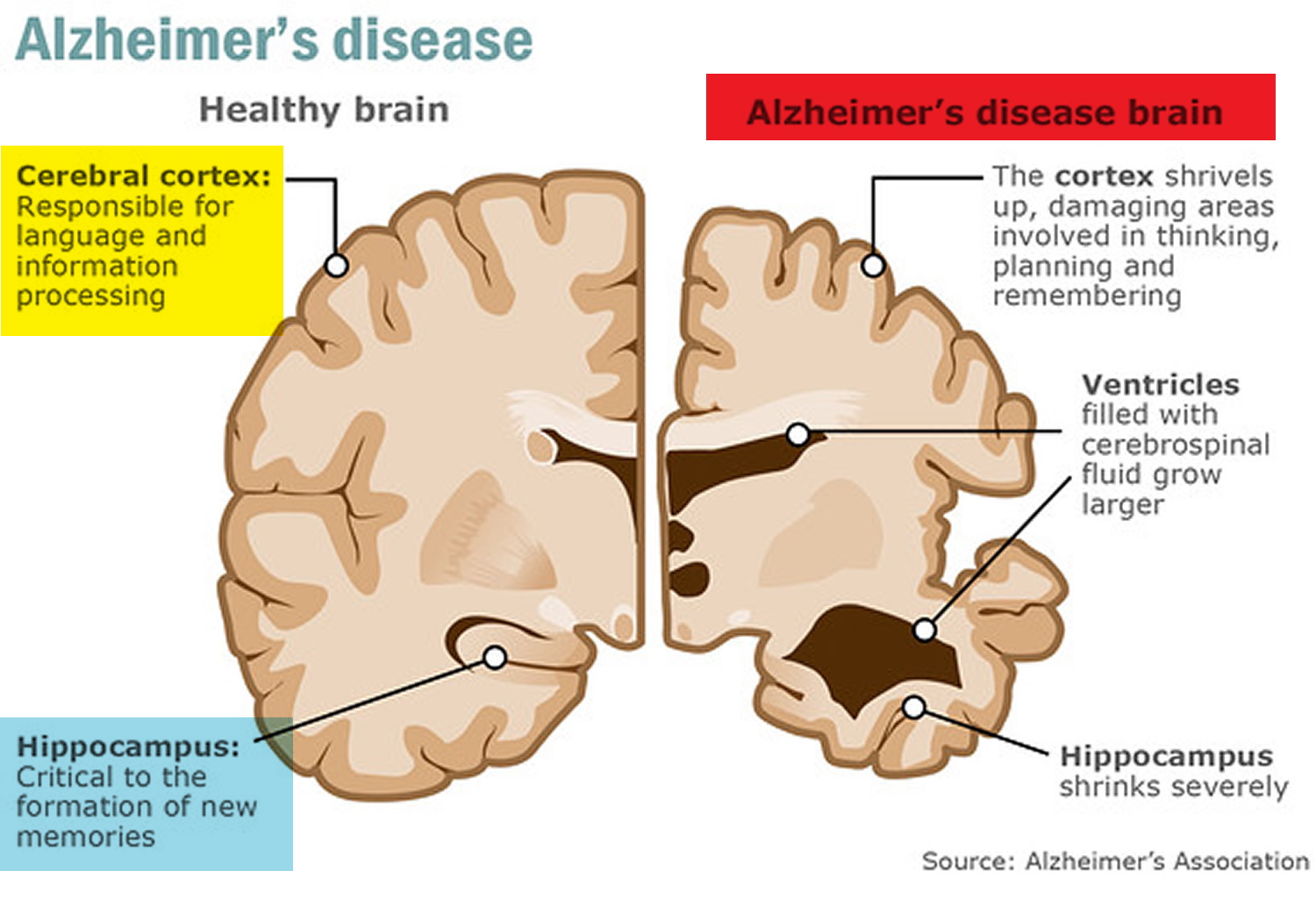
Figure 2. Alzheimer’s disease brain with amyloid plaques and neurofibrillary or tau, tangles
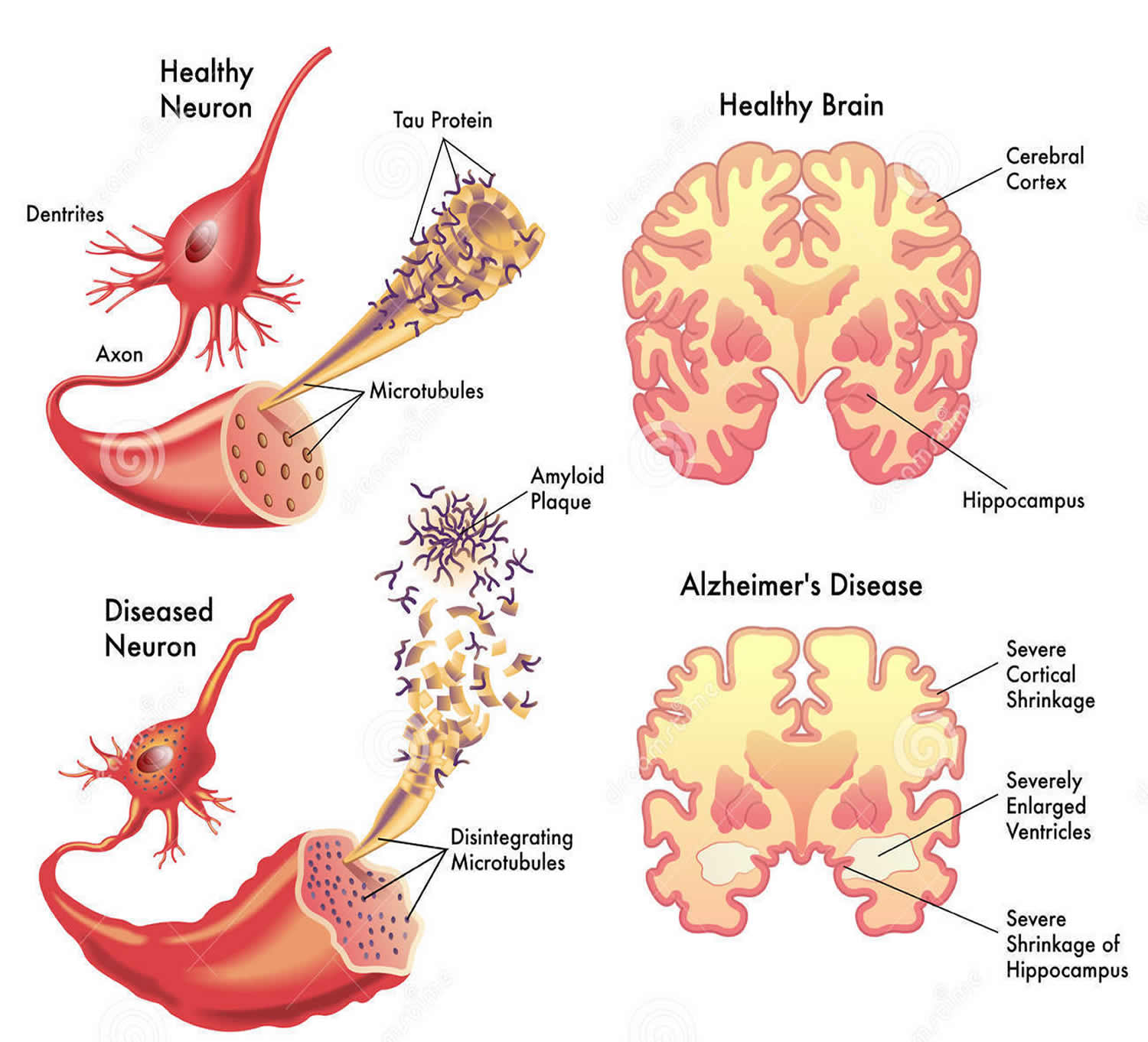
Figure 3. Glial (neuroglial) cells in the brain (astrocytes, oligodendrocytes, and microglial cells)
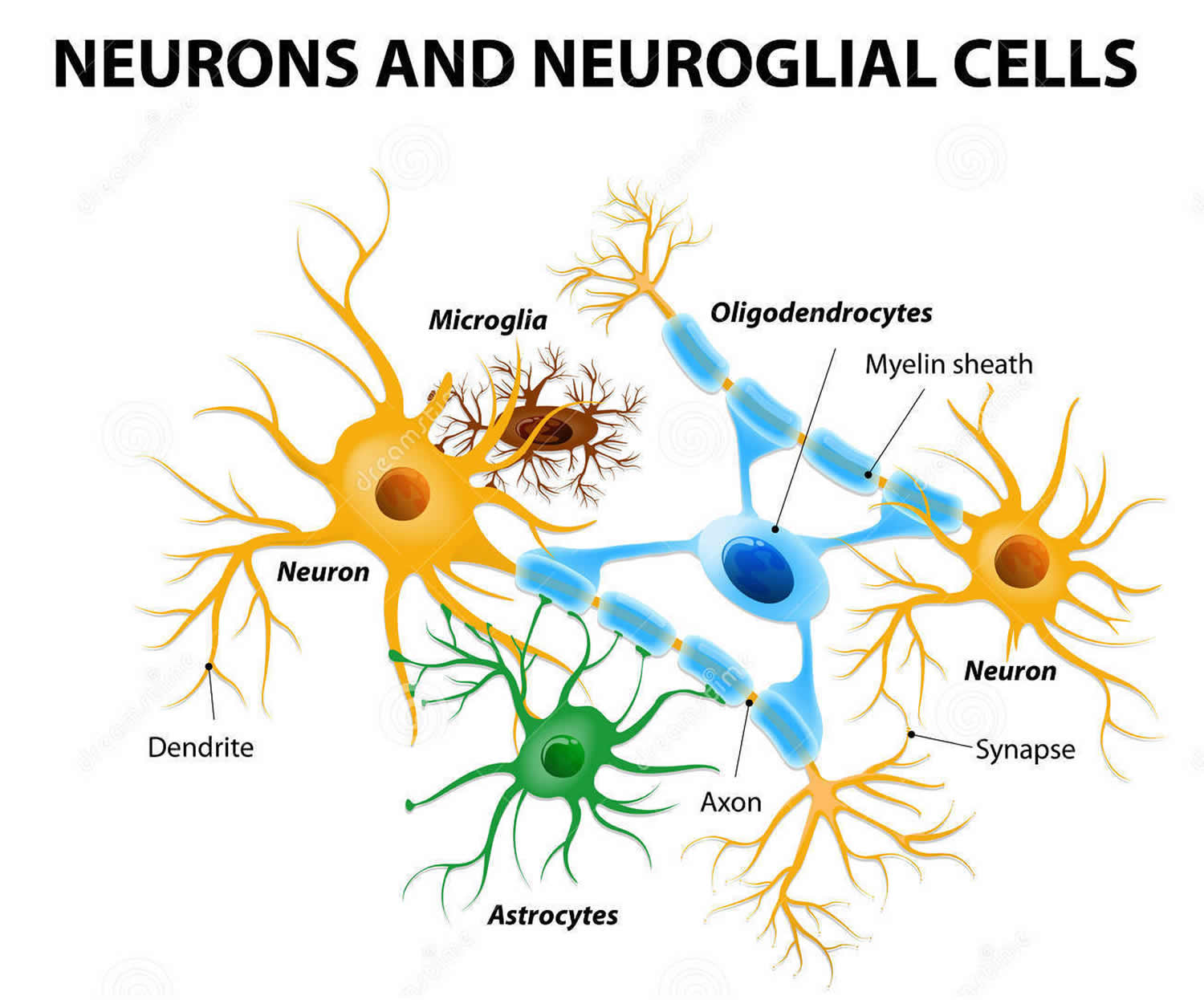
Footnote: Glial (Neuroglial) cells do not conduct nerve impulses, but, instead, support, nourish, and protect the neurons. Glial cells (glia) are far more numerous than neurons and, unlike neurons, are capable of mitosis. Glial roles that are well-established include maintaining the ionic milieu of nerve cells, modulating the rate of nerve signal propagation, modulating synaptic action by controlling the uptake of neurotransmitters, providing a scaffold for some aspects of neural development, and aiding in (or preventing, in some instances) recovery from neural injury.
[Source 22 ]How Does Alzheimer’s Disease Affect the Brain?
The brain typically shrinks to some degree in healthy aging but, surprisingly, does not lose neurons in large numbers. In Alzheimer’s disease, however, damage is widespread, as many neurons stop functioning, lose connections with other neurons, and die. Alzheimer’s disrupts processes vital to neurons and their networks, including communication, metabolism, and repair 23.
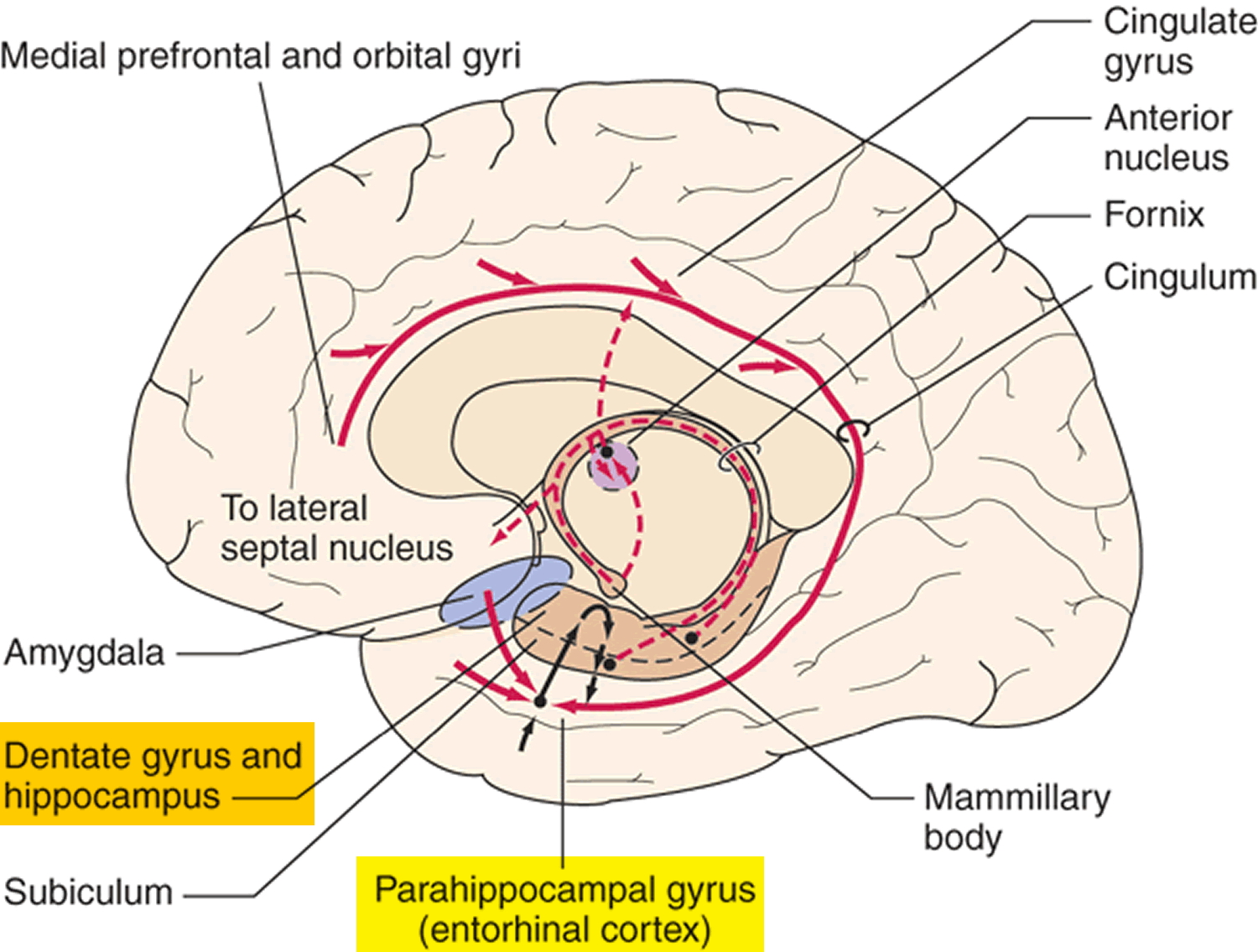
At first, Alzheimer’s disease typically destroys neurons and their connections in parts of the brain involved in memory, including the entorhinal cortex and hippocampus. It later affects areas in the cerebral cortex responsible for language, reasoning, and social behavior. Eventually, many other areas of the brain are damaged. Over time, a person with Alzheimer’s gradually loses his or her ability to live and function independently. Ultimately, the disease is fatal 23.
Figure 4. Brain showing Entorhinal cortex (parahippocampal gyrus) and Hippocampus
What are the main characteristics of the brain with Alzheimer’s disease?
Many molecular and cellular changes take place in the brain of a person with Alzheimer’s disease. These changes can be observed in brain tissue under the microscope after death. Investigations are underway to determine which changes may cause Alzheimer’s and which may be a result of the disease.
Amyloid Plaques
The beta-amyloid protein involved in Alzheimer’s comes in several different molecular forms that collect between neurons. It is formed from the breakdown of a larger protein, called amyloid precursor protein. One form, beta-amyloid 42, is thought to be especially toxic. In the Alzheimer’s brain, abnormal levels of this naturally occurring protein clump together to form plaques that collect between neurons and disrupt cell function. Research is ongoing to better understand how, and at what stage of the disease, the various forms of beta-amyloid influence Alzheimer’s.
Neurofibrillary Tangles
Neurofibrillary tangles are abnormal accumulations of a protein called tau that collect inside neurons. Healthy neurons, in part, are supported internally by structures called microtubules, which help guide nutrients and molecules from the cell body to the axon and dendrites. In healthy neurons, tau normally binds to and stabilizes microtubules. In Alzheimer’s disease, however, abnormal chemical changes cause tau to detach from microtubules and stick to other tau molecules, forming threads that eventually join to form tangles inside neurons. These tangles block the neuron’s transport system, which harms the synaptic communication between neurons.
Emerging evidence suggests that Alzheimer’s-related brain changes may result from a complex interplay among abnormal tau and beta-amyloid proteins and several other factors. It appears that abnormal tau accumulates in specific brain regions involved in memory. Beta-amyloid clumps into plaques between neurons. As the level of beta-amyloid reaches a tipping point, there is a rapid spread of tau throughout the brain.
Chronic Inflammation
Research suggests that chronic inflammation may be caused by the buildup of glial cells normally meant to help keep the brain free of debris. One type of glial cell, microglia, engulfs and destroys waste and toxins in a healthy brain. In Alzheimer’s, microglia fail to clear away waste, debris, and protein collections, including beta-amyloid plaques. Researchers are trying to find out why microglia fail to perform this vital function in Alzheimer’s.
One focus of study is a gene called TREM2. Normally, TREM2 tells the microglia cells to clear beta-amyloid plaques from the brain and helps fight inflammation in the brain. In the brains of people where this gene does not function normally, plaques build up between neurons. Astrocytes—another type of glial cell—are signaled to help clear the buildup of plaques and other cellular debris left behind. These microglia and astrocytes collect around the neurons but fail to perform their debris-clearing function. In addition, they release chemicals that cause chronic inflammation and further damage the neurons they are meant to protect.
Vascular Contributions to Alzheimer’s Disease
People with dementia seldom have only Alzheimer’s-related changes in their brains. Any number of vascular issues—problems that affect blood vessels, such as beta-amyloid deposits in brain arteries, atherosclerosis (hardening of the arteries), and mini-strokes—may also be at play.
Vascular problems may lead to reduced blood flow and oxygen to the brain, as well as a breakdown of the blood-brain barrier, which usually protects the brain from harmful agents while allowing in glucose and other necessary factors. In a person with Alzheimer’s, a faulty blood-brain barrier prevents glucose from reaching the brain and prevents the clearing away of toxic beta-amyloid and tau proteins. This results in inflammation, which adds to vascular problems in the brain. Because it appears that Alzheimer’s is both a cause and consequence of vascular problems in the brain, researchers are seeking interventions to disrupt this complicated and destructive cycle.
Loss of Neuronal Connections and Cell Death
In Alzheimer’s disease, as neurons are injured and die throughout the brain, connections between networks of neurons may break down, and many brain regions begin to shrink. By the final stages of Alzheimer’s, this process—called brain atrophy—is widespread, causing significant loss of brain volume.
How many Americans have Alzheimer’s disease?
Estimates vary, but experts suggest that more than 5 million Americans may have Alzheimer’s 20. Unless the disease can be effectively treated or prevented, the number of people with it will increase significantly if current population trends continue. This is because increasing age is the most important known risk factor for Alzheimer’s disease 20.
What does Alzheimer’s disease look like?
Memory problems are typically one of the first signs of Alzheimer’s, though initial symptoms may vary from person to person. A decline in other aspects of thinking, such as finding the right words, vision/spatial issues, and impaired reasoning or judgment, may also signal the very early stages of Alzheimer’s disease. Mild cognitive impairment is a condition that can be an early sign of Alzheimer’s, but not everyone with mild cognitive impairment will develop the disease.
People with Alzheimer’s have trouble doing everyday things like driving a car, cooking a meal, or paying bills. They may ask the same questions over and over, get lost easily, lose things or put them in odd places, and find even simple things confusing. As the disease progresses, some people become worried, angry, or violent. Later on, they may become anxious or aggressive, or wander away from home. Eventually, they need total care. This can cause great stress for family members who must care for them.
No treatment can stop the disease. However, some drugs may help keep symptoms from getting worse for a limited time.
How long can a person live with Alzheimer’s disease?
The time from diagnosis to death varies—as little as 3 or 4 years if the person is older than 80 when diagnosed, to as long as 10 or more years if the person is younger 20.
Alzheimer’s disease is currently ranked as the sixth leading cause of death in the United States, but recent estimates indicate that the disorder may rank third, just behind heart disease and cancer, as a cause of death for older people 20.
Although treatment can help manage symptoms in some people, currently there is no cure for this devastating disease 20.
Alzheimer’s disease causes
Alzheimer’s disease is an irreversible, progressive brain disorder that slowly destroys memory and thinking skills, and eventually the ability to carry out the simplest tasks 24.
Scientists believe that many factors influence when Alzheimer’s disease begins and how it progresses.
Increasing age is the most important known risk factor for Alzheimer’s disease 24. The number of people with the disease doubles every 5 years beyond age 65. About one-third of all people age 85 and older may have Alzheimer’s disease 24.
The causes of late-onset Alzheimer’s, the most common form of the disease, probably include a combination of genetic, lifestyle, and environmental factors 24. The importance of any one of these factors in increasing or decreasing the risk of development Alzheimer’s may differ from person to person.
Scientists are learning how age-related changes in the brain may harm nerve cells and contribute to Alzheimer’s damage. These age-related changes include atrophy (shrinking) of certain parts of the brain, inflammation, production of unstable molecules called free radicals, and breakdown of energy production within cells.
As scientists learn more about this devastating disease, they realize that genes also play an important role.
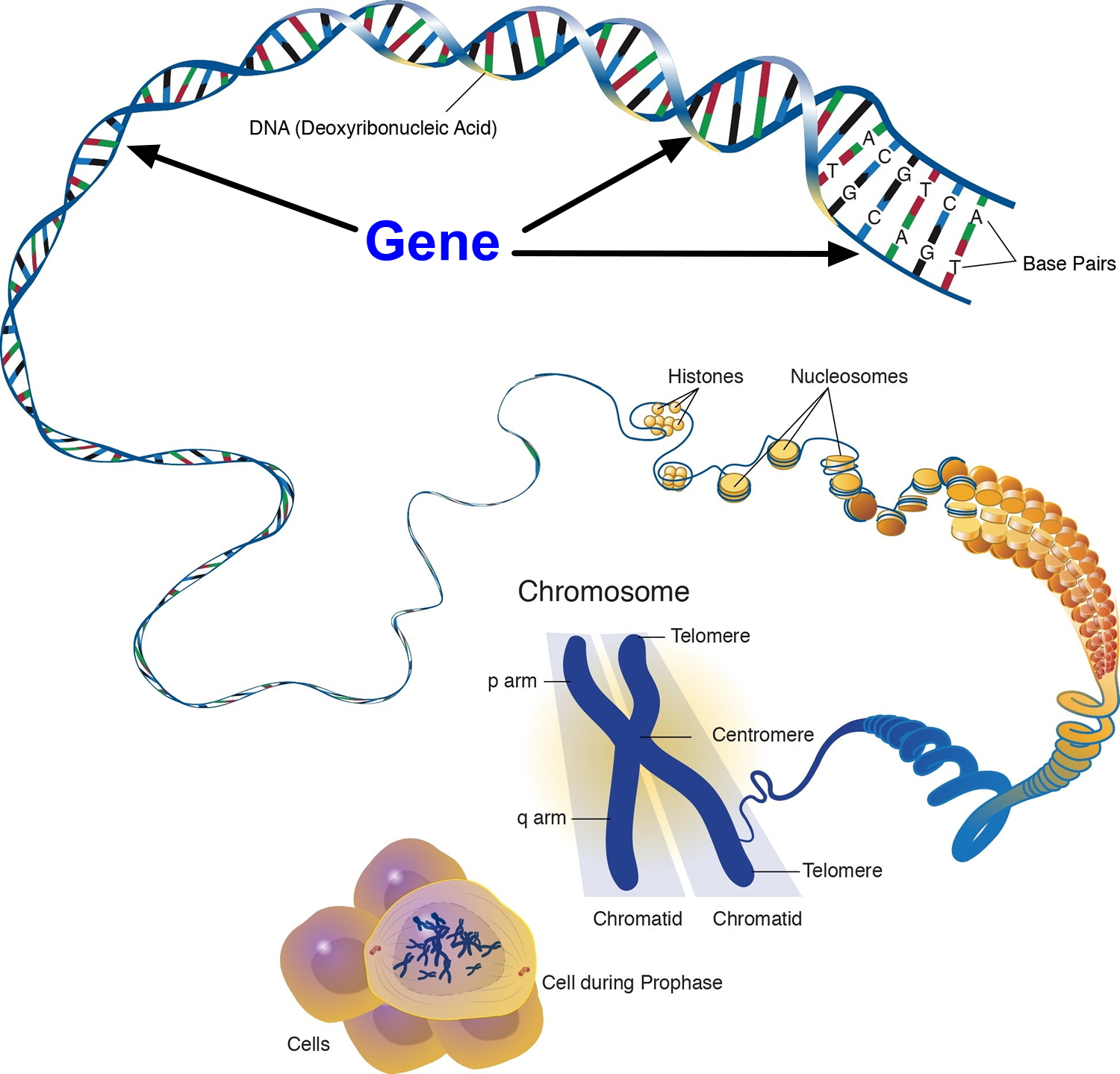
Each human cell contains the instructions a cell needs to do its job. These instructions are made up of DNA, which is packed tightly into structures called chromosomes. Each chromosome has thousands of segments called genes.
Genes are passed down from a person’s birth parents. They carry information that defines traits such as eye color and height. Genes also play a role in keeping the body’s cells healthy. Problems with genes—even small changes to a gene—can cause diseases like Alzheimer’s disease.
Figure 5. Genes
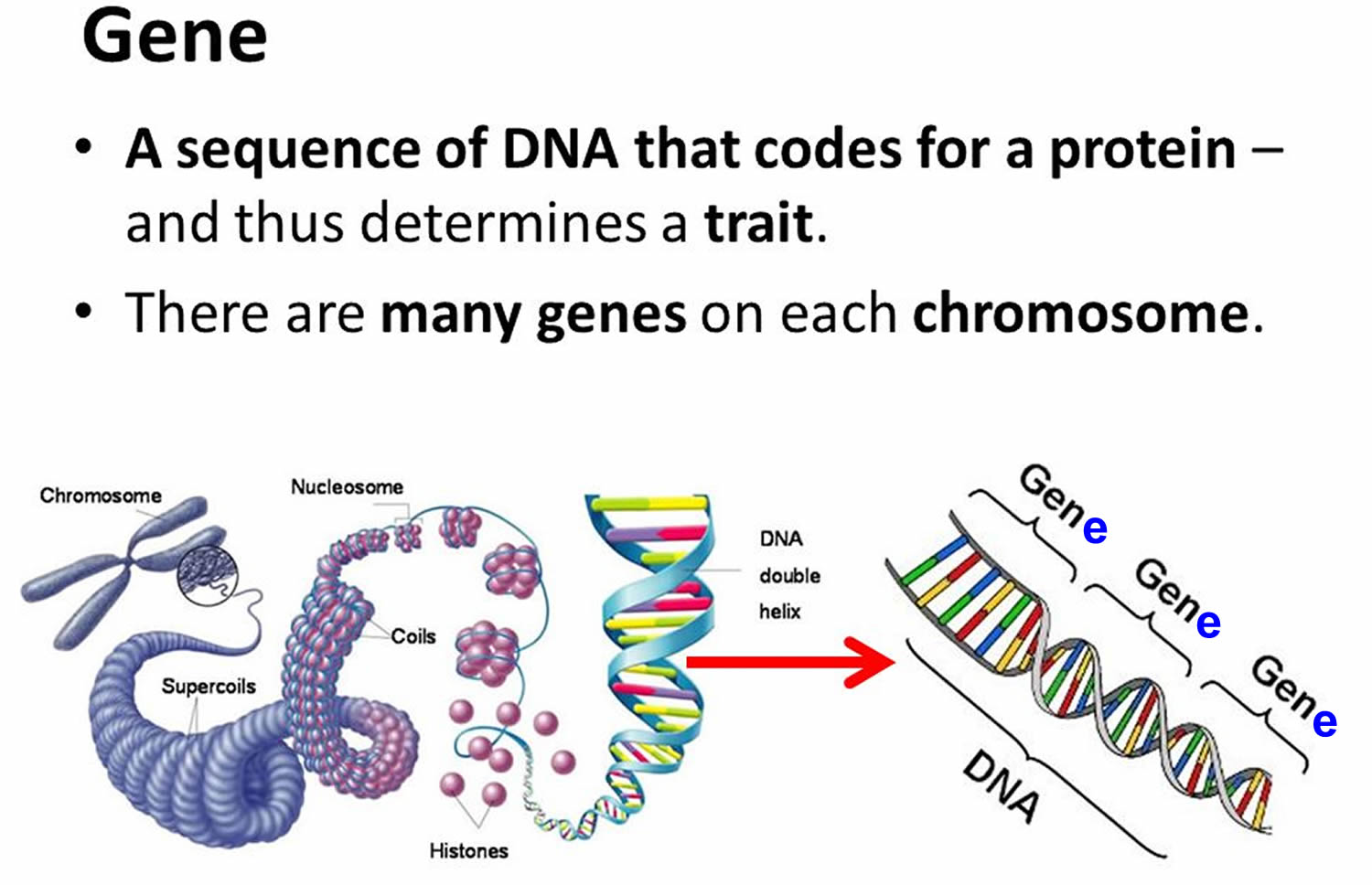
Figure 6. Genes (DNA) come from Chromosomes that reside inside your cells nucleus
Some diseases are caused by a genetic mutation, or permanent change in one or more specific genes. If a person inherits from a parent a genetic mutation that causes a certain disease, then he or she will usually get the disease. Sickle cell anemia, cystic fibrosis, and early-onset familial Alzheimer’s disease are examples of inherited genetic disorders.
In other diseases, a genetic variant may occur. A single gene can have many variants. Sometimes, this difference in a gene can cause a disease directly. More often, a variant plays a role in increasing or decreasing a person’s risk of developing a disease or condition. When a genetic variant increases disease risk but does not directly cause a disease, it is called a genetic risk factor.
Alzheimer’s Disease Genetics
There are two types of Alzheimer’s—early-onset and late-onset. Both types have a genetic component.
Table 1. Some differences between Late-Onset and Early-Onset Alzheimer’s disease
| Late-Onset Alzheimer’s | Early-Onset Alzheimer’s |
|---|---|
| Signs first appear in a person’s mid-60s | Signs first appear between a person’s 30s and mid-60s |
| Most common type (over 90% of Alzheimer’s) | Very rare (under 10% of Alzheimer’s) |
| May involve a gene called APOE ɛ4 | Usually caused by gene changes passed down from parent to child |
Late-Onset Alzheimer’s Disease
Most people with Alzheimer’s disease have the late-onset form of the disease, in which symptoms become apparent in the mid-60s.
Researchers have not found a specific gene that directly causes the late-onset form of the disease 24. However, one genetic risk factor—having one form of the apolipoprotein E (APOE) gene on chromosome 19—does increase a person’s risk. APOE comes in several different forms, or alleles:
- APOE ɛ2 is relatively rare and may provide some protection against the disease. If Alzheimer’s disease occurs in a person with this allele, it usually develops later in life than it would in someone with the APOE ɛ4 gene.
- APOE ɛ3, the most common allele, is believed to play a neutral role in the disease—neither decreasing nor increasing risk.
- APOE ɛ4 increases risk for Alzheimer’s disease and is also associated with an earlier age of disease onset. A person has zero, one, or two APOE ɛ4 alleles. Having more APOE ɛ4 alleles increases the risk of developing Alzheimer’s.
APOE ɛ4 is called a risk-factor gene because it increases a person’s risk of developing the disease. However, inheriting an APOE ɛ4 allele does not mean that a person will definitely develop Alzheimer’s disease. Some people with an APOE ɛ4 allele never get the disease, and others who develop Alzheimer’s disease do not have any APOE ɛ4 alleles.
Early-Onset Alzheimer’s Disease
Early-onset Alzheimer’s disease occurs between a person’s 30s to mid-60s and represents less than 10 percent of all people with Alzheimer’s disease 24. Some cases are caused by an inherited change in one of three genes, resulting in a type known as early-onset Familial Alzheimer’s disease. For other cases of early-onset Alzheimer’s disease, research suggests there may be a genetic component related to factors other than these three genes.
A child whose biological mother or father carries a genetic mutation for early-onset Familial Alzheimer’s disease has a 50/50 chance of inheriting that mutation. If the mutation is in fact inherited, the child has a very strong probability of developing early-onset Familial Alzheimer’s disease.
Early-onset Familial Alzheimer’s disease is caused by any one of a number of different single gene mutations on chromosomes 21, 14, and 1 24. Each of these mutations causes abnormal proteins to be formed. Mutations on chromosome 21 cause the formation of abnormal amyloid precursor protein (APP) 24. A mutation on chromosome 14 causes abnormal presenilin 1 to be made, and a mutation on chromosome 1 leads to abnormal presenilin 2 24.
Each of these mutations plays a role in the breakdown of APP, a protein whose precise function is not yet fully understood. This breakdown is part of a process that generates harmful forms of amyloid plaques, a hallmark of Alzheimer’s disease.
Health, Environmental, and Lifestyle Factors
Research suggests that a host of factors beyond genetics may play a role in the development and course of Alzheimer’s disease. There is a great deal of interest, for example, in the relationship between cognitive decline and vascular conditions such as heart disease, stroke, and high blood pressure, as well as metabolic conditions such as diabetes and obesity. Ongoing research will help us understand whether and how reducing risk factors for these conditions may also reduce the risk of Alzheimer’s.
A nutritious diet, physical activity, social engagement, and mentally stimulating pursuits have all been associated with helping people stay healthy as they age. These factors might also help reduce the risk of cognitive decline and Alzheimer’s disease. Clinical trials are testing some of these possibilities.
Alzheimer’s disease diagnosis
Doctors use several methods and tools to help determine whether a person who is having memory problems has “possible Alzheimer’s dementia” (dementia may be due to another cause), “probable Alzheimer’s dementia” (no other cause for dementia can be found), or some other problem 25.
To diagnose Alzheimer’s, doctors may 25:
- Ask the person and a family member or friend questions about overall health, use of prescription and over-the-counter medicines, diet, past medical problems, ability to carry out daily activities, and changes in behavior and personality
- Conduct tests of memory, problem solving, attention, counting, and language
- Carry out standard medical tests, such as blood and urine tests, to identify other possible causes of the problem
- Perform brain scans, such as computed tomography (CT), magnetic resonance imaging (MRI), or positron emission tomography (PET), to rule out other possible causes for symptoms
These tests may be repeated to give doctors information about how the person’s memory and other cognitive functions are changing over time. They can also help diagnose other causes of memory problems, such as stroke, tumor, Parkinson’s disease, sleep disturbances, side effects of medication, an infection, mild cognitive impairment, or a non-Alzheimer’s dementia, including vascular dementia. Some of these conditions may be treatable and possibly reversible.
People with memory problems should return to the doctor every 6 to 12 months 25.
- It’s important to note that Alzheimer’s disease can be definitively diagnosed only after death, by linking clinical measures with an examination of brain tissue in an autopsy 25.
What Happens Next?
If a primary care doctor suspects mild cognitive impairment or possible Alzheimer’s, he or she may refer the patient to a specialist who can provide a detailed diagnosis or further assessment. Specialists include:
- Geriatricians, who manage health care in older adults and know how the body changes as it ages and whether symptoms indicate a serious problem
- Geriatric psychiatrists, who specialize in the mental and emotional problems of older adults and can assess memory and thinking problems
- Neurologists, who specialize in abnormalities of the brain and central nervous system and can conduct and review brain scans
- Neuropsychologists, who can conduct tests of memory and thinking
Memory clinics and centers, including Alzheimer’s Disease Research Centers, offer teams of specialists who work together to diagnose the problem. Tests often are done at the clinic or center, which can speed up diagnosis.
What are the benefits of Early Alzheimer’s Disease Diagnosis?
Early, accurate diagnosis is beneficial for several reasons. Beginning treatment early in the disease process may help preserve daily functioning for some time, even though the underlying Alzheimer’s process cannot be stopped or reversed.
Having an early diagnosis helps people with Alzheimer’s Disease and their families:
- Plan for the future
- Take care of financial and legal matters
- Address potential safety issues
- Learn about living arrangements
- Develop support networks
In addition, an early diagnosis gives people greater opportunities to participate in clinical trials that are testing possible new treatments for Alzheimer’s disease or in other research studies.
Alzheimer’s disease treatment
Alzheimer’s disease is complex, and it is unlikely that any one drug or other intervention will successfully treat it 26. Current approaches focus on helping people maintain mental function, manage behavioral symptoms, and slow or delay the symptoms of disease.
Several prescription drugs are currently approved by the U.S. Food and Drug Administration (FDA) to treat people who have been diagnosed with Alzheimer’s disease. Treating the symptoms of Alzheimer’s can provide patients with comfort, dignity, and independence for a longer period of time and can encourage and assist their caregivers as well.
Most medicines work best for people in the early or middle stages of Alzheimer’s. For example, they can keep memory loss from getting worse over time. It is important to understand that none of these medications stops the disease itself.
Treatment for Mild to Moderate Alzheimer’s disease
Medications called cholinesterase inhibitors are prescribed for mild to moderate Alzheimer’s disease. These drugs may help delay or prevent symptoms from becoming worse for a limited time and may help control some behavioral symptoms. The medications are Razadyne® (galantamine), Exelon® (rivastigmine), and Aricept® (donepezil) 26.
Scientists do not yet fully understand how cholinesterase inhibitors work to treat Alzheimer’s disease, but research indicates that they prevent the breakdown of acetylcholine, a brain chemical believed to be important for memory and thinking 26. As Alzheimer’s progresses, the brain produces less and less acetylcholine; therefore, cholinesterase inhibitors may eventually lose their effect.
No published study directly compares these drugs. Because they work in a similar way, switching from one of these drugs to another probably will not produce significantly different results. However, an Alzheimer’s patient may respond better to one drug than another.
Treatment for Moderate to Severe Alzheimer’s disease
A medication known as Namenda® (memantine), an N-methyl D-aspartate (NMDA) antagonist, is prescribed to treat moderate to severe Alzheimer’s disease 26. This drug’s main effect is to delay progression of some of the symptoms of moderate to severe Alzheimer’s. It may allow patients to maintain certain daily functions a little longer than they would without the medication. For example, Namenda® may help a patient in the later stages of the disease maintain his or her ability to use the bathroom independently for several more months, a benefit for both patients and caregivers.
The FDA has also approved Aricept® and Namzaric®, a combination of Namenda® and Aricept®, for the treatment of moderate to severe Alzheimer’s disease 26.
Namenda® is believed to work by regulating glutamate, an important brain chemical. When produced in excessive amounts, glutamate may lead to brain cell death. Because NMDA antagonists work very differently from cholinesterase inhibitors, the two types of drugs can be prescribed in combination 26.
Table 2. Medicines used to treat Alzheimer’s Disease
| Drug Name | Drug Type and Use | Delivery Method | How It Works | Manufacturer’s Recommended Dosage | Common Side Effects | Prescribing information |
|---|---|---|---|---|---|---|
| Aducanumab (Aduhelm®) | Disease-modifying immunotherapy prescribed to treat mild cognitive impairment or mild Alzheimer’s | Intravenous (IV): Dose is determined by a person’s weight; given over one hour every four weeks; most people will start with a lower dose and over a period of time increase the amount of medicine to reach the full prescription dose | Removes abnormal beta-amyloid to help reduce the number of plaques in the brain |
| Amyloid-related imaging abnormalities (ARIA), which can lead to fluid buildup or bleeding in the brain; also headache, dizziness, falls, diarrhea, confusion | https://www.biogencdn.com/us/aduhelm-pi.pdf |
| Donepezil (Aricept®) | Cholinesterase inhibitor prescribed to treat symptoms of mild, moderate, and severe Alzheimer’s | Tablet: Once a day; dosage may be increased over time if well tolerated Orally disintegrating tablet: Same dosing regimen as above | Prevents the breakdown of acetylcholine in the brain |
| Nausea, vomiting, diarrhea, muscle cramps, fatigue, weight loss | http://www.aricept.com |
| Rivastigmine (Exelon®) | Cholinesterase inhibitor prescribed to treat symptoms of mild, moderate, and severe Alzheimer’s | Capsule: Twice a day; dosage may be increased over time, at minimum two-week intervals, if well tolerated Patch: Once a day; dosage amount may be increased over time, at minimum four-week intervals, if well tolerated | Prevents the breakdown of acetylcholine and butyrylcholine (a brain chemical similar to acetylcholine) in the brain |
| Nausea, vomiting, diarrhea, weight loss, indigestion, muscle weakness | https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/022083s008lbl.pdf |
| Memantine (Namenda®) | N-methyl D-aspartate (NMDA) antagonist prescribed to treat symptoms of moderate to severe Alzheimer’s | Tablet: Once a day; dosage may be increased in amount and frequency (up to twice a day) if well tolerated Oral solution: Same dosage as tablet Extended-release capsule: Once a day; dosage may increase in amount over time, at minimum one-week intervals, if well tolerated | Blocks the toxic effects associated with excess glutamate and regulates glutamate activation |
| Dizziness, headache, diarrhea, constipation, confusion | https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/021487s010s012s014,021627s008lbl.pdf |
| Manufactured combination of memantine and donepezil (Namzaric®) | NMDA antagonist and cholinesterase inhibitor prescribed to treat symptoms of moderate to severe Alzheimer’s | Extended-release capsule: Once a day; initial dosage depends on whether the person is already on a stable dose of memantine and/or donepezil; dosage may increase over time, at minimum one-week intervals, if well tolerated | Blocks the toxic effects associated with excess glutamate and prevents the breakdown of acetylcholine in the brain |
| Headache, nausea, vomiting, diarrhea, dizziness, anorexia | https://www.rxabbvie.com/pdf/namzaric_pi.pdf |
| Galantamine (Razadyne®) | Cholinesterase inhibitor prescribed to treat symptoms of mild to moderate Alzheimer’s | Tablet: Twice a day; dosing may increase over time, at minimum four-week intervals, if well tolerated Extended-release capsule: Same dosage as tablet but taken once a day | Prevents the breakdown of acetylcholine and stimulates nicotinic receptors to release more acetylcholine in the brain |
| Nausea, vomiting, diarrhea, decreased appetite, dizziness, headache | https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/021615s021lbl.pdf |
Footnote: * Available as a generic drug.
[Source: National Institutes of Health. National Institute on Aging 26 ]Dosage and Side Effects
Doctors usually start patients at low drug doses and gradually increase the dosage based on how well a patient tolerates the drug. There is some evidence that certain patients may benefit from higher doses of the cholinesterase inhibitors. However, the higher the dose, the more likely side effects are to occur.
Patients should be monitored when a drug is started. All of these medicines have possible side effects, including nausea, vomiting, diarrhea, and loss of appetite. Report any unusual symptoms to the prescribing doctor right away. It is important to follow the doctor’s instructions when taking any medication, including vitamins and herbal supplements. Also, let the doctor know before adding or changing any medications.
Managing Behavior
Common behavioral symptoms of Alzheimer’s include sleeplessness, wandering, agitation, anxiety, aggression, restlessness, and depression 26. Scientists are learning why these symptoms occur and are studying new treatments—drug and nondrug—to manage them. Research has shown that treating behavioral symptoms can make people with Alzheimer’s disease more comfortable and makes things easier for caregivers.
Examples of medicines used to help with depression, aggression, restlessness, and anxiety include:
- Celexa® (citalopram)
- Remeron® (mirtazapine)
- Zoloft® (sertraline)
- Wellbutrin® (bupropion)
- Cymbalta® (duloxetine)
- Tofranil® (imipramine)
Experts agree that medicines to treat these behavior problems should be used only after other strategies that don’t use medicine have been tried 26.
Medicines to be Used with Caution
There are some medicines, such as sleep aids, anti-anxiety drugs, anticonvulsants, and antipsychotics, that a person with Alzheimer’s disease should take only:
- After the doctor has explained all the risks and side effects of the medicine
- After other, safer non-medication options have not helped treat the problem
You will need to watch closely for side effects from these medications.
Sleep aids are used to help people get to sleep and stay asleep. People with Alzheimer’s disease should NOT use these drugs regularly because they make the person more confused and more likely to fall. Examples of these medicines include:
- Ambien® (zolpidem)
- Lunesta® (eszopiclone)
- Sonata® (zaleplon)
Anti-anxiety drugs are used to treat agitation. These drugs can cause sleepiness, dizziness, falls, and confusion. For this reason, doctors recommend using them only for short periods of time. Examples of these medicines include:
- Ativan® (lorazepam)
- Klonopin® (clonazepam)
Anticonvulsants are drugs sometimes used to treat severe aggression. Side effects may cause sleepiness, dizziness, mood swings, and confusion. Examples of these medicines include:
- Depakote® (sodium valproate)
- Tegretol® (carbamazepine)
- Trileptal® (oxcarbazepine)
Antipsychotics are drugs used to treat paranoia, hallucinations, agitation, and aggression. Side effects of using these drugs can be serious, including increased risk of death in some older people with dementia. They should only be given to people with Alzheimer’s disease when the doctor agrees that the symptoms are severe. Examples of these medicines include:
- Risperdal® (risperidone)
- Seroquel® (quetiapine)
- Zyprexa® (olanzapine)
Tips for Dealing with Memory Problems and Forgetfulness
We’ve all forgotten a name, where we put our keys, or if we locked the front door. It’s normal to forget things once in a while. But serious memory problems make it hard to do everyday things. Forgetting how to make change, use the telephone, or find your way home may be signs of a more serious memory problem.
For some older people, memory problems are a sign of mild cognitive impairment, Alzheimer’s disease, or a related dementia. People who are worried about memory problems should see a doctor. Signs that it might be time to talk to a doctor include:
- Asking the same questions over and over again.
- Getting lost in places a person knows well.
- Not being able to follow directions.
- Becoming more confused about time, people, and places.
- Not taking care of oneself—eating poorly, not bathing, or being unsafe.
People with memory complaints should make a follow-up appointment to check their memory after 6 months to a year. They can ask a family member, friend, or the doctor’s office to remind them if they’re worried they’ll forget.
People with some forgetfulness can use a variety of techniques that may help them stay healthy and deal with changes in their memory and mental skills 27. Here are some tips:
- Learn a new skill.
- Stay involved in activities that can help both the mind and body.
- Volunteer in your community, at a school, or at your place of worship.
- Spend time with friends and family.
- Use memory tools such as big calendars, to-do lists, and notes to yourself.
- Put your wallet or purse, keys, and glasses in the same place each day.
- Get lots of rest.
- Exercise and eat well.
- Don’t drink a lot of alcohol.
- Get help if you feel depressed for weeks at a time.
Take Care of Your Health
Taking care of your physical health may help your cognitive health. Cognitive health—the ability to clearly think, learn, and remember—is an important component of your brain health. You can:
- Get recommended health screenings.
- Manage chronic health problems like diabetes, high blood pressure, depression, and high cholesterol.
- Consult with your healthcare provider about the medicines you take and possible side effects on memory, sleep, and brain function.
- Reduce risk for brain injuries due to falls and other accidents.
- Limit use of alcohol (some medicines can be dangerous when mixed with alcohol).
- Quit smoking, if you smoke.
- Get enough sleep, generally 7-8 hours each night.
Eat Healthy Foods
A healthy diet can help reduce the risk of many chronic diseases, such as heart disease or diabetes. It may also help keep your brain healthy.
In general, a healthy diet consists of fruits and vegetables; whole grains; lean meats, fish, and poultry; and low-fat or non-fat dairy products. You should also limit solid fats, sugar, and salt. Be sure to control portion sizes and drink enough water and other fluids.
Researchers are looking at whether a healthy diet can help preserve cognitive function or reduce the risk of Alzheimer’s. For example, there is some evidence that people who eat a “Mediterranean diet” have a lower risk of developing mild cognitive impairment.
Researchers have developed and are testing another diet, called MIND, a combination of the Mediterranean and DASH (Dietary Approaches to Stop Hypertension) diets. One study suggests that a combination of the Mediterranean and DASH (Dietary Approaches to Stop Hypertension) diets may affect the risk of Alzheimer’s disease 28.
Be Physically Active
Being physically active—through regular exercise, household chores, or other activities—has many benefits. It can help you:
- Keep and improve your strength.
- Have more energy.
- Improve your balance.
- Prevent or delay heart disease, diabetes, and other diseases.
- Perk up your mood and reduce depression.
Studies link ongoing physical activity with benefits for the brain, too. In one study, exercise stimulated the human brain’s ability to maintain old network connections and make new ones that are vital to cognitive health 28. Other studies 29 have shown that exercise increased the size of a brain structure important to memory and learning, improving spatial memory.
Aerobic exercise, such as brisk walking, is thought to be more beneficial to cognitive health than non-aerobic stretching and toning exercise. Studies are ongoing.
Federal guidelines recommend that all adults get at least 150 minutes of physical activity each week. Aim to move about 30 minutes on most days. Walking is a good start. You can also join programs that teach you to move safely and prevent falls, which can lead to brain and other injuries. Check with your healthcare provider if you haven’t been active and want to start a vigorous exercise program.
Keep Your Mind Active
Being intellectually engaged may benefit the brain. People who engage in meaningful activities, like volunteering or hobbies, say they feel happier and healthier. Learning new skills may improve your thinking ability, too. For example, one study found that older adults who learned quilting or digital photography had more memory improvement than those who only socialized or did less cognitively demanding activities.
Lots of activities can keep your mind active. For example, read books and magazines. Play games. Take or teach a class. Learn a new skill or hobby. Work or volunteer. These types of mentally stimulating activities have not been proven to prevent serious cognitive impairment or Alzheimer’s disease, but they can be fun!
Scientists think that such activities may protect the brain by establishing “cognitive reserve.” They may help the brain become more adaptable in some mental functions, so it can compensate for age–related brain changes and health conditions that affect the brain.
Formal cognitive training also seems to have benefits. In the Advanced Cognitive Training for Independent and Vital Elderly (ACTIVE) trial, healthy adults 65 and older participated in 10 sessions of memory training, reasoning training, or processing–speed training. The sessions improved participants’ mental skills in the area in which they were trained. Most of these improvements persisted 10 years after the training was completed.
Be wary of claims that playing certain computer and online games can improve your memory and other types of thinking. Evidence to back up such claims is evolving. National Institute of Aging and others are supporting research to determine if different types of cognitive training have lasting effects.
Stay Connected
Connecting with other people through social activities and community programs can keep your brain active and help you feel less isolated and more engaged with the world around you. Participating in social activities may lower the risk for some health problems and improve well-being.
So, visit with family and friends. Join programs through your Area Agency on Aging , senior center, or other community organizations.
We don’t know for sure yet if any of these actions can prevent or delay Alzheimer’s disease and age–related cognitive decline. But some of them have been associated with reduced risk of cognitive impairment and dementia.
Lewy body dementia
Lewy body dementia also known as dementia with Lewy bodies, is the second most common type of progressive dementia behind Alzheimer’s disease 30. Lewy body dementia affects approximately 1.4 million Americans, most often after the age of 50. Protein deposits, called Lewy bodies, which are a microscopic aggregation of a specific abnormal protein called alpha-synuclein (α-synuclein), that develop in nerve cells in the brain regions involved in thinking, memory and movement (motor control) 31. Lewy body dementia causes a progressive decline in mental abilities. Lewy body dementia is a progressive disease, meaning symptoms start slowly and worsen over time. People with Lewy body dementia might have visual hallucinations and changes in alertness and attention.
There are two types of Lewy body dementia 32:
- Dementia with Lewy bodies (DLB). Dementia with Lewy bodies causes problems with thinking ability that seem similar to Alzheimer’s disease. Later, it also causes other symptoms, such as movement symptoms, visual hallucinations, and certain sleep disorders. It also causes more trouble with mental activities than with memory.
- Parkinson’s disease dementia (PDD). Parkinson’s disease dementia starts as a movement disorder. It first causes the symptoms of Parkinson’s disease: slowed movement, muscle stiffness, tremor, and a shuffling walk. Later on, it causes dementia.
The earliest signs of these two diseases differ but reflect the same biological changes in the brain. And, over time, they can cause similar symptoms. The main difference is in when the cognitive (thinking) and movement symptoms start. The two diseases are demarcated clinically from one another by the so-called 1-year rule, based on the temporal onset of motor relative to cognitive symptoms (ie, in Parkinson’s disease dementia [PDD] the motor symptoms precede the onset of dementia by at least one year) 33. Individuals with dementia with Lewy bodies (DLB) present with dementia as the early disabling symptom, plus other Lewy body dementia symptoms, one of which may be parkinsonism. Others will present with motor symptoms resulting in a diagnosis of Parkinson’s disease and may also have some mild cognitive impairment (MCI) initially or in the early stages of the disease; over time, usually several years or more, some will progress to Parkinson’s disease dementia (PDD) 32.
The most common features of dementia with Lewy bodies (DLB) are progressive cognitive impairment leading eventually to full-blown dementia, parkinsonian motor symptoms (tremor, slowed
mobility, stiffness of muscles, stooped posture, shuffling gait), visual hallucinations, and fluctuations in levels of alertness and cognitive acuity. Other symptoms include acting out dreams (REM sleep behavior disorder) and disturbances of autonomic function (low blood pressure, constipation and urinary frequency) 33. Severe sensitivity or over-reaction to antipsychotic drugs (neuroleptics) are also common.
Lewy body dementia is often misdiagnosed as Alzheimer’s disease, especially in those individuals who have few, if any signs of motor parkinsonism 34.
Lewy body dementia diagnosis is challenging because the order of symptom appearance, their relative severity and the combination of features present varies among individuals 32.
Lewy body dementia signs and symptoms
Lewy body dementia is the most misdiagnosed form of dementia. Lewy body dementia is the second most common cause of progressive dementia after Alzheimer’s disease. Other effects include Parkinson’s disease signs and symptoms such as rigid muscles, slow movement, walking difficulty and tremors.
Lewy body dementia signs and symptoms can include:
- Visual hallucinations. Hallucinations — seeing things that aren’t there — might be one of the first symptoms, and they often recur. People with Lewy body dementia might hallucinate shapes, animals or people. Sound (auditory), smell (olfactory) or touch (tactile) hallucinations are possible.
- Movement disorders. Signs of Parkinson’s disease (parkinsonian signs), such as slowed movement, rigid muscles, tremor or a shuffling walk can occur. This can lead to falling.
- Poor regulation of body functions (autonomic nervous system). Blood pressure, pulse, sweating and the digestive process are regulated by a part of the nervous system that is often affected by Lewy body dementia. This can result in sudden drops in blood pressure upon standing (orthostatic hypotension), dizziness, falls, loss of bladder control (urinary incontinence) and bowel issues such as constipation.
- Cognitive problems. You might have thinking (cognitive) problems similar to those of Alzheimer’s disease, such as confusion, poor attention, visual-spatial problems and memory loss.
- Sleep difficulties. You might have rapid eye movement (REM) sleep behavior disorder, which can cause you to physically act out your dreams while you’re asleep. This might involve behavior such as punching, kicking, yelling and screaming while sleeping.
- Fluctuating attention. Episodes of drowsiness, long periods of staring into space, long naps during the day or disorganized speech are possible.
- Depression. You might develop depression.
- Apathy. You might lose motivation.
Early, suggestive indicators of Lewy body dementia:
- Often recognized only in retrospect, possibly extending back 1-3 years. Occasional minor episodes of forgetfulness, sometimes described as lapses of concentration or ‘switching off’.
- An initial brief period of delirium in association with genuine physical illness and/or surgical procedures, return to baseline, followed by a subsequent mental and physical decline.
Lewy body dementia is progressive. Signs and symptoms worsen, causing:
- Severe dementia
- Aggressive behavior. Increasing behavioral disturbances, including shouting, aggression on approach and evidence of persisting delirium.
- Depression
- Increased risk of falling and injury
- Worsening of parkinsonian signs and symptoms, such as tremors
- Death, on average about seven to eight years after symptoms start. Death is usually due to respiratory or cardiac disease, or injuries sustained in falls.
Lewy body dementia causes
The cause of Lewy body dementia is still unknown 35. Lewy body dementia is characterized by the abnormal buildup of proteins into masses known as Lewy bodies, which are a microscopic aggregation of a specific abnormal protein called alpha-synuclein (α-synuclein). This protein is also associated with Parkinson’s disease. People who have Lewy bodies in their brains also have the plaques and tangles associated with Alzheimer’s disease.
Brain pathological changes in Lewy body dementia involve selective damage and loss of nerve cells in certain regions of the brain (example: substantia nigra in the brainstem). Affected, but less damaged cells contain Lewy bodies. The Lewy body is the pathological signature of Lewy body dementia that overwhelms the cell’s normal biological functions and causes it to die 36.
There are many possible causes of Lewy body dementia but researchers are just beginning to understand the reasons why some people are more susceptible to developing Lewy body dementia. One important reason that has recently come to light is the discovery of an increasing number of genetic variants that increase the likelihood that a person will develop Lewy body dementia. Dementia with Lewy bodies (DLB) and Parkinson’s disease dementia (PDD) are clinically similar, except for the timing of onset of cognitive impairment, but the pathology of the two is almost identical. This is both a surprise and a mystery, since there is no good explanation for the variability of the motor-cognitive interval among people with Lewy body dementia. In other words, why do some people develop serious cognitive impairment at the earliest stage of a Lewy body disorder, whereas others remain cognitively normal for many years before impairment develops or never develop dementia?
Another puzzling fact is the frequent coexistence of the pathology of Alzheimer’s disease (amyloid plaques and neurofibrillary tangles) in dementia with Lewy bodies (DLB) compared with Parkinson’s disease dementia (PDD). Alzheimer’s disease differs from Lewy body dementia clinically because of its distinctive cognitive profile (mostly a disorder of memory without the other features typical of Lewy body dementia) and its lack of parkinsonian features except in late stages. The clinical overlap between Alzheimer’s disease and dementia with Lewy bodies (DLB) in the absence of a specific diagnostic test leads to misdiagnosis in a significant minority of patients. Currently, the only way to definitively diagnose Lewy body dementia is with an autopsy 32. These facts underscore the current concept of a neurodegenerative continuum with boundaries that are frequently blurred. It is only through research that these and other fundamental questions will be answered.
Risk factors for developing Lewy body dementia
A few factors seem to increase the risk of developing Lewy body dementia, including:
- Older age. Older age is the greatest risk factor for Lewy body dementia, with most diagnoses being made in individuals over the age of 50. There is some evidence that the age of onset of the symptoms of dementia with Lewy bodies (DLB) is younger than in Parkinson’s disease dementia (PDD) and the rate of progression/duration of disease is slightly faster in dementia with Lewy bodies (DLB) 37.
- Sex. Lewy body dementia affects more men than women.
- Family history. Those who have a family member with Lewy body dementia or Parkinson’s disease are at greater risk. Mutations in over a dozen genes have been shown to “cause” Parkinson’s disease 38. Individuals with such rare genetic variants have a very high risk of developing Parkinson’s disease during their lifetime, and many of them will later develop dementia. Mutations in one of these genes (SNCA) can occasionally result in a clinical picture that resembles dementia with Lewy bodies (DLB) 39. However, no more than 2% of patients with Parkinson’s disease, and likely even fewer with dementia with Lewy bodies (DLB), carry a disease-causing mutation in a known gene. In most instances Parkinson’s disease and dementia with Lewy bodies (DLB) are thought to arise through a complex interaction between common genetic and environmental factors, each one with a small-to-modest effect. Two important common genetic risk factors that have recently come to light are variants in the APOE and GBA genes. The
APOE ε4 allele has long been known to increase the risk of developing Alzheimer’s disease, but there is now strong evidence that it does the same for dementia with Lewy bodies (DLB) 40. Furthermore, patients with Parkinson’s disease who carry APOE ε4 have (on average) more severe cognitive problems 41. A number of variants in the GBA gene have been shown to increase risk for
both Parkinson’s disease and dementia with Lewy bodies (DLB) 42. In addition, patients with Parkinson’s disease who have one of these GBA variants have a more rapid cognitive decline and are more likely to develop dementia 43. Since mutations that cause Lewy body dementia are rare, and no treatments have been discovered to reverse the effects of known genetic risk factors, genetic testing is not currently recommended for routine screening. However, if a family has multiple individuals with Parkinson’s disease (with or without dementia) and/or dementia with Lewy bodies (DLB), it is reasonable to consider genetic testing for some or all of the known genes. The rationale for considering such testing would be to (1) confirm a diagnosis and (2) provide genetic counseling for family members, if the results are positive. These decisions need to be made carefully with family members and the individual’s healthcare provider. It is prudent to undergo pre- and post-testing counseling so that the individual fully understands the risks and benefits of learning about their genetic status. In addition, certain research centers at academic institutions and the National Institutes of Health are investigating genetic risk and are actively seeking people who would like to volunteer as research subjects. - Rapid eye movement (REM) sleep behavior disorder (RBD), a condition characterized by dream enactment, is a common risk factor for dementia with Lewy bodies (DLB), Parkinson’s disease and other synucleinopathies, often occurring many years before the onset of parkinsonism or cognitive impairment 44. Pre-Parkinson’s sleep behavior disorder is thought to increase the risk of cognitive impairment when the motor phase of Parkinson’s disease evolves, compared with Parkinson’s disease that has no REM sleep behavior disorder prodrome. Parkinson’s disease is a risk factor for developing dementia, since the majority of those with Parkinson’s disease will eventually suffer from cognitive impairment.
Lewy body dementia diagnosis
Lewy body dementia diagnosis is challenging because the order of symptom appearance, their relative severity and the combination of features present varies among individuals 32. Currently, the only way to definitively diagnose Lewy body dementia is with an autopsy 32.
A diagnosis of Lewy body dementia requires a progressive decline in your ability to think, as well as at least two of the following:
- Fluctuating alertness and thinking function
- Repeated visual hallucinations
- Parkinsonian symptoms
- REM sleep behavior disorder, in which people act out their dreams during sleep
Autonomic dysfunction, which involves instability in blood pressure and heart rate, poor regulation of body temperature, sweating, and related signs and symptoms, supports a Lewy body dementia diagnosis. So does sensitivity to antipsychotic drugs, particularly first-generation antipsychotics such as haloperidol (Haldol). Medications like haloperidol aren’t used for people with Lewy body dementia because they can cause a severe reaction.
No single test can diagnose Lewy body dementia. The diagnosis is based the on signs and symptoms you have and ruling out other conditions that can cause similar signs and symptoms. Tests might include:
- Neurological and physical examination
- Your doctor may check for signs of Parkinson’s disease, strokes, tumors or other medical conditions that can affect the brain and physical function. A neurological examination tests:
- Reflexes
- Strength
- Walking
- Muscle tone
- Eye movements
- Balance
- Sense of touch
- Your doctor may check for signs of Parkinson’s disease, strokes, tumors or other medical conditions that can affect the brain and physical function. A neurological examination tests:
- Assessment of mental abilities
- A short form of this test, which assesses your memory and thinking skills, can be done in less than 10 minutes in your doctor’s office. It’s not generally useful in distinguishing Lewy body dementia from Alzheimer’s disease but can determine whether cognitive impairment is present. Longer tests that take several hours help identify Lewy body dementia.
- Blood tests
- These can rule out physical problems that can affect brain function, such as vitamin B-12 deficiency or an underactive thyroid gland.
- Brain scans
- Your doctor might order an MRI or CT scan to identify a stroke or bleeding and to rule out a tumor. While dementias are diagnosed based on the medical history and physical examination, certain features on imaging studies can suggest different types of dementia, such as Alzheimer’s or Lewy body dementia.
- If the diagnosis is unclear or the signs and symptoms aren’t typical, your doctor might suggest additional imaging tests, including these that can support a diagnosis of Lewy body dementia:
- Fluorodeoxyglucose PET brain scans, which assess brain function.
- Single-photon emission computerized tomography (SPECT) or PET imaging, which can determine whether dopamine transporter uptake is reduced in the brain.
- Sleep evaluation
- Your doctor might order a sleep evaluation called a polysomnogram to check for REM sleep behavior disorder or an autonomic function test to look for signs of heart rate and blood pressure instability.
- Heart test
- In some countries, doctors might also order a heart test called myocardial scintigraphy to check the blood flow to your heart for indications of Lewy body dementia. However, the test isn’t used in the United States.
Emerging biomarkers
Research is ongoing into other indicators of Lewy body dementia. These biomarkers might eventually enable early diagnosis of Lewy body dementia before the full disease develops.
Lewy body dementia treatment
Currently there’s no cure for Lewy body dementia but many of the symptoms can improve with targeted treatments.
Lewy body dementia medications
- Cholinesterase inhibitors. These Alzheimer’s disease medications, such as rivastigmine (Exelon), donepezil (Aricept) and galantamine (Razadyne), work by increasing the levels of chemical messengers in the brain (neurotransmitters) believed to be important for memory, thought and judgment. This can help improve alertness and cognition and might reduce hallucinations and other behavioral problems. Possible side effects include gastrointestinal upset, muscle cramps and frequent urination. It can also increase the risk of certain cardiac arrhythmias. In some people with moderate or severe dementia, an N-methyl-d-aspartate (NMDA) receptor antagonist called memantine (Namenda) might be added to the cholinesterase inhibitor.
- Parkinson’s disease medications. These medications, such as carbidopa-levodopa (Sinemet, Rytary, Duopa) can help reduce parkinsonian signs and symptoms, such as rigid muscles and slow movement. However, these medications can also increase confusion, hallucinations and delusions.
- Medications to treat other symptoms. Your doctor might prescribe medications to treat other signs and symptoms associated with Lewy body dementia, such as sleep or movement problems.
Certain medications can worsen memory. Try to avoid over-the-counter sleep aids that contain diphenhydramine (Advil PM, Aleve PM) and medications used to treat urinary urgency such as oxybutynin (Ditropan XL).
Also limit sedatives and sleeping tablets, and talk to your doctor about whether any of the drugs you take might make your memory worse.
Antipsychotic drugs can cause severe confusion, severe parkinsonism, sedation and sometimes death. Very rarely, certain second-generation antipsychotics, such as quetiapine (Seroquel) or clozapine (Clozaril, Versacloz) might be prescribed for a short time at a low dose but only if the benefits outweigh the risks.
Lewy body dementia therapies
Because antipsychotic drugs can worsen Lewy body dementia symptoms, it might be helpful to first try nondrug approaches, such as:
- Tolerating the behavior. Some people with Lewy body dementia aren’t distressed by the hallucinations. In these cases, the side effects of medication might be worse than the hallucinations themselves.
- Modifying the environment. Reducing clutter and noise can make it easier for someone with dementia to function. Caregivers’ responses sometimes worsen behavior. Avoid correcting and quizzing a person with dementia. Offer reassurance and validation of his or her concerns.
- Creating daily routines and keeping tasks simple. Break tasks into easier steps and focus on successes, not failures. Structure and routine during the day can be less confusing.
Alternative medicine
Frustration and anxiety can worsen dementia symptoms. To promote relaxation, consider:
- Music therapy, which involves listening to soothing music
- Pet therapy, which involves the use of animals to improve moods and behaviors in people with dementia
- Aromatherapy, which uses fragrant plant oils
- Massage therapy
Lewy body dementia home remedies
Symptoms and progression are different for everyone with Lewy body dementia. Caregivers and care partners may need to adapt the following tips to individual situations:
- Speak clearly and simply. Maintain eye contact and speak slowly, in simple sentences, and don’t rush the response. Present only one idea or instruction at a time. Use gestures and cues, such as pointing to objects.
- Encourage exercise. Benefits of exercise include improvements in physical function, behavior and depression. Some research shows exercise might slow cognitive decline in people with dementia.
- Provide mind stimulation. Participating in games, crossword puzzles and other activities that involve thinking skills might help slow mental decline in people with dementia. Encourage artistic and creative activities, such as painting, singing or making music.
- Create opportunities for social activity. Talk to friends. Participate in religious services.
- Establish bedtime rituals. Behavior issues can worsen at night. Create calming bedtime rituals without the distraction of television, meal cleanup and active family members. Leave night lights on to prevent disorientation.
Limiting caffeine during the day, discouraging daytime napping and offering opportunities for daytime exercise might help prevent nighttime restlessness.
Lewy body dementia coping and support
People with Lewy body dementia often have a mixture of emotions, such as confusion, frustration, anger, fear, uncertainty, grief and depression. Offer support by listening, reassuring the person that he or she still can enjoy life, being positive, and doing your best to help the person retain dignity and self-respect.
If you’re a caregiver or care partner for someone with Lewy body dementia, watch the person closely to make sure he or she doesn’t fall, lose consciousness or react negatively to medications. Provide reassurance during times of confusion, delusions or hallucinations.
Looking after yourself
Caring for a person with Lewy body dementia can be exhausting physically and emotionally. You may have anger, guilt, frustration, discouragement, worry, grief or social isolation. Help prevent caregiver burnout by:
- Asking friends or other family members for help when you need it. Consider in-home health services to help you care for the person with Lewy body dementia.
- Exercising regularly and eating a healthy diet.
- Learning about the disease. Ask questions of doctors, social workers and others on the care team.
- Joining a support group.
Many people with Lewy body dementia and their families can benefit from counseling or local support groups. Contact your local agencies on health or aging to get connected with support groups, doctors, resources, referrals, home care agencies, supervised living facilities, a telephone help line and educational seminars.
Lewy body dementia prognosis
Like Alzheimer’s disease, the rate of progression of Lewy body dementia is highly variable. The average life expectancy after diagnosing Lewy body dementia is about 5 to 8 years 45. This also can be due to a lack of knowledge regarding Lewy body dementia among physicians and the population and difficulty in differentiating it from other similar conditions which leads to a delay in diagnosis which delays the onset of specific therapy. Anecdotal experience suggests that the trajectory of Lewy body dementia progression may mirror the rate of early symptom onset and progression. Patients die from multiple complications like falls, immobility, cardiac complications, medication side effects, pneumonia, swallowing problems, and depression leading to suicide 35.
There are no formally defined stages of Lewy body dementia like there are in Alzheimer’s disease. Until then, expert insights may be useful to both the clinician and Lewy body dementia family. Efforts are underway to define the mild cognitive impairment (MCI) stage of dementia with Lewy bodies (DLB) to allow for an earlier diagnosis. Criteria have been developed for mild cognitive impairment (MCI) in Parkinson’s disease 46.
Vascular dementia
Vascular dementia is dementia caused by conditions that damage blood vessels in the brain that disrupt blood flow to the brain and lead to problems with memory, thinking, and behavior. Vascular dementia is defined as a reduced or lack of blood flow to the brain that causes dementia 47. Vascular dementia is caused by a blocked or reduced blood flow to the brain which will deprive neurons of critical nutrients 48. This deprivation eventually causes the neurons to die, and the brain tissue starts to shrink. Some common contributors to vascular dementia include stroke, cardiovascular disease, diabetes, hyperlipidemia, and hypertension 49.
Vascular dementia is the second most common form of dementia after Alzheimer’s disease, affecting almost a third of people over age 70 and can occur alone or alongside another form of dementia 50. The brains of people with vascular dementia often show evidence of prior strokes, thickening blood vessel walls, and thinning white matter — the brain’s connecting “wires” that relay messages between regions. However, not everyone who has had a stroke will develop vascular dementia. A person’s risk for dementia after stroke depends on the size and number of strokes and the brain regions affected. Vascular dementia can also result from other conditions that impede blood flow and delivery of oxygen to the brain, such as narrowing of the arteries. High blood pressure, problems with the heartbeat’s rhythm, diabetes, and high cholesterol can increase a person’s risk of vascular dementia. By controlling or managing risk factors, you may lower your chance of developing cognitive impairment and dementia.
A growing number of experts prefer the term “vascular contributions to cognitive impairment and dementia” or “multi-infarct dementia” 47. Vascular contributions to cognitive impairment and dementia is the condition arising from stroke and other vascular brain injuries that cause significant changes to memory, thinking, and behavior 47 and vascular dementia is the most severe stage while multi-infarct dementia, which is produced by the synergistic effects caused by multiple mini strokes in the brain irrespective of specific location or volume 47.
Vascular dementia and vascular cognitive impairment arise as a result of risk factors that similarly increase the risk for cerebrovascular disease (such as stroke), including atrial fibrillation, hypertension (high blood pressure), diabetes, and high cholesterol. The symptoms of vascular dementia can be similar to those of Alzheimer’s disease and both conditions can occur at the same time (a condition called “mixed dementia”). Symptoms of vascular dementia can begin suddenly and worsen or improve during one’s lifetime.
Vascular dementia is often managed with drugs to prevent strokes. The aim is to reduce the risk of additional brain damage. Some studies suggest that drugs that improve memory in Alzheimer’s disease might benefit people with early vascular dementia. Interventions that address risk factors may be incorporated into the management of vascular dementia.
Figure 7. Brain Blood Supply (Vascular Anatomy) – looking through a brain sliced in the center
Figure 8. Brain Blood Supply (Vascular Anatomy) – looking from the base of the brain up
Types of vascular dementia
Multi-infarct dementia
This type of dementia occurs when a person has had many small strokes that damage brain cells. One side of the body may be disproportionally affected, and multi-infarct dementia may impair language or other functions, depending on the region of the brain that is affected. In classic multi-infarct dementia, the cognitive deterioration is stepwise rather than smoothly progressive 51. The cognitive changes vary, but memory loss is usually much less prominent than in Alzheimer’s disease. With each event (stroke) the patient suddenly worsens but then improves either completely or partially. As the disease progresses, the patient develops an accretion of abnormal neurologic signs such as asymmetric reflexes, pseudobulbar changes (i.e. swallowing and speech difficulties along with emotional lability), pathologic reflexes (e.g. Babinski signs), and sensory abnormalities. This condition is usually seen in hypertensive individuals and is caused by multiple small infarcts in the white matter of the brain as well as the basal ganglia and cortex. A variant of multi-infarct dementia is Binswanger subcortical arteriosclerotic encephalopathy also called subcortical vascular dementia, in which the disease is confined to the white matter of the brain hemispheres and is usually reported as a fairly rapidly progressing dementia with significant neurologic and cognitive changes.
When the strokes occur on both sides of the brain, dementia is more likely than when stroke occurs on one side of the brain. In some cases, a single stroke can damage the brain enough to cause dementia. This so-called single-infarct dementia is more common when stroke affects the left side of the brain—where speech centers are located—and/or when it involves the hippocampus, the part of the brain that is vital for memory.
Multi-infarct dementia is the second most common cause of dementia (after Alzheimer’s disease) in people over age 65 52. Multi-infarct dementia affects men more often than women. The disorder usually affects people between ages 55 and 75 52.
Multi-infarct dementia causes
Multi-infarct dementia is caused by a series of small strokes. A stroke is an interruption in or blockage of the blood supply to any part of the brain. A stroke is sometimes called an infarct. “Multi-infarct” means that many areas in the brain have been injured due to a lack of blood. If blood flow is stopped for longer than a few seconds, the brain cannot get oxygen. Brain cells can die, causing permanent damage. When these strokes affect a small area, there may be no symptoms of a stroke. These are often called silent strokes. Over time, as more areas of the brain are damaged, the symptoms of multi-infarct dementia begin to appear. Not all strokes need to be “silent.” Larger strokes that have clear affects on strength, sensation, or other brain and nervous system (neurologic) function also can lead to multi-infarct dementia.
Risk factors for multi-infarct dementia
Risk factors for multi-infarct dementia include a history of:
- Diabetes
- Hardening of the arteries (atherosclerosis)
- High blood pressure (hypertension)
- Smoking
- Stroke
See also: Stroke risk factors and prevention
Symptoms of dementia in any one person may be caused by either Alzheimer’s disease or multi-infarct dementia. The symptoms for each problem are very similar, and multi-infarct dementia may be a risk factor for Alzheimer’s disease.
Multi-infarct dementia prevention
Control conditions that increase the risk of hardening of the arteries (atherosclerosis) by:
- Controlling high blood pressure
- Controlling weight
- Reducing saturated fats and salt in the diet
- Treating related disorders.
Multi-infarct dementia signs and symptoms
Symptoms may develop gradually or may progress after each small stroke.
The symptoms of the disorder may begin suddenly after each stroke. Some people with multi-infarct dementia may appear to improve for short periods of time, then decline after having more silent strokes.
The early symptoms of dementia can include:
- Difficulty performing tasks that used to come easily, such as balancing a checkbook, playing games (such as bridge), and learning new information or routines
- Getting lost on familiar routes
- Language problems, such as trouble finding the name of familiar objects
- Losing interest in things you previously enjoyed, flat mood
- Misplacing items
- Personality changes and loss of social skills.
As the dementia becomes worse, symptoms are more obvious and interfere with the ability to take care of yourself. The symptoms may include:
- Change in sleep patterns, often waking up at night
- Difficulty doing basic tasks, such as preparing meals, choosing proper clothing, or driving
- Forgetting details about current events
- Forgetting events in your own life history, losing awareness of who you are
- Having delusions, depression, or agitation
- Having hallucinations, arguments, striking out, or violent behavior
- Having more difficulty reading or writing
- Having poor judgment and loss of ability to recognize danger
- Using the wrong word, not pronouncing words correctly, or speaking in confusing sentences
- Withdrawing from social contact
Any of the neurologic problems that occur with a stroke may also be present.
Multi-infarct dementia possible complications
Complications include the following:
- Future strokes
- Heart disease
- Loss of ability to function or care for self
- Loss of ability to interact
- Pneumonia, urinary tract infections, skin infections
- Pressure sores
Multi-infarct dementia diagnosis
Tests may be ordered to help determine whether other medical problems could be causing dementia or making it worse, such as:
- Anemia
- Brain tumor
- Chronic infection
- Drug and medication intoxication
- Severe depression
- Thyroid disease
- Vitamin deficiency
Neuropsychological testing is often helpful to find out what parts of thinking have been affected, and to guide other tests.
Tests that can show evidence of previous strokes in the brain may include:
- Head CT scan
- MRI of the brain
Multi-infarct dementia treatment
There is no treatment to turn back damage to the brain caused by small strokes.
An important goal is to control symptoms and correct risk factors such as high blood pressure, smoking, and high cholesterol to prevent future strokes.
- Avoid fatty foods. Follow a healthy, low-fat diet.
- Do not drink more than 1 – 2 alcoholic drinks a day.
- Keep blood pressure less than 130/80 mm/Hg (ask your doctor what your blood pressure reading should be).
- Keep LDL “bad” cholesterol lower than 70 mg/dL.
- Quit smoking.
- Your doctor may suggest taking aspirin or another drug called clopidogrel (Plavix) to help prevent blood clots from forming in the arteries. These medicines are called antiplatelet drugs. DO NOT take aspirin without talking to your doctor first.
The goals of helping someone with dementia in the home environment are to:
- Manage behavior problems, confusion, sleep problems, and agitation
- Modify the home environment
- Support family members and other caregivers
- Hearing aids, glasses, or cataract surgery may be needed if the person has sensory problems.
See home care plan for information about taking care of a loved one with dementia.
Medications
Medications may be needed to control aggressive, agitated, or dangerous behaviors. The health care provider will usually prescribe these medicines in very low doses and adjust the dose as needed. Such medications may include:
- Antipsychotics (olanzapine, quetiapine)
- Serotonin-affecting drugs (trazodone, buspirone, or fluoxetine)
Medications used to treat Alzheimer’s disease have not been shown to work for multi-infarct dementia.
Multi-infarct dementia prognosis
Some improvement may occur for short periods of time, but the disorder will generally get worse over time.
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL)
CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy) also known as hereditary multi-infarct dementia, is an inherited form of cardiovascular disease that results in a thickening of the walls of small- and medium-sized blood vessels, eventually stemming the flow of blood to the brain. The muscle cells surrounding these blood vessels (vascular smooth muscle cells) are abnormal and gradually die 53. In the brain, the resulting blood vessel damage (arteriopathy) can cause migraines, often with visual sensations or auras, or recurrent seizures (epilepsy).
CADASIL is likely a rare condition; however, its prevalence is unknown 53. CADASIL affects approximately 2 to 5 of 100,000 people. Research suggests that the disorder often goes undiagnosed or misdiagnosed making it difficult to determine the true frequency of CADASIL in the general population. CADASIL affects males and females in equal numbers. The disorder is found worldwide and affects all races.
In individuals with CADASIL, a stroke can occur at any time from childhood to late adulthood, but typically happens during mid-adulthood (U.S. National Library of Medicine. Genetics Home Reference. cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. https://ghr.nlm.nih.gov/condition/cerebral-autosomal-dominant-arteriopathy-with-subcortical-infarcts-and-leukoencephalopathy)). People with CADASIL often have more than one stroke in their lifetime. Recurrent strokes can damage the brain over time. Strokes that occur in the subcortical region of the brain, which is involved in reasoning and memory, can cause progressive loss of intellectual function (dementia) and changes in mood and personality.
Many people with CADASIL also develop leukoencephalopathy, which is a change in a type of brain tissue called white matter that can be seen with magnetic resonance imaging (MRI) (U.S. National Library of Medicine. Genetics Home Reference. cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. https://ghr.nlm.nih.gov/condition/cerebral-autosomal-dominant-arteriopathy-with-subcortical-infarcts-and-leukoencephalopathy)).
The age at which the signs and symptoms of CADASIL first begin varies greatly among affected individuals, as does the severity of these features (U.S. National Library of Medicine. Genetics Home Reference. cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. https://ghr.nlm.nih.gov/condition/cerebral-autosomal-dominant-arteriopathy-with-subcortical-infarcts-and-leukoencephalopathy)).
CADASIL is not associated with the common risk factors for stroke and heart attack, such as high blood pressure and high cholesterol, although some affected individuals might also have these health problems (U.S. National Library of Medicine. Genetics Home Reference. cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. https://ghr.nlm.nih.gov/condition/cerebral-autosomal-dominant-arteriopathy-with-subcortical-infarcts-and-leukoencephalopathy)).
CADASIL is associated with mutations of a gene called Notch3. CADASIL is associated with multi-infarct dementia, stroke, migraine with aura (migraine preceded by visual symptoms), and mood disorders. The first symptoms can appear in people between ages 20 and 40. Many people with CADASIL are undiagnosed. People with first-degree relatives who have CADASIL can be tested for genetic mutations to the Notch3 gene to determine their own risk of developing CADASIL.
CADASIL signs and symptoms
Hallmark symptoms of CADASIL may include:
- Recurrent strokes,
- Cognitive impairment,
- Migraine with aura, and
- Psychiatric disturbances.
These symptoms are caused by damage to small blood vessels, especially those within the brain. The specific symptoms and severity of the disorder can vary greatly among affected individuals, even among members of the same family.
Despite this variability, most individuals (approximately three out of four patients) experience recurrent stroke or transient ischemic attacks (TIAs), beginning at 40-50 years of age. Strokes cause weakness and/or loss of feeling of one part of the body, speech difficulties, visual loss or lack of coordination. TIAs result in similar symptoms as strokes but resolve in less than 24 hours. Repeated strokes can cause progression of symptoms listed above and also cause cognitive disturbances, loss of bladder control (urinary incontinence) or loss of balance.
Although strokes are the most common symptom associated with CADASIL, some affected individuals never have strokes. It is not uncommon for CADASIL patients to have evidence of stroke on MRI without any history of stroke-like symptoms (silent strokes).
Cognitive impairment eventually develops in many affected individuals on average between the ages of 50-60, although the progression of the disease will vary. Symptoms may include slowly progressive difficulty with concentration, deficits in attention span or memory dysfunction, difficulty making decisions or solving problems, and general loss of interest (apathy). With age, continued cognitive decline may result in dementia, a progressive loss of memory and decline in intellectual abilities that interferes with performing routine tasks of daily life.
Migraine with aura may be a predominant symptom in some affected individuals, occurring in at least half of CADASIL patients. Migraines are severe headaches that often cause excruciating pain and can be disabling. In individuals with CADASIL, abnormal feelings or warning signs called “aura” often precede these headaches. These additional symptoms usually affect vision and may consist of the sudden appearance of a bright light in the center of the field of vision (scintillating scotoma) or, less frequently, disturbances in all or part of the field of vision. The auras preceding the migraine usually last 20 to 30 minutes but are sometimes longer. Some patients suffer from severe attacks with unusual symptoms such as confusion, fever or coma in very rare situations. Finally, many individuals with CADASIL develop psychiatric abnormalities ranging from personality and behavioral changes to severe anxiety and depression.
CADASIL causes
CADASIL is caused by changes (mutations) in the NOTCH3 gene. The NOTCH3 gene contains instructions to create a protein that is predominantly expressed in smooth muscle cells in the walls of small arteries. Mutations in the NOTCH3 gene result in abnormal accumulation of this protein at the surface of smooth muscle cells. Ultimately, NOTCH3 mutations lead to progressive damage to the small blood vessels in the brain, premature destruction of smooth muscle cells, and narrowing of the lumen and thickening the vessel wall of the small blood vessels. Microscopic protein accumulations of debris called granular osmiophilic material (GOM) accumulate in blood vessels of CADASIL patients. As a consequence of these changes, there is reduction of blood flow to the brain causing small strokes (or lacunes), small bleeds (microbleeds), dilated spaces surrounded the vessels (dilated perivascular spaces) and tissue loss in the surface of the brain (cortex) as well underneath the cortex (subcortical region).
CADASIL is inherited in an autosomal dominant fashion. Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother. Dominant genetic disorders occur when only a single copy of an abnormal gene is necessary for the appearance of the disease. Most individuals with CADASIL have a parent with the disorder, but CADASIL can be due to a spontaneous genetic mutation that occurs for unknown reasons (de novo mutation). In these rare cases, there is no previous family history of the disorder. The risk of passing the abnormal gene from affected parent to offspring is 50 percent for each pregnancy. The risk is the same for males and females.
CADASIL diagnosis
CADASIL is suspected based on symptoms, family history, and brain MRI lesions compatible with the disease. Although MRI can identify characteristic changes in the brains of individuals with CADASIL, such changes are not unique to CADASIL and can occur with other disorders. As such, the CADASIL diagnosis can only be confirmed by DNA testing of blood samples for characteristic mutations in the NOTCH3 gene or by identifying granular osmiophilic material (GOM) inclusions on a skin biopsy.
CADASIL treatment
At the present, there is no treatment that can cure CADASIL or prevent its onset. Patients should be treated for factors that can further damage blood vessels, such as hypertension, and should be encouraged to abstain from smoking. The efficacy of tPA for treatment of acute strokes in CADASIL patients is uncertain; although no contraindication to tPA has been established for this specific population, careful evaluation of prior microbleeds is suggested. Migraines can be treated with traditional analgesics such as acetaminophen or NSAIDs. Other medicines commonly used to treat acute migraine attack such as vasoconstrictors: especially triptans or ergot derivates, are not recommended for patients with CADASIL. Medications such as anti-hypertensive, anti-convulsants, and anti-depressants may be used for prevention of migraines in CADASIL patients. Drug therapy for depression or other psychiatric abnormalities are sometimes needed. Psychological support is often essential, and genetic counseling is recommended for affected individuals and their families.
Subcortical vascular dementia or Binswanger’s disease
Binswanger’s disease also called subcortical vascular dementia, is a rare type of dementia caused by widespread, microscopic areas of damage to the deep layers of white matter in the brain 54. The damage is the result of the thickening and narrowing (atherosclerosis) of arteries that feed the subcortical areas of the brain. Atherosclerosis (commonly known as “hardening of the arteries”) is a systemic process that affects blood vessels throughout the body. It begins late in the fourth decade of life and increases in severity with age. As the arteries become more and more narrowed, the blood supplied by those arteries decreases and brain tissue dies. A characteristic pattern of Binswanger’s disease-damaged brain tissue can be seen with modern brain imaging techniques such as CT scans or magnetic resonance imaging (MRI). The symptoms associated with Binswanger’s disease are related to the disruption of subcortical neural circuits that control what neuroscientists call executive cognitive functioning: short-term memory, organization, mood, the regulation of attention, the ability to act or make decisions, and appropriate behavior. The most characteristic feature of Binswanger’s disease is psychomotor slowness – an increase in the length of time it takes, for example, for the fingers to turn the thought of a letter into the shape of a letter on a piece of paper. Other symptoms include forgetfulness (but not as severe as the forgetfulness of Alzheimer’s disease), changes in speech, an unsteady gait, clumsiness or frequent falls, changes in personality or mood (most likely in the form of apathy, irritability, and depression), and urinary symptoms that aren’t caused by urological disease. Brain imaging, which reveals the characteristic brain lesions of Binswanger’s disease, is essential for a positive diagnosis.
The symptoms of Binswanger’s disease are related to the disruption of subcortical neural circuits involving short-term memory, organization, mood, attention, decision making, and appropriate behavior. A characteristic feature of this disease is psychomotor slowness, such as an increase in the time it takes for a person to think of a letter and then write it on a piece of paper.
Binswanger disease causes
Binswanger disease is caused by arteriosclerosis, thromboembolism and other diseases that obstruct blood vessels that supply the deep structures of the brain. Hypertension, smoking, hypercholesterolemia, heart disease and diabetes mellitus are risk factors for Binswanger disease. Rare hereditary diseases such as CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy) also cause Binswanger disease. Thus, Binswanger disease is actually a clinical syndrome of vascular dementia with multiple causes, not a specific disease. The reduced blood flow in brain tissue appears to produce secondary inflammation that may be a target for treatment.
Binswanger disease signs and symptoms
Affected individuals often become depressed, uncaring (apathetic), inactive, and unable to act or make decisions (abulic). They become withdrawn, and exhibit poor judgement, reduced planning and organizational skills, and less spontaneous communication. In addition, affected individuals may have difficulty with speech (dysarthria), swallowing (dysphagia), and urinary bladder control (incontinence). Some patients exhibit abnormalities that are similar to those seen in Parkinson disease, such as slowness, poor balance and short, shuffling steps (Parkinsonism). Tremor is usually not a feature.
Many individuals with Binswanger disease have a history of strokes or transient ischemic attacks. Consequently, the symptoms and signs of this disease develop in a stuttering or stepwise fashion; in contrast to the insidious, gradually progressive course of neurodegenerative diseases.
Binswanger disease diagnosis
The diagnosis of Binswanger disease is usually based on a thorough clinical evaluation, including a detailed patient history, physical examination, and magnetic resonance imaging (MRI) or computerized tomography (CT) scanning of the brain. MRI and CT reveal nerve fiber (white matter) degeneration and multiple small strokes in the deep structures of the brain.
Binswanger’s disease treatment
There is no specific course of treatment for Binswanger’s disease because ischemic brain damage in Binswanger disease is not reversible. Because there is no cure, the best treatment is preventive, early in the adult years, by controlling risk factors such as hypertension, diabetes, and smoking. Binswanger disease treatment is symptomatic and is focused on reducing risk factors for stroke, thereby retarding progression of the disease. The successful management of hypertension and diabetes can slow the progression of atherosclerosis, and subsequently slow the progress of Binswanger’s disease. Treatment usually involves the use of anti-hypertensive drugs to control blood pressure, antiplatelet drugs (e.g., aspirin) or warfarin to reduce thromboembolism, statins to reduce atherosclerosis, smoking cessation and diabetic control. People with depression or anxiety may require antidepressant medications such as the serotonin-specific reuptake inhibitors (SSRI) sertraline or citalopram. Atypical antipsychotic drugs, such as risperidone and olanzapine, can be useful in individuals with agitation and disruptive behavior. Recent drug trials with the drug memantine have shown improved cognition and stabilization of global functioning and behavior.
Binswanger’s disease prognosis
Binswanger’s disease is a progressive disease; there is no cure. Changes may be sudden or gradual and then progress in a stepwise manner. Binswanger’s disease can often coexist with Alzheimer’s disease. Behaviors that slow the progression of high blood pressure, diabetes, and atherosclerosis — such as eating a healthy diet and keeping healthy wake/sleep schedules, exercising, and not smoking or drinking too much alcohol — can also slow the progression of Binswanger’s disease.
Vascular dementia signs and symptoms
Vascular dementia can start suddenly or come on slowly over time.
People with vascular dementia may experience:
- Difficulty performing tasks that used to be easy, such as paying bills
- Trouble following instructions or learning new information and routines
- Forgetting current or past events
- Misplacing items
- Getting lost on familiar routes
- Problems with language, such as finding the right word or using the wrong word
- Changes in sleep patterns
- Difficulty reading and writing
- Loss of interest in things or people
- Changes in personality, behavior, and mood, such as depression, agitation, and anger
- Hallucinations or delusions (believing something is real that is not)
- Poor judgment and loss of ability to perceive danger
Symptoms may depend on the size, location, and number of damaged areas of the brain.
These problems can make daily activities increasingly difficult and someone with the condition may eventually be unable to look after themselves.
Early symptoms of vascular dementia
Early signs of vascular dementia can include mild:
- slowness of thought
- difficulty with planning
- trouble with understanding
- problems with concentration
- mood or behavioral changes
- problems with memory and language (but these aren’t as common as they are in people with Alzheimer’s disease)
As this point, these problems may be barely noticeable or mistaken for something else, such as depression. But they indicate some brain damage has occurred and that treatment is needed.
Later symptoms of vascular dementia
The symptoms often continue to get worse over time. This may happen slowly, or in sudden steps every few months or years.
The symptoms depend on the part of the brain that’s affected, but can include:
- significant slowness of thought
- feeling disorientated and confused
- memory loss and difficulty concentrating
- difficulty finding the right words
- severe personality changes, such as becoming aggressive
- depression, mood swings and lack of interest or enthusiasm
- finding it difficult to walk and keep balance, with frequent falls
- loss of bladder control (incontinence)
- increasing difficulty with daily activities
Some people also have some symptoms of Alzheimer’s disease.
If it’s spotted at an early stage, treatment may be able to stop the vascular dementia getting worse, or at least slow it down.
If you’re worried about someone else, encourage them to make an appointment with their doctor and perhaps suggest that you go with them.
Symptoms of dementia can have several causes. Your medical practitioner can do some simple checks to try to find out the cause and may refer you to a specialist for further tests.
Vascular dementia causes
Vascular dementia is caused by conditions that damage blood vessels in the brain and interrupt the flow of blood and oxygen to the brain, which damage and eventually kills the brain cells. However, not everyone who has had a stroke will develop vascular dementia. A person’s risk for dementia after stroke depends on the size and number of strokes and the brain regions affected. Vascular dementia can also result from other conditions that impede blood flow and delivery of oxygen to the brain, such as narrowing of the arteries. High blood pressure, problems with the heartbeat’s rhythm, diabetes, and high cholesterol can increase a person’s risk of vascular dementia. By controlling or managing risk factors, you may lower your chance of developing cognitive impairment and dementia.
In the research community, these conditions are known as vascular contributions to cognitive impairment and dementia (VCID). The brains of people with vascular dementia often show evidence of prior strokes, thickening blood vessel walls, and thinning white matter — the brain’s connecting “wires” that relay messages between regions.
Vascular dementia is usually due to:
- narrowing of the small blood vessels deep inside the brain – known as subcortical vascular dementia or small vessel disease.
- a stroke (where the blood supply to part of the brain is suddenly cut off, usually as the result of a blood clot) – called post-stroke dementia or single-infarct dementia.
- lots of “mini strokes” that cause tiny but widespread damage to the brain – known as multi-infarct dementia.
Some people with vascular dementia also have brain damage caused by Alzheimer’s disease. This is known as mixed dementia.
Things that can increase your chances of getting vascular dementia in later life include:
- high blood pressure (hypertension)
- smoking
- an unhealthy diet
- high blood cholesterol
- lack of exercise
- being overweight or obese
- diabetes
- excessive alcohol consumption
- atrial fibrillation (a type of irregular heartbeat) and other types of heart disease
These problems increase the risk of damage to the blood vessels in and around the brain, or cause blood clots to develop inside them.
Vascular dementia prevention
By making healthy lifestyle changes – such as stopping smoking, lose excess body weight and exercising regularly – and treating any health conditions you have, you may be able to reduce your risk of vascular dementia. Tackling these might reduce your risk of vascular dementia in later life, although it’s not yet clear exactly how much your risk of dementia can be reduced.
This may also help slow down or stop the progression of vascular dementia if you’re diagnosed in the early stages. See treating vascular dementia for more information.
But there are some things you can’t change that can increase your risk of vascular dementia, such as:
- your age – the risk of vascular dementia increases as you get older, with people over 65 most at risk
- your family history – your risk of problems such as strokes is higher if a close family member has had them
- your ethnicity – if you have a south Asian, African or Caribbean background, your risk of vascular dementia is higher, as related problems such as diabetes and high blood pressure are more common in these groups
In rare cases, unavoidable genetic conditions can also increase your risk of vascular dementia.
Vascular dementia diagnosis
There’s no single test for vascular dementia. To diagnose vascular dementia, a doctor may ask about problems with daily activities, conduct memory or thinking tests, and speak with someone who knows the person well to see if symptoms of dementia are present. Medical history, lifestyle, and brain imaging tests are often used to help determine whether vascular dementia is the cause of symptoms.
The following are needed to make a diagnosis:
- An assessment of symptoms – for example, whether there are typical symptoms of vascular dementia
- A full medical history, including asking about a history of conditions related to vascular dementia, such as strokes or high blood pressure
- An assessment of mental abilities –this will usually involve a number of tasks and questions
- Brain scan, such as an MRI scan, CT scan or a single photon-emission computed tomography (SPECT) scan – this can detect signs of dementia and damage to the blood vessels in the brain.
- Transcranial Doppler – transcranial doppler sonography studies can now provide valuable information on cerebrovascular resistance, cerebrovascular reserve, and cerebral perfusion 55. Vascular resistance is calculated from the pulsative index (systolic/diastolic ratio); increased pulsative index indicates increased cerebrovascular resistance. Cerebrovascular reserve is calculated from the response of the cerebral vessels to a vasodilatory challenge either with CO2 elevation, as tested by breath holding, or with acetazolamide injection 56. Cerebral perfusion is assessed as a velocity measure of the individual vessels. Patients with vascular dementia secondary to small vessel disease have a significant increase in vascular resistance and a decrease in vascular reserve. In Alzheimer’s disease, vascular resistance and reserve are normal, and there is a decrease in perfusion through the middle cerebral artery secondary to the atrophic brain tissue that it supplies. Therefore, Doppler studies can be very helpful in sorting out the vascular factors and establishing the diagnosis in dementia patients with abnormal MRIs and a history compatible with cerebrovascular disease.
Vascular dementia treatment
Currently there are no treatments are available to reverse brain damage that has been caused by a stroke. Treatment for vascular dementia focuses on preventing future strokes. A healthy lifestyle is important to help reduce risk factors of vascular dementia. This includes eating well, limiting alcohol, not smoking, exercising, and managing stress. Medications to prevent strokes, such as blood thinners, may help decrease the risk of further damage to the brain. Medications that help treat the symptoms of Alzheimer’s disease might benefit people with early vascular dementia. A doctor may also recommend treating risk factors, such as high blood pressure or high cholesterol, through medications and lifestyle changes.
Lifestyle changes
The main aim of treatment for vascular dementia is to treat the underlying cause to help stop the condition getting worse.
This will usually involve making healthy lifestyle changes, such as:
- eating healthily, for example, you may be advised to follow a low-salt diet to manage high blood pressure
- losing weight if you’re overweight
- stopping smoking
- getting fit
- cutting down on alcohol
Medication
Medication may also be offered to treat the underlying cause of vascular dementia and help stop it getting worse.
These include:
- medication for high blood pressure
- statins to treat high cholesterol
- medicines such as aspirin or clopidogrel to reduce the risk of blood clots and further strokes
- anticoagulant medication, such as warfarin, which can also reduce the risk of blood clots and further strokes
- medication for diabetes
Some medicines may also help with some of the symptoms of vascular dementia. For example, antidepressants can help relieve depression.
Alzheimer’s disease medications such as donepezil (Aricept), galantamine (Reminyl) or rivastigmine (Exelon) aren’t used to treat vascular dementia, but may be used in people with a combination of vascular dementia and Alzheimer’s disease.
Vascular dementia life expectancy
Vascular dementia will usually get worse over time. This can happen in sudden steps, with periods in between where the symptoms don’t change much, but it’s difficult to predict when this will happen.
Home-based help will usually be needed, and some people will eventually need care in a nursing home.
Although treatment can help, vascular dementia can significantly shorten life expectancy. But this is highly variable and many people live for a number of years with the condition or die from some other cause.
Frontotemporal dementia
Frontotemporal dementia also known as frontotemporal lobe dementia, frontotemporal dementia with parkinsonism or Pick’s disease, is an umbrella term for a group of brain disorders that primarily affect the frontal and temporal lobes of the brain 57. The frontal and temporal lobes of the brain are generally associated with personality, behavior and language. The frontal lobe is the largest lobe in the brain and is located right behind the forehead. The frontal lobe is critical for thinking, planning, decision making and other higher mental processes. The frontal lobe helps people to manage and control emotional responses. The temporal lobes are located below and to both sides of the frontal lobe. The temporal lobes are believed to be involved in semantic memory, or your knowledge of objects, people, words and faces. The temporal lobes also play a role in language and emotion. In frontotemporal dementia, the frontal and temporal lobes of the brain shrink (atrophy). Signs and symptoms vary, depending on which part of the brain is affected. Some people with frontotemporal dementia have dramatic changes in their personalities and become socially inappropriate, impulsive or emotionally indifferent, while others lose the ability to use language properly. Presently, the term frontotemporal dementia encompasses clinical disorders that include changes in behavior, language, executive function and often motor symptoms 58. Frontotemporal dementia usually causes changes in behavior or language problems at first. These come on gradually and get worse slowly over time. Eventually, most people will experience problems in both of these areas. Some people also develop physical problems and difficulties with their mental abilities. Frontotemporal dementia can be misdiagnosed as a psychiatric problem or as Alzheimer’s disease.
Frontotemporal dementia is the third most common form of dementia across all age groups, after Alzheimer’s disease and Lewy body dementia and is the second most common cause of early-onset dementia (age <65) and usually involves patients with age ranges from 45 to 65 59, 60. Frontotemporal dementia can occur in people as young as 20. But it usually begins between ages 40 and 60. The average age at which it begins is 54. People with frontotemporal dementia have abnormal substances called tangles, Pick bodies, and Pick cells and 3R tau proteins inside nerve cells in the damaged areas of the brain.
The first description of a patient with frontotemporal dementia was made by Arnold Pick in 1892 61; the patient had aphasia (aphasia affects a person’s ability to express and understand written and spoken language), lobar atrophy, and presenile dementia. In 1911, Alois Alzheimer recognized the characteristic association with Pick bodies and named the clinicopathological entity Pick’s disease 62, which led to the use of Pick’s disease as a synonym for frontotemporal dementia. In 1982, Mesulam described a language subtype of the disorder, later defined as primary progressive aphasia 63. It was later replaced by the term frontotemporal dementia based on distinct clinical and histopathological criteria, assigned by a research group from the United Kingdom and Sweden in 1994 64.
Frontotemporal dementia is classified into two distinct clinical types; behavior variant frontotemporal dementia (bvFTD) and language type frontotemporal dementia 60. The language type of frontotemporal dementia is further classified into non-fluent variant primary progressive aphasia (nfvPPA), and semantic variant primary progressive aphasia (svPPA) depending on the areas of neuronal loss in frontal and temporal lobes 65.
Frontotemporal dementia symptoms include marked changes in social behavior and personality, and/or problems with language. People with behavior changes may have disinhibition (with socially inappropriate behavior), apathy and loss of empathy, hyperorality (eating excessive amounts of food or attempting to consume inedible things), agitation, compulsive behavior, and various other changes 66. Examples of problems with language include difficulty speaking or understanding speech. Some people with frontotemporal dementia also develop a motor syndrome such as parkinsonism or motor neuron disease (which may be associated with various additional symptoms).
The exact cause of frontotemporal dementia is unknown, but it’s mainly a sporadic disease. Genetics also plays a key role where approximately 30-40% of cases are passed down through families 67, 68. Among them, 13.4% of cases have an autosomal dominant inheritance. Mutations in over 20 genes have been identified in the possible development of frontotemporal dementia 60. Commonly involved genes are MAPT gene (microtubules associated protein tau gene located on chromosome 17q21.32), GRN gene (granulin gene on chromosome 17q21.32) and hexanucleotide repeat expansion of the chromosome 9 open-reading-frame 72 (C9orf72) gene on chromosomal location 9p21.2 (responsible for cytoskeleton organization) 69, 70. It is also postulated that C9orf72 gene mutation is a continuum between frontotemporal dementia and amyotrophic lateral sclerosis (ALS or Lou Gehrig’s disease) and is responsible for the occurrence of both entities. If you have a family history of frontotemporal dementia, you may want to consider talking to your doctor about being referred to a geneticist and possibly having a genetic test to see if you’re at risk. Head trauma and thyroid disease have been linked with the development of frontotemporal dementia with a 3.3-fold higher risk and 2.5-fold higher risk, respectively 60.
Frontotemporal dementia is rare with estimation of 1.61 to 4.1 cases per one hundred thousand people annually 71. One study showed the prevalence of frontotemporal dementia, progressive supranuclear palsy and corticobasal syndrome was 10.8 per 100,000 with peak prevalence at the age range of 65 to 69 years 71. There is an estimated twenty to thirty thousand people in the United States with frontotemporal dementia at one time 72.
Types of frontotemporal dementia
Frontotemporal degeneration is the umbrella term for a range of disorders that impact the frontal and temporal lobes of the brain. Each disorder can be identified according to the symptoms that appear first and most prominently, whether in behavior (behavioral variant frontotemporal degeneration [bvFTD]), changes in the ability to speak and understand language (primary progressive aphasia [PPA]) or in movement (corticobasal syndrome, progressive supranuclear palsy). The clinical syndrome where FTD and amyotrophic lateral sclerosis (ALS) occur in the same person is referred to as ALS-Frontotemporal Spectrum Disorder (ALS-FTSD).
The most common types of FTD are:
- Behavioral variant frontotemporal dementia (bvFTD). Progressive behavior/personality decline—characterized by changes in personality, behavior, emotions, and judgment (called
behavioral variant frontotemporal dementia). - Primary progressive aphasia (PPA). Progressive language decline — Aphasia means difficulty communicating. This form has three subtypes:
- Progressive Nonfluent/Agrammatic variant PPA (nfvPPA) also called progressive nonfluent aphasia, which affects the ability to speak.
- Semantic variant PPA also called semantic dementia, which affects the ability to use and understand language.
- Logopenic variant PPA also known as PPA-L have difficulty finding words when they are speaking.
- Frontotemporal Dementia with Motor Neuron Disease (FTD-MND) also called Frontotemporal Degeneration with ALS (FTD-ALS).
- Frontotemporal degeneration and the motor neuron disease ALS can occur in the same individual. ALS also known as Lou Gehrig’s disease in the US, is sometimes used interchangeably with the term motor neuron disease (MND). Motor neuron disease is also used as a general term for a group of progressive neurological disorders in which there is a loss of nerve cells called motor neurons. These cells control voluntary muscle activity such as speaking, walking, and talking. Individuals may exhibit weakness, muscle loss (atrophy), tiny involuntary muscle twitches (fasciculation), difficulty speaking (dysarthria) and difficult swallowing (dysphagia). Involuntary control of muscle activity such as breathing and increased tightness and stiffness of muscles (spasticity) is also affected. When people have symptoms of frontotemporal degeneration and motor neuron disease, they may be described as having FTD associated with motor neuron disease (FTD-MND), or FTD associated with amyotrophic lateral sclerosis (FTD-ALS).
- Corticobasal Syndrome (CBS)
- Progressive Supranuclear Palsy (PSP)
In the early stages it can be hard to know which of these disorders a person has because symptoms and the order in which they appear can vary widely from one person to the next. Also, the same symptoms can appear later in different disorders. For example, language problems are most typical of primary progressive aphasia but can also appear later in the course of behavioral variant frontotemporal dementia (bvFTD)
Of the clinical syndromes, frontotemporal dementia with motor neuron disease (FTD-MND) is the most heritable and svPPA is the least heritable (Goldman et al., 2005).
Table 3. Types of Frontotemporal Dementia (FTD) or Frontotemporal Lobar Degeneration (FLTD)
Frontotemporal dementia signs and symptoms
Signs and symptoms of frontotemporal dementia can be different from one individual to the next. Signs and symptoms get progressively worse over time, usually over years. Clusters of symptom types tend to occur together, and people may have more than one cluster of symptom types.
Signs of frontotemporal dementia can include:
- Personality and behavior changes – acting inappropriately or impulsively, appearing selfish or unsympathetic, neglecting personal hygiene, overeating, or loss of motivation
- Language problems – speaking slowly, struggling to make the right sounds when saying a word, getting words in the wrong order, or using words incorrectly
- Problems with mental abilities – getting distracted easily, struggling with planning and organisation
- Memory problems – these only tend to occur later on, unlike more common forms of dementia, such as Alzheimer’s disease
There may also be physical problems, such as slow or stiff movements, loss of bladder or bowel control (usually not until later on), muscle weakness or difficulty swallowing.
These problems can make daily activities increasingly difficult, and the person may eventually be unable to look after themselves.
Behavior and personality changes
Many people with frontotemporal dementia develop a number of unusual behaviors they’re not aware of. The most common signs of frontotemporal dementia involve extreme changes in behavior and personality. These can include:
- being insensitive or rude
- increasingly inappropriate social behavior
- acting impulsively or rashly
- loss of inhibitions
- lack of judgment
- seeming subdued
- losing interest (apathy) in people and things, which can be mistaken for depression
- losing drive and motivation
- inability to empathise with others, seeming cold and selfish
- repetitive compulsive behaviors, such as humming, smacking lips, hand-rubbing, clapping and foot-tapping, or routines such as walking exactly the same route repetitively
- a change in food preferences, such as suddenly liking sweet foods, overeating and poor table manners
- eating inedible objects
- compulsively wanting to put things in the mouth
- compulsive eating, alcohol drinking and/or smoking
- neglecting personal hygiene
As the condition progresses, people with frontotemporal dementia may become socially isolated and withdrawn.
Speech and language problems
Some subtypes of frontotemporal dementia lead to language problems or impairment or loss of speech. Primary progressive aphasia, semantic dementia and progressive agrammatic (nonfluent) aphasia are all considered to be frontotemporal dementia.
Problems caused by these conditions include:
- Increasing difficulty in using and understanding written and spoken language, such as having trouble finding the right word to use in speech or naming objects
- Trouble naming things, possibly replacing a specific word with a more general word such as “it” for pen
- Using words incorrectly – for example, calling a sheep a dog
- No longer knowing word meanings
- Forgetting the meaning of common words
- Loss of vocabulary
- Having hesitant speech that may sound telegraphic
- Making mistakes in sentence construction
- Difficulty making the right sounds to say words
- Repeating a limited number of phrases
- Slow, hesitant speech
- Getting words in the wrong order
- Automatically repeating things other people have said
Some people gradually lose the ability to speak, and can eventually become completely mute.
Problems with mental abilities
Problems with thinking do not tend to occur in the early stages of frontotemporal dementia, but these often develop as the condition progresses. These can include:
- difficulty working things out and needing to be told what to do
- poor planning, judgement and organization
- becoming easily distracted
- thinking in a rigid and inflexible way
- losing the ability to understand abstract ideas
- difficulty recognizing familiar people or objects
- memory difficulties, although this is not common early on
Motor disorders
Rarer subtypes of frontotemporal dementia are characterized by problems with movement similar to those associated with Parkinson’s disease or amyotrophic lateral sclerosis (ALS).
Motor-related problems may include:
- Tremor
- Rigidity
- Muscle spasms or twitches
- Slow, stiff movements, similar to Parkinson’s disease
- Poor coordination
- Difficulty swallowing
- Loss of bladder control
- Loss of bowel control
- Muscle weakness
- Inappropriate laughing or crying
- Falls or walking problems
Some people have frontotemporal dementia overlapping with other neurological (nerve and brain) problems, including:
- motor neurone disease – causes increasing weakness, usually with muscle wasting
- corticobasal degeneration – causes problems controlling limbs, loss of balance and co-ordination, slowness and reduced mobility
- progressive supranuclear palsy – causes problems with balance, movement, eye movements and swallowing.
Frontotemporal dementia complications
According to survey-based studies, sleep, or nighttime behavior disturbance is demonstrated by 33%-76% of patients with frontotemporal dementia. these are more likely to be reported in patients with primary progressive aphasia than behavior variant frontotemporal dementia (bvFTD). Similar is the case in the caregivers of primary progressive aphasia patients. All sleep disorders require sleep hygiene and routine maintenance. Also, there is a risk of an overlapping syndrome with frontotemporal dementia, such as frontotemporal dementia-motor neuron disease, frontotemporal dementia-progressive supranuclear palsy, and frontotemporal dementia-amyotrophic lateral sclerosis. All require both pharmacologic and non-pharmacologic approaches as well as treatment of underlying disease.
- Insomnia: reported in 48% of cases
- Sleep-disordered breathing: reported in 68% of cases 74
- Excessive daytime sleepiness: reported in 64% of cases
- Restless leg syndrome: reported in 8% of cases
- Risk of falls 75
- Hypersexuality
- Eating disorders, (such as hyperphagia/binge eating, lack of appetite, a particular food preference) 76
Frontotemporal dementia causes
In frontotemporal dementia, the frontal and temporal lobes of the brain shrink. In addition, there is an abnormal buildup of altered brain proteins in the frontal and temporal lobes of the brain. What causes these changes is usually unknown. In about 60% of people, they are often the first person is their family to be affected by frontotemporal degeneration. These people are classified as having a sporadic or nonfamilial form of the frontotemporal dementia. Researchers do not know why these people have developed frontotemporal degeneration. Their family members do not appear to be at an increased risk of developing the disorder. However, there is a strong genetic component to frontotemporal degeneration. In about 40% percent of cases there is a history of neurodegeneration or psychiatric illness in the family 68. In some of these people, the cause of the disorder is unknown. Researchers do not know whether there is an altered gene in these families, but the disorder occurs with more frequency than it otherwise would by chance. One theory is that, in these families, frontotemporal degeneration occurs because of the interaction of multiple factors (multifactorial cause). This might mean that there is a gene or genes that cause people to be more likely (predisposed) to develop a disorder, but the disorder will not develop unless these gene(s) are triggered or “activated” under certain circumstances such as particular environmental factors. However, despite research, no evidence has been found linking any potential environmental factor to the development of frontotemporal degeneration. There is no known risk factor for the disorder other than a family history of frontotemporal degeneration or a similar disorder. Recently, researchers have confirmed shared genetics and molecular pathways between frontotemporal dementia and amyotrophic lateral sclerosis (ALS). More research needs to be done to understand the connection between these conditions, however.
The two most common proteins involved are the tau protein and the transactive response DNA binding protein-43 (TDP-43). These proteins are normally found in the brain. Their specific functions are complex and not fully understood. However, these proteins are critical to the proper health and function of nerve cells (neurons). In individuals with frontotemporal dementia these proteins are misfolded (misshapen); as a result, they clump together. As these protein clumps, or aggregates, build up in the brain, they interfere with or disrupt the normal functioning of nerve cells, which ultimately die. Nerve cell death within the frontal and temporal lobes causes these areas of the brain shrink (atrophy).
These clumps of misfolded proteins may be referred to as “bodies” or “inclusions”. So the terms, tau bodies or tau inclusions, refer to nerve cells that contain these clumps of misfolded proteins. These inclusions may have distinctive features even when the same protein is involved. For examples, Pick’s disease refers to distinctive, round, silver-staining inclusions called Pick’s bodies. Most inclusions associated with frontotemporal dementia contain either variations of tau protein or TDP-43.
In about 10-25% of people diagnosed with frontotemporal dementia, an altered gene has been identified. Mutations in over 20 genes have been identified in the possible development of frontotemporal dementia 60. Genes provide instructions for creating proteins that play a critical role in many functions of the body. When an alteration of a gene occurs, the protein product may be faulty, inefficient, or absent. Depending upon the functions of the particular protein, this can affect many organ systems of the body, including the brain.
The major genes that have been linked to frontotemporal degeneration include the MAPT gene (microtubules associated protein tau gene located on chromosome 17q21.32), GRN gene (granulin gene on chromosome 17q21.32) and hexanucleotide repeat expansion of the chromosome 9 open-reading-frame 72 (C9orf72) gene on chromosomal location 9p21.2 (responsible for cytoskeleton organization) 69, 70. The MAPT gene provides instructions for making a protein called tau. This protein is found throughout the nervous system, including in nerve cells (neurons) in the brain. It is involved in assembling and stabilizing microtubules, which are rigid, hollow fibers that make up the cell’s structural framework (the cytoskeleton). Microtubules help cells maintain their shape, assist in the process of cell division, and are essential for the transport of materials within cells. The GRN gene produces the brain protein known as progranulin, which is important for optimal functioning of the protein TDP-43. Progranulin appears to be critical for the survival of nerve cells (neurons). The C9ORF72 gene also produces a protein that is found in nerve cells although its role in not well understood. It is also postulated that C9orf72 gene mutation is a continuum between frontotemporal dementia and amyotrophic lateral sclerosis and is responsible for the occurrence of both entities. These three genes account for the majority of people with inherited frontotemporal degeneration. In rare instances, additional genes have been identified as causing frontotemporal degeneration including the VCP, TARDBP, FUS, CHMP2B and TBK1 genes.
The altered genes that cause frontotemporal degeneration are usually inherited in an autosomal dominant manner. Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother. Dominant genetic disorders occur when only a single copy of an abnormal gene is necessary for the appearance of the disease. The altered gene can be inherited from either parent, or can be the result of a new mutation (gene change) in the affected individual. The risk of passing the abnormal gene from affected parent to offspring is 50% for each pregnancy regardless of the sex of the resulting child. If you have a family history of frontotemporal dementia, you may want to consider talking to your doctor about being referred to a geneticist and possibly having a genetic test to see if you’re at risk.
Head trauma and thyroid disease have been linked with the development of frontotemporal dementia with a 3.3-fold higher risk and 2.5-fold higher risk, respectively 60.
Frontotemporal dementia diagnosis
There’s no single test for frontotemporal dementia. A diagnosis of frontotemporal degeneration is based upon identification of characteristic symptoms, a detailed patient and family history, a thorough clinical evaluation and a variety of specialized tests. The disorder can be especially challenging to diagnose early because symptoms of frontotemporal dementia often overlap with those of other conditions. In addition, early in the course of the disorder, people with the behavioral variant of frontotemporal degeneration tend to score very well on neuropsychological testing.
Blood tests
Blood tests to help rule out other conditions, such as liver or kidney disease, your doctor may order blood tests.
Sleep study
Some symptoms of obstructive sleep apnea (memory and thinking problems and behavioral changes) can be similar to those of frontotemporal dementia. If you also have symptoms of sleep apnea (loud snoring and pauses in breathing while sleeping), your doctor may have you undergo a sleep study to rule out obstructive sleep apnea as a cause of your symptoms.
Neuropsychological testing
A neuropsychological evaluation involves an interview and certain tests, often pencil and paper type tests. This evaluation will allow a physician to assess behavior, language, memory, visual-spatial and other cognitive functions. This type of testing is especially helpful in determining the type of dementia at an early stage. The pattern of testing abnormality may help distinguish frontotemporal dementia from other causes of dementia.
Brain scans
By looking at images of the brain, doctors may be able to pinpoint any visible conditions — such as clots, bleeding or tumors — that may be causing signs and symptoms.
- Magnetic resonance imaging (MRI). An MRI machine uses radio waves and a strong magnetic field to produce detailed images of the brain.
- Fluorodeoxyglucose positron emission tracer (FDG-PET) scan. This test uses a low-level radioactive tracer attached to glucose (blood sugar) that’s injected into the blood. This allows physicians to see the chemical activity (metabolism) in the brain. The tracer can help show areas of the brain where nutrients are poorly metabolized. Areas of low metabolism can show where degeneration has occurred in the brain, which can help doctors diagnose the type of dementia. Individuals with frontotemporal degeneration exhibit reduced chemical activity (hypometabolism) in the frontal and temporal areas of the brain, a finding that can distinguish the disorder from Alzheimer disease. More recently, an amyloid PET tracer has been developed that can bind to misfolded amyloid proteins accumulated in the brain. This can support or rule out a diagnosis of Alzheimer’s disease which can then be ruled out during the diagnostic process for frontotemporal dementia. A spinal tap (lumbar puncture) can be performed to measure amyloid and tau proteins. This test can also support or decrease the likelihood of an Alzheimer’s diagnosis.
- Single-photon emission computed tomography (SPECT) scan. In SPECT, physicians use a radioactive substance and a special camera to create 3-D images of internal areas of the body, including the brain. SPECT can reveal characteristic changes such as reduced blood flow in certain areas of the brain.
Lumbar puncture
Lumbar puncture (spinal tap) to test the spinal fluid (fluid that surrounds and supports the brain and spine); this may be useful to rule out Alzheimer’s disease as the cause of symptoms.
Molecular genetic testing
Molecular genetic testing can confirm a diagnosis of frontotemporal degeneration in certain people. Molecular genetic testing can detect mutations in the MAPT, GRN or C9ORF72 genes known to cause frontotemporal dementia, but testing is currently available only as a diagnostic service at specialized laboratories or as part of a clinical research study. Genetic testing should only be performed after genetic counseling.
Frontotemporal dementia treatment
There’s currently no cure for frontotemporal dementia or any treatment that will slow it down. Drugs used to treat or slow Alzheimer’s disease don’t seem to be helpful for people with frontotemporal dementia, and some may worsen the symptoms of frontotemporal dementia. But certain medications and speech therapy can help manage symptoms of frontotemporal dementia, possibly for several years.
Frontotemporal dementia treatments include:
- medicines – to control some of the behavioural problems
- therapies – such as physiotherapy, occupational therapy, and speech and language therapy for problems with movement, everyday tasks and communication
- dementia activities – such as memory cafes, which are drop-in sessions for people with memory problems and their carers to get support and advice
- support groups – who can offer tips on managing symptoms from dementia experts and people living with frontotemporal dementia, and their families
Medications
- Antidepressants. Some types of antidepressants, such as trazodone, may reduce the behavioral problems associated with frontotemporal dementia. Selective serotonin reuptake inhibitors (SSRIs) — such as citalopram (Celexa), paroxetine (Paxil) or sertraline (Zoloft) — also have been effective in some people.
- Antipsychotics. Antipsychotic medications, such as olanzapine (Zyprexa) or quetiapine (Seroquel), are sometimes used to treat the behavioral problems of frontotemporal dementia. However, these medications must be used with caution in people with dementia due to the risk of serious side effects, including an increased risk of death.
Therapy
People experiencing language difficulties may benefit from speech therapy to learn alternate strategies for communication.
Home remedies
You’ll need to have caregivers, as your condition progresses, to assist with daily life activities, maintain your safety, provide transportation and help with finances. Your doctor will discuss lifestyle changes with you, such as when you may need to stop driving a car or let someone you trust take over your finances. Regular cardiovascular exercise may help improve your mood and thinking skills.
It may be helpful to make some adjustments in your home to make daily living tasks easier and reduce your chance of injuries, such as removing rugs or raising toilets.
In some cases, caregivers can reduce behavioral problems by changing the way they interact with people with dementia. Ask your loved one’s doctor about any available resources that provide training in caring for someone with dementia. Possible changes in interaction include:
- Avoiding events or activities that trigger the undesirable behavior
- Removing negative environmental cues, such as the car keys
- Maintaining a calm environment
- Providing structured routines
- Simplifying daily tasks
- Distracting and redirecting attention from problem behaviors
Frontotemporal dementia prognosis
How quickly frontotemporal dementia gets worse varies from person to person and is very difficult to predict. People with frontotemporal dementia can become socially isolated as the illness progresses. They may not want to spend time in the company of others, or may behave in rude or insulting ways. Home-based help will usually be needed at some stage, and some people will eventually need care in a nursing home.
The average survival time after frontotemporal dementia symptoms start is around 8 to 10 years. But this is highly variable and some people live much longer than this.
The mortality risk associated with frontotemporal dementia is six-fold as compared to Alzheimer’s disease, which has a four-fold risk 70.
HIV associated dementia
When someone has the human immunodeficiency virus (HIV) and acquired immune deficiency syndrome (AIDS) they may develop a complication to the disease which is known as HIV associated dementia, or as AIDS Dementia Complex 77.
AIDS Dementia Complex is a complicated syndrome made up of different nervous system and mental symptoms that can develop in some people with HIV disease. The incidence of ADC is uncommon in people with the early stages of the disease, but may increase as the disease advances to around 7% in people not taking anti-HIV drugs.
Not everyone who has HIV/AIDS will develop AIDS Dementia Complex, but some will.
Who gets AIDS Dementia Complex ?
People who have HIV/AIDS are at risk of developing AIDS Dementia Complex. Because HIV/AIDS affects so many young people who are enjoying a full and independent lifestyle, there are particular issues such as employment, identity and sexuality which may have to be addressed. HIV/AIDS is still a disease that has a stigma attached to it by many people, and the effect of dementia on top of that can be enormously difficult for all concerned. AIDS Dementia Complex can cause great isolation and loneliness which adds to the daily struggles with the disease.
What is the progress of AIDS Dementia Complex ?
The rate of progression varies from person to person. However the disease can lead to complete dependence and death.
HIV associated dementia causes
While it has been shown that HIV does not directly infect nerve cells, it is thought that it can somehow infect them indirectly. Immune cells that are present in the brain act as HIV reservoirs and are the primary source of indirect damage to nerve cells.
HIV associated dementia signs and symptoms
The following is a list of possible AIDS Dementia Complex symptoms that could also be related to other problems that are not AIDS Dementia Complex.
Possible symptoms of Early Stage AIDS Dementia Complex are:
- Difficulty concentrating
- Difficulty remembering phone numbers or appointments
- Slowed thinking
- Taking longer to complete complicated tasks
- Difficulty keeping track of daily activities
- Irritability
- Unsteady gait or difficulty keeping balance
- Poor coordination and a change in handwriting
- Depression
Results of mental status tests and other mental capabilities may be normal in the early stages. Symptoms usually develop slowly. In the later stages of ADC they become worse. They may also worsen temporarily when the person is sick with other illnesses.
Possible symptoms of Middle Stage AIDS Dementia Complex are:
- Symptoms of motor dysfunction, such as muscle weakness
- Poor performance on regular tasks
- Increased concentration and attention required
- Reversing of numbers or words
- Slower responses and frequently dropping objects
- General feelings of indifference or apathy
- Slowness in normal activities, such as eating and writing
- Walking, balance, and coordination requires an increased effort.
Possible symptoms of Late Stage AIDS Dementia Complex are:
- Loss of bladder or bowel control
- Spastic gait, making walking increasingly difficult
- Loss of initiative or interest
- Withdrawal
- Psychosis or mania
- Confinement to bed.
These symptoms can leave the person confused and unable to make sense of the world. This frequently results in depression.
HIV associated dementia diagnosis
AIDS Dementia Complex should be diagnosed by someone with knowledge and experience with HIV patients, such as an HIV general practitioner or a medical specialist. The diagnosis of AIDS Dementia Complex is usually made by excluding other possible causes of the symptoms.
However, the main way to diagnose and evaluate AIDS Dementia Complex is a test called the mental status examination. Also, certain laboratory tests, including an examination of cerebrospinal fluid (CSF) can be useful. In addition, the amount of HIV in the CSF seems to correlate with progressive dementia in children. Other tests which can help in the differential diagnosis of AIDS Dementia Complex are CT scans, MRI scans and SPECT scans. These tests help differentiate AIDS Dementia Complex from other brain disorders such as cryptococcal meningitis, toxoplasmosis, lymphoma. An early diagnosis is important as many of the symptoms can be caused by other conditions and illnesses common to people with HIV/AIDS, many of which may be treatable. Some of the symptoms typical of AIDS Dementia Complex are also seen in psychiatric illnesses such as anxiety or depression. If an early diagnosis of AIDS Dementia Complex is made, appropriate treatment and management can be started.
AIDS Dementia Complex treatment
AIDS Dementia Complex can be treated to some degree and may even be preventable. The best treatments seem to be the anti-HIV drugs. Initially it was feared that highly active antiretroviral therapy (HAART) would not be effective against HIV in the brain, because many of these drugs do not cross the blood-brain barrier. However recent research has shown evidence of improvements in dementia and other neurological problems due to HAART. Despite these encouraging results, there is evidence that HAART is not as effective against dementia as it is against other opportunistic infections, as dementia is related more to tissue damage rather than removal of an infective organism.
Much of the evidence for the effectiveness of anti-HIV drugs against dementia relates to the drug AZT, mainly because for many years it was the only available anti-HIV drug which crossed the blood-brain barrier to any appreciable extent.
Some of the newer drugs such as d4T, abacavir, nevirapine, indinavir and efavirenz also cross the blood-brain barrier and reduce the amount of HIV in the CSF. However, by treating the HIV outside of the brain, the immune system can recover and fight the HIV inside the brain to help reduce or prevent AIDS Dementia Complex.
Medications that can also relieve some of the symptoms of AIDS Dementia Complex include antipsychotics, antidepressants and anticonvulsants.
Alcoholic dementia
Alcohol related dementia or alcohol induced dementia, as the name suggests, is a form of dementia related to the excessive drinking of alcohol. This affects memory, learning and other mental functions. Korsakoff’s syndrome and Wernicke/Korsakoff syndrome are particular forms of alcohol related brain injury which may be related to alcohol related dementia.
Who gets alcohol induced dementia?
Anyone who drinks excessive amounts of alcohol over a period of years may get alcohol related dementia. Males who drink more than six standard alcoholic drinks a day, and women who drink more than four, seem to be at increased risk of developing alcohol related dementia. The risk clearly increases for people who drink high levels of alcohol on a regular basis. The U.S. Department of Health and Human Services Dietary Guidelines for Americans recommends that for health reasons related to the prevention of brain and liver damage adult males should drink no more than two standard drinks per day and adult females should drink no more than one standard drinks per day. One standard drink-equivalent contains 14 grams of pure alcohol.
Some people who drink at high levels do not develop alcohol related dementia, but it is not currently possible to understand and predict who will and who won’t develop alcohol related dementia. Some people who develop alcohol related dementia might also show some degree of recovery over time if they reduce alcohol intake to safe levels or abstain from alcohol and maintain good health. Alcohol related dementia can affect both men and women of any age.
Alcohol induced dementia causes
It is currently unclear as to whether alcohol has a direct toxic effect on the brain cells, or whether the damage is due to lack of thiamine, vitamin B1. Nutritional problems, which often accompany consistent or episodic heavy use of alcohol, are thought to be contributing factors. Key parts of the brain may suffer damage through vitamin deficiencies, particularly marked levels of thiamine deficiency and the direct effect that alcohol has on the absorption and use of thiamine.
Alcohol induced dementia signs and symptoms
This can vary from person to person, but generally symptoms will include:
- Impaired ability to learn things
- Personality changes
- Problems with memory
- Difficulty with clear and logical thinking on tasks which require planning, organising, common sense judgement and social skills
- Problems with balance
- Decreased initiative and spontaneity.
Generally skills learned earlier in life and old habits such as language and gestures tend to be relatively unaffected.
Alcohol induced dementia treatment
At an early stage alcoholic dementia, problems may be reduced or reversed if the person abstains from alcohol, improves their diet and replace vitamins especially thiamine (vitamin B1). Thiamine is important to limit some of the toxic effects of alcohol, and is an important supplement for heavy drinkers.
Community support is available for the person with dementia, their family and carers. This support can make a positive difference to managing dementia.
Many people who develop alcohol related dementia are young, and this can mean that they and their family and carers will need extra consideration.
Creutzfeldt-Jakob disease
Creutzfeldt-Jakob disease (CJD) is a rare, degenerative, invariably fatal brain disorder that is characterized by rapidly progressive dementia 78. It affects about one person in every one million people per year worldwide; in the United States there are about 300 cases per year. Creutzfeldt-Jakob disease usually appears in later life and runs a rapid course. Typically, onset of symptoms occurs about age 60, and about 90 percent of individuals die within 1 year. In the early stages of disease, people may have failing memory, behavioral changes, lack of coordination and visual disturbances. As the illness progresses, mental deterioration becomes pronounced and involuntary movements, blindness, weakness of extremities, and coma may occur 79. This condition often leads to death within a few weeks or months after symptoms begin. About 90 percent of patients do not survive for more than one year.
There are three major categories of Creutzfeldt-Jakob disease:
- In sporadic Creutzfeldt-Jakob disease, the disease appears even though the person has no known risk factors for the disease. This is by far the most common type of Creutzfeldt-Jakob disease and accounts for at least 85 percent of cases.
- In hereditary Creutzfeldt-Jakob disease, the person has a family history of the disease and/or tests positive for a genetic mutation associated with Creutzfeldt-Jakob disease. About 5 to 10 percent of cases of Creutzfeldt-Jakob disease in the United States are hereditary.
- In acquired Creutzfeldt-Jakob disease, the disease is transmitted by exposure to brain or nervous system tissue, usually through certain medical procedures. There is no evidence that Creutzfeldt-Jakob disease is contagious through casual contact with a Creutzfeldt-Jakob disease patient. Since Creutzfeldt-Jakob disease was first described in 1920, fewer than 1 percent of cases have been acquired Creutzfeldt-Jakob disease.
Creutzfeldt-Jakob disease belongs to a family of human and animal diseases known as the transmissible spongiform encephalopathies (TSEs). Spongiform refers to the characteristic appearance of infected brains, which become filled with holes until they resemble sponges under a microscope. Creutzfeldt-Jakob disease is the most common of the known human TSEs. Other human TSEs include kuru, fatal familial insomnia (FFI), and Gerstmann-Straussler-Scheinker disease (GSS). Kuru was identified in people of an isolated tribe in Papua New Guinea and has now almost disappeared. FFI and GSS are extremely rare hereditary diseases, found in just a few families around the world. Other TSEs are found in specific kinds of animals. These include bovine spongiform encephalopathy (BSE), which is found in cows and is often referred to as “mad cow” disease; scrapie, which affects sheep and goats; mink encephalopathy; and feline encephalopathy. Similar diseases have occurred in elk, deer, and exotic zoo animals.
Creutzfeldt-Jakob disease can be very difficult to diagnose because it is similar to other forms of dementia 79. The only way to confirm the diagnosis is to test a small sample of brain tissue, which can be done by brain biopsy or autopsy. Creutzfeldt-Jakob disease is caused by the build up of abnormal prion proteins in the brain. For most patients, the reason for the abnormal prions is unknown (sporadic Creutzfeldt-Jakob disease). About 5 to 10 percent of cases are due to an inherited genetic mutation associated with Creutzfeldt-Jakob disease (familial Creutzfeldt-Jakob disease). This condition can also be acquired through contact with infected brain tissue (iatrogenic Creutzfeldt-Jakob disease) or consuming infected beef (variant Creutzfeldt-Jakob disease). There is no specific treatment for Creutzfeldt-Jakob disease, so the goal is to make a person as comfortable as possible 78, 80.
How is Creutzfeldt-Jakob disease transmitted ?
CJD cannot be transmitted through the air or through touching or most other forms of casual contact. Spouses and other household members of sporadic CJD patients have no higher risk of contracting the disease than the general population. However, exposure to brain tissue and spinal cord fluid from infected individuals should be avoided to prevent transmission of the disease through these materials.
In some cases, CJD has spread to other people from grafts of dura mater (a tissue that covers the brain), transplanted corneas, implantation of inadequately sterilized electrodes in the brain, and injections of contaminated pituitary growth hormone derived from human pituitary glands taken from cadavers. Doctors call these cases that are linked to medical procedures iatrogenic cases. Since 1985, all human growth hormone used in the United States has been synthesized by recombinant DNA procedures, which eliminates the risk of transmitting CJD by this route.
The appearance of the new variant of Creutzfeldt-Jakob disease (nv-CJD or v-CJD) in several younger than average people in Great Britain and France has led to concern that BSE may be transmitted to humans through consumption of contaminated beef. Although laboratory tests have shown a strong similarity between the prions causing BSE and v-CJD, there is no direct proof to support this theory.
Many people are concerned that it may be possible to transmit Creutzfeldt-Jakob disease through blood and related blood products such as plasma. Some animal studies suggest that contaminated blood and related products may transmit the disease, although this has never been shown in humans. If there are infectious agents in these fluids, they are probably in very low concentrations. Scientists do not know how many abnormal prions a person must receive before he or she develops CJD, so they do not know whether these fluids are potentially infectious or not. They do know that, even though millions of people receive blood transfusions each year, there are no reported cases of someone contracting CJD from a transfusion. Even among people with hemophilia, who sometimes receive blood plasma concentrated from thousands of donors, there are no reported cases of CJD.
While there is no evidence that blood from people with sporadic CJD is infectious, studies have found that infectious prions from BSE and vCJD may accumulate in the lymph nodes (which produce white blood cells), the spleen, and the tonsils. These findings suggest that blood transfusions from people with vCJD might transmit the disease. The possibility that blood from people with vCJD may be infectious has led to a policy preventing people in the United States from donating blood if they have resided for more than 3 months in a country or countries where BSE is common.`
How can people avoid spreading Creutzfeldt-Jakob disease ?
To reduce the already very low risk of CJD transmission from one person to another, people should never donate blood, tissues, or organs if they have suspected or confirmed CJD, or if they are at increased risk because of a family history of the disease, a dura mater graft, or other factor.
Normal sterilization procedures such as cooking, washing, and boiling do not destroy prions. Caregivers, healthcare workers, and undertakers should take the following precautions when they are working with a person with CJD:
- Cover cuts and abrasions with waterproof dressings.
- Wear surgical gloves when handling a patient’s tissues and fluids or dressing the patient’s wounds.
- Avoid cutting or sticking themselves with instruments contaminated by the patient’s blood or other tissues.
- Use disposable bedclothes and other cloth for contact with the patient. If disposable materials are not available, regular cloth should be soaked in undiluted chlorine bleach for an hour or more, and then washed in a normal fashion after each use.
- Use face protection if there is a risk of splashing contaminated material such as blood or cerebrospinal fluid.
- Soak instruments that have come in contact with the patient in undiluted chlorine bleach for an hour or more, then use an autoclave (pressure cooker) to sterilize them in distilled water for at least one hour at 132 – 134 degrees Centigrade.
Creutzfeldt-Jakob disease causes
Some researchers believe an unusual “slow virus” or another organism causes Creutzfeldt-Jakob disease. However, they have never been able to isolate a virus or other organism in people with the disease. Furthermore, the agent that causes Creutzfeldt-Jakob disease has several characteristics that are unusual for known organisms such as viruses and bacteria. It is difficult to kill, it does not appear to contain any genetic information in the form of nucleic acids (DNA or RNA), and it usually has a long incubation period before symptoms appear. In some cases, the incubation period may be as long as 50 years. The leading scientific theory at this time maintains that Creutzfeldt-Jakob disease and the other TSEs are caused by a type of protein called a prion.
Prion proteins occur in both a normal form, which is a harmless protein found in the body’s cells, and in an infectious form, which causes disease. The harmless and infectious forms of the prion protein have the same sequence of amino acids (the “building blocks” of proteins) but the infectious form of the protein takes a different folded shape than the normal protein. Sporadic Creutzfeldt-Jakob disease may develop because some of a person’s normal prions spontaneously change into the infectious form of the protein and then alter the prions in other cells in a chain reaction.
Once they appear, abnormal prion proteins aggregate, or clump together. Investigators think these protein aggregates may lead to the neuron loss and other brain damage seen in Creutzfeldt-Jakob disease. However, they do not know exactly how this damage occurs.
About 5 to 10 percent of all Creutzfeldt-Jakob disease cases are inherited. These cases arise from a mutation, or change, in the gene that controls formation of the normal prion protein. While prions themselves do not contain genetic information and do not require genes to reproduce themselves, infectious prions can arise if a mutation occurs in the gene for the body’s normal prion protein. If the prion protein gene is altered in a person’s sperm or egg cells, the mutation can be transmitted to the person’s offspring. All mutations in the prion protein gene are inherited as dominant traits. Therefore, family history is helpful in considering the diagnosis. Several different mutations in the prion gene have been identified. The particular mutation found in each family affects how frequently the disease appears and what symptoms are most noticeable. However, not all people with mutations in the prion protein gene develop Creutzfeldt-Jakob disease.
Creutzfeldt-Jakob disease signs and symptoms
Initially, patients experience problems with muscular coordination; personality changes, including impaired memory, judgment, and thinking; and impaired vision. People with the disease also may experience insomnia, depression, or unusual sensations. Creutzfeldt-Jakob disease does not cause a fever or other flu-like symptoms. As the illness progresses, the patients’ mental impairment becomes severe. They often develop involuntary muscle jerks called myoclonus, and they may go blind. They eventually lose the ability to move and speak and enter a coma. Pneumonia and other infections often occur in these patients and can lead to death 78.
There are several known variants of Creutzfeldt-Jakob disease. These variants differ somewhat in the symptoms and course of the disease. For example, a variant form of the disease-called new variant or variant (nv-Creutzfeldt-Jakob disease, v-Creutzfeldt-Jakob disease), described in Great Britain and France-begins primarily with psychiatric symptoms, affects younger patients than other types of Creutzfeldt-Jakob disease, and has a longer than usual duration from onset of symptoms to death. Another variant, called the panencephalopathic form, occurs primarily in Japan and has a relatively long course, with symptoms often progressing for several years. Scientists are trying to learn what causes these variations in the symptoms and course of the disease 78.
Some symptoms of Creutzfeldt-Jakob disease can be similar to symptoms of other progressive neurological disorders, such as Alzheimer’s or Huntington’s disease. However, Creutzfeldt-Jakob disease causes unique changes in brain tissue which can be seen at autopsy. It also tends to cause more rapid deterioration of a person’s abilities than Alzheimer’s disease or most other types of dementia.
Creutzfeldt-Jakob disease diagnosis
There is currently no single diagnostic test for Creutzfeldt-Jakob disease. When a doctor suspects Creutzfeldt-Jakob disease, the first concern is to rule out treatable forms of dementia such as encephalitis (inflammation of the brain) or chronic meningitis. A neurological examination will be performed and the doctor may seek consultation with other physicians. Standard diagnostic tests will include a spinal tap to rule out more common causes of dementia and an electroencephalogram (EEG) to record the brain’s electrical pattern, which can be particularly valuable because it shows a specific type of abnormality in Creutzfeldt-Jakob disease. Computerized tomography of the brain can help rule out the possibility that the symptoms result from other problems such as stroke or a brain tumor. Magnetic resonance imaging (MRI) brain scans also can reveal characteristic patterns of brain degeneration that can help diagnose Creutzfeldt-Jakob disease.
The only way to confirm a diagnosis of Creutzfeldt-Jakob disease is by brain biopsy or autopsy. In a brain biopsy, a neurosurgeon removes a small piece of tissue from the patient’s brain so that it can be examined by a neuropathologist. This procedure may be dangerous for the individual, and the operation does not always obtain tissue from the affected part of the brain. Because a correct diagnosis of Creutzfeldt-Jakob disease does not help the person, a brain biopsy is discouraged unless it is needed to rule out a treatable disorder. In an autopsy, the whole brain is examined after death. Both brain biopsy and autopsy pose a small, but definite, risk that the surgeon or others who handle the brain tissue may become accidentally infected by self-inoculation. Special surgical and disinfection procedures can minimize this risk. A fact sheet with guidance on these procedures is available from the National Institute of Neurological Disorders and Stroke and the World Health Organization.
Scientists are working to develop laboratory tests for Creutzfeldt-Jakob disease. One such test, developed at the National Institute of Neurological Disorders and Stroke, is performed on a person’s cerebrospinal fluid and detects a protein marker that indicates neuronal degeneration. This can help diagnose Creutzfeldt-Jakob disease in people who already show the clinical symptoms of the disease. This test is much easier and safer than a brain biopsy. The false positive rate is about 5 to 10 percent. Scientists are working to develop this test for use in commercial laboratories. They are also working to develop other tests for this disorder.
Creutzfeldt-Jakob disease treatment
There is no treatment that can cure or control Creutzfeldt-Jakob disease. Researchers have tested many drugs, including amantadine, steroids, interferon, acyclovir, antiviral agents, and antibiotics. Studies of a variety of other drugs are now in progress. However, so far none of these treatments has shown any consistent benefit in humans.
Current treatment for Creutzfeldt-Jakob disease is aimed at alleviating symptoms and making the individual as comfortable as possible. Opiate drugs can help relieve pain if it occurs, and the drugs clonazepam and sodium valproate may help relieve myoclonus. During later stages of the disease, changing the person’s position frequently can keep him or her comfortable and helps prevent bedsores. A catheter can be used to drain urine if the individual cannot control bladder function, and intravenous fluids and artificial feeding also may be used.
Huntington’s disease
Huntington’s disease is an inherited disorder that causes degeneration of brain cells, called neurons, in motor control regions of the brain, as well as other areas 81. Huntington’s disease is characterized by the gradual development of involuntary muscle movements affecting the hands, feet, face, and trunk and progressive deterioration of cognitive processes and memory (dementia). Dementia is typically associated with progressive disorientation and confusion, personality disintegration, impairment of memory control, restlessness, agitation, and other symptoms and findings. Symptoms of Huntington’s disease, which gets progressively worse, include uncontrolled, irregular, rapid, jerky movements (called chorea) and athetosis, a condition characterized by relatively slow, writhing involuntary movements, abnormal body postures, and changes in behavior, emotion, judgment, and cognition. People with Huntington’s disease also develop impaired coordination, slurred speech, and difficulty feeding and swallowing. Huntington’s disease typically begins between ages 30 and 50 81. An earlier onset form called juvenile Huntington’s disease, occurs under age 20 81. Symptoms of juvenile Huntington’s disease differ somewhat from adult onset Huntington’s disease and include unsteadiness, rigidity, difficulty at school, and seizures. In individuals with Huntington’s disease, the disease duration may range from approximately 10 years up to 25 years or more. Huntington’s disease progresses slowly and a person may live for another 15-20 years after the onset of symptoms. Life-threatening complications may result from pneumonia or other infections, injuries related to falls, or other associated developments.
More than 30,000 Americans have Huntington’s disease and another 200,000 are at risk of developing the condition. Symptoms commonly develop between ages 30 and 50 81. Genetic counseling will be of benefit for affected individuals and their families. Family members of affected individuals should also receive clinical evaluations to detect any symptoms and physical characteristics that may be potentially associated with Huntington’s disease.
Huntington’s disease is caused by a mutation in the gene for a protein called “huntingtin” located on the short arm (p) of chromosome 4 (4p16.3). The defect causes the cytosine, adenine, and guanine (CAG) building blocks of DNA to repeat many more times than is normal. The length of the expanded repeats may affect the age at symptom onset. The specific symptoms and physical features associated with Huntington’s disease result from degeneration of nerve cells (neurons) within certain areas of the brain (e.g., basal ganglia, cerebral cortex). Huntington’s disease is transmitted as an autosomal dominant trait. Each child of a parent with Huntington’s disease has a 50-50 chance of inheriting the Huntington’s disease gene 81. If a child does not inherit the Huntington’s disease gene, he or she will not develop the disease and generally cannot pass it to subsequent generations. There is a small risk that someone who has a parent with the mutated gene but who did not inherit the Huntington’s disease gene may pass a possibly harmful genetic sequence to her/his children. A person who inherits the Huntington’s disease gene will eventually develop the disease 81. A genetic test, coupled with a complete medical history and neurological and laboratory tests, helps physicians diagnose Huntington’s disease 81.
Huntington’s disease signs and symptoms
Huntington’s disease is characterized by rapid uncontrollable muscle movements such as tics or muscle jerks (choreiform movements or chorea). This disorder causes a loss of coordination and personality changes. As the disease progresses, the ability to speak may be impaired, memory may fade, and the involuntary jerky muscle movements (chorea) become more severe.
Huntington’s disease runs a ten to 25 year progressive course. As the disorder progresses, the chorea may subside and there may be an absence of movement (akinesia). Dementia gradually develops. Patients with Huntington’s disease are at high risk of developing pneumonia as a result of being bedridden and undernourished.
Huntington’s disease causes
Huntington’s disease is inherited as an autosomal dominant trait. Human traits, including the classic genetic diseases, are the product of the interaction of two genes, one received from the father and one from the mother. In dominant disorders, a single copy of the disease gene (received from either the mother or father) will be expressed “dominating” the other normal gene and resulting in the appearance of the disease. The risk of transmitting the disorder from affected parent to offspring is 50 percent for each pregnancy regardless of the sex of the resulting child.
Huntington’s disease is caused by changes (mutations) of a gene that is located on the short arm (p) of chromosome 4 (4p16.3). Chromosomes are found in the nucleus of all body cells. They carry the genetic characteristics of each individual. Pairs of human chromosomes are numbered from 1 through 22, with an unequal 23rd pair of X and Y chromosomes for males and two X chromosomes for females. Each chromosome has a short arm designated as “p” and a long arm identified by the letter “q.” Chromosomes are further subdivided into bands that are numbered.
Known as huntingtin, the gene controls the production of a protein found in nerve cells (neurons) throughout the brain. However, the specific function of the huntingtin protein is not known. In individuals with the disorder, the huntingtin gene contains errors in the coded “building blocks” that make up its specific genetic instructions. The instructions within every gene consist of different arrangements of four basic chemicals (nucleotide bases) called adenine (A), cytosine (C), guanine (G), and thymine (T). In those with Huntington’s Disease, the huntingtin gene contains abnormally long repeats of coded instructions consisting of cytosine, adenine, and guanine (CAG trinucleotide repeat expansion). For example, individuals with the disease have over 35 CAG repeats within the huntingtin gene, with most having more than 39. However, individuals without the disorder tend to have about 20 repeats in the gene. Expanded CAG repeats are unstable and may expand further over time and with successive generations. This is thought to be responsible for genetic anticipation, a phenomenon in which an individual with Huntington’s disease may have symptom onset at a significantly earlier age than his or her affected parent. In addition, some researchers suggest that expanded CAG repeats of the huntingtin gene may become more unstable when the gene is transmitted from the father.
The specific symptoms associated with Huntington’s disease are caused by degenerative changes of nerve cells (neurons) within certain regions of the brain, including the basal ganglia and cerebral cortex. The basal ganglia are specialized nerve cells deep within the brain that play a role in regulating movements. The cerebral cortex, the outer region of the brain, is responsible for conscious thought and movement.
Huntington’s disease diagnosis
The diagnosis of Huntington’s disease may be confirmed by a thorough clinical evaluation, detailed patient history, and a variety of specialized tests. Specialized x-ray studies such as computerized tomography (CT) scanning, magnetic resonance imaging (MRI), or electroencephalography (EEG) may help confirm the diagnosis of Huntington’s Disease. During CT scanning, a computer and x-rays are used to create a file showing cross-sectional images of the brain. During MRI, a magnetic field and radio waves are used to create cross-sectional images of the brain. During an EEG, an instrument records electrical activity of the brain. Neuropsychological and/or genetic tests are also used to aid the diagnosis of Huntington’s disease.
Huntington’s disease treatment
There is no treatment that can stop or reverse the course of Huntington’s disease. Tetrabenazine and deuterabenazine are prescribed for treating the chorea associated with Huntington’s disease. Antipsychotic drugs may help to alleviate chorea and may also be used to help control hallucinations, delusions, and violent outbursts. Neuroleptic medication such as haloperidon can partially suppress the involuntary movement, especially in the early stages. Other medications may be prescribed to treat depression and anxiety. Drugs used to treat the symptoms of Huntington’s disease may have side effects such as fatigue, sedation, decreased concentration, restlessness, or hyperexcitability, and should be only used when symptoms create problems for the individual.
Special high calorie food preparations may help an affected individual maintain weight and avoid choking during the later stages of Huntington’s disease.
Huntington’s disease prognosis
Huntington’s disease causes disability that gets worse over time. People with this disease usually die within 15 to 20 years following diagnosis. At this time, no treatment is available to slow, stop or reverse the course of Huntington’s disease.
Dementia Care Plans
A care plan sometimes called a care and support plan, or support plan if you’re a carer, sets out how your care and support needs will be met. Before treatment starts, your current and future health and social care needs will be assessed and a care plan drawn up.
This is a way of ensuring you receive the right treatment for your needs. It involves identifying areas where you may need some assistance, such as:
- what support you or your carer need for you to remain as independent as possible – including whether you might need care at home or in a nursing home
- whether there are any changes that need to be made to your home to make it easier to live in
- whether you need any financial assistance
You should be fully involved in the preparation of your care plan, and you and anyone else you request should also get a written copy. The care plan must set out:
- the needs identified by the assessment
- whether, and to what extent, the needs meet the eligibility criteria
- the needs that the authority is going to meet, and how it intends to do so
- for a person needing care, for which of the desired outcomes care and support could be relevant
- for a carer, the outcomes the carer wishes to achieve, and their wishes around providing care, work, education and recreation where support could be relevant
- the personal budget
- information and advice on what can be done to reduce the needs in question, and to prevent or delay the development of needs in the future
- where needs are being met via a direct payment, the needs to be met via the direct payment and the amount and frequency of the payments
Your care plan should be individual to you, and you should be allowed to have as much involvement in the development of your plan as you wish.
Care and support should help you to:
- live independently
- have as much control over your life as possible
- participate in society on an equal level, with access to employment and a family life
- have the best possible quality of life
- keep as much dignity and respect as possible
It’s worth remembering that if there are different options that would meet your assessed needs equally well, the local authority can choose what it believes are the most cost-effective options.
Reviews of your care plan
Your care plan should be reviewed by social services within the first three months, and then at least annually.
The review looks at whether the outcomes identified in the care plan are being met. It should also review these goals to make sure they’re still appropriate (and for instance, that your care and support needs haven’t changed), and check that any risk assessments are up to date.
If, after the review, it is clear that things have changed that affect the detail within the care plan, then the local authority will conduct a revision of the plan. This may also involve a needs assessment and financial assessment.
If it’s decided that you no longer qualify for local authority support, you should receive written reasons for this, with information about other help available, including funding your own care.
Support and other therapies
There are also a number of therapies and practical measures that can help make everyday living easier for someone with dementia.
These include:
- occupational therapy to identify problem areas in everyday life, such as getting dressed, and help work out practical solutions
- speech and language therapy to help improve any communication problems
- physiotherapy to help with movement difficulties
- psychological therapies, such as cognitive stimulation (activities and exercises designed to improve memory, problem-solving skills and language ability)
- relaxation techniques, such as massage and music or dance therapy
- social interaction, leisure activities and other dementia activities, such as memory cafés (drop-in sessions for people with memory problems and their carers to get support and advice)
- home modifications, such as removing loose carpets and potential trip hazards, ensuring the home is well lit, and adding grab bars and handrails
It may also be helpful to get in touch with a support group, such as the Alzheimer’s Society or Dementia Organization.
End of life and legal issues
If you’ve been diagnosed with dementia, you might want to make arrangements for your care that take into account the decline in your mental abilities.
This may include ensuring that your wishes are upheld if you’re not able to make decisions for yourself.
You may want to consider:
- drawing up an advance decision – this makes your treatment preferences known in case you’re unable to do this in the future
- having a preferred place of care plan – this outlines where you would like to receive treatment
- giving a relative lasting power of attorney – this is the power to make decisions about you if you’re unable to.
Looking after someone with dementia
If you have dementia, or you are looking after someone who has dementia, you are likely to face many practical issues in your daily life.
The symptoms of dementia will usually get gradually worse. How quickly this occurs will depend on the general health of the person with dementia and on the type of dementia they have.
Over time, people with dementia will need help to cope at home, and they may even need residential care in a nursing home eventually. It is natural to feel worried about the future, but you are not alone – whether you have dementia or you care for someone with the condition. The National Institutes of Health, National Institute on Aging social services and voluntary organisations can all provide advice and support to help you and your family.
People with dementia can feel vulnerable as their condition progresses and they increasingly rely on other people to do things for them. It is important that people who have dementia feel reassured and supported, while retaining some level of independence.
Although some symptoms are common to many people with dementia, each person’s experience of the disease and how they cope with it will be different.
Staying independent with dementia
Being diagnosed with dementia will have a big impact on your life. You and your family may worry about how long you can care for yourself, particularly if you live alone. People with dementia can remain independent for some time, but will need support from family and friends.
Living at home when you have dementia
In the early stages of dementia, many people are able to look after their homes in the same way as before their diagnosis. However, as the illness gets worse, it is likely that someone who has dementia will find it difficult to look after their home and they may need help with daily activities, such as housework and shopping. The home of a person with dementia may also need to be adapted to enable them to stay safe, mobile and independent.
Living alone with dementia
It’s good to stay independent for as long as possible. Many people with dementia continue to live successfully on their own for some time. However, be aware that, as your condition progresses, you will need extra support to help you cope, and it’s better to get this in place early.
Talk to family, friends and health professionals about how they can help you to stay independent. They can advise on how to cope with practical tasks, such as shopping. Find out about the local support services that can help you manage in your home – for example, by doing laundry and supervising meals.
Working when you have dementia
Coping at work can be worrying for people with dementia. You should speak to your employer as soon as you feel ready. You can also get advice from the disability employment adviser at your local job centre, your trade union or your local Citizens Advice Bureau. If you decide to leave work, seek advice about your pensions and benefits.
You could continue to work or return to work by asking your employer if you can change your workload. Your local disability employment adviser can help and advise you.
Driving
Some people with dementia prefer to give up driving because they find it stressful, but others continue driving for some time. To continue driving, you must inform the Driver and Vehicle Licensing Authority (DMV) that you have dementia. The DMV will ask for medical reports and possibly a special driving assessment to decide whether you can continue driving.
People with dementia must give up driving when their symptoms become bad enough to make them unsafe on the road. This is to protect themselves, their passengers and other road users.
Assistive technology for people with dementia
Assistive technology is available for people with dementia or other conditions that affect memory. AT Dementia is an organization that provides access to technology aimed specifically at people with dementia, including:
- daily living aids – special utensils to help people eat and drink
- stand-alone devices – aids that can be used without being linked to a monitoring centre or carer
- telecare – sensors or detectors that automatically send a signal to a carer or monitoring centre by telephone.
Helping someone with dementia with everyday tasks
When a person with dementia finds that their mental abilities are declining, they’re likely to feel anxious, stressed and scared. They may be aware of their increasing clumsiness and inability to remember things, and this can be very frustrating and upsetting for them.
If you are looking after someone with dementia, you can help them feel more secure by creating a regular daily routine in a relaxed environment, where they’re encouraged and not criticised.
Involving the person you look after in everyday tasks may make them feel useful and improve their sense of self-worth. They could help with the shopping, laying the table or sweeping leaves in the garden, for example.
As the illness progresses, these tasks may become harder for them to manage independently, and you may need to give them more support.
How you can help
The main way you can help someone with dementia is by offering support sensitively and try not to be critical of what they do. It can be very important for the person with dementia to feel that they’re still useful.
In the early stages, memory aids can be used around the home to help the person remember where things are.
For example, you could put pictures on cupboard doors of what’s inside, such as cups and saucers. This may help to trigger their memory and enable them to retain their independence a little longer.
Keeping up hobbies and interests when someone has dementia
Many people with dementia will still enjoy their hobbies or interests. For example, if they like cooking, they may be able to help make a meal. Going for a walk or gardening is a simple way to get some exercise and a sense of achievement. Or they may prefer listening to music or playing a board game. Caring for a pet cat or dog can bring a lot of pleasure to some people.
If the person you care for was very sociable and outgoing, or if they have a large family, they may really enjoy visits from one or two family members or friends. However, they may struggle to keep up with conversations if they have a lot of visitors at the same time.
Maintaining good health and nutrition in someone with dementia
It’s important that the person you care for has a healthy, balanced diet and gets some exercise. The longer they stay fit and healthy, the better their quality of life will be. If you want some easy exercises, try these sitting exercises.
If the person you care for doesn’t eat enough or eats unhealthy food, they can become susceptible to other illnesses. People with dementia can become more confused if they get ill.
Common food-related problems for people with dementia include:
- not recognising foods
- forgetting what food they like
- refusing or spitting out food
- resisting being fed
- asking for strange food combinations
This behaviour is usually due to confusion, or irritation in the mouth caused by dental problems, rather than wanting to be awkward. If you’re concerned about the person’s eating behaviour, speak to your family doctor.
How you can help
Involve the person you care for. For example, if they cannot feed themselves, you could put the cutlery in their hand and help guide it to their mouth. You could also involve them in preparing food, if they are able to.
Try to stay calm. If you feel stressed at mealtimes, the person you care for will probably be stressed too. Make sure you have plenty of time for meals, so you can deal with any problems that arise.
Try to accommodate behaviour changes. It’s likely that the person you care for will change their eating patterns and habits over time. Being aware of this and trying to be flexible will make mealtimes less stressful for both of you.
If you think the person you care for may have health or dental problems, get help from your doctor or dentist. You could also contact a local carers’ group to speak to other people who may have experienced similar difficulties.
If the person with dementia smokes, replace matches with disposable lighters to lower the risk of them accidentally causing a fire.
If the person you care for drinks alcohol, check if this is recommended alongside any medication they make take. If in doubt, ask your doctor.
Dealing with incontinence in someone with dementia
Incontinence can be difficult to deal with and can be very upsetting for the person you care for. It’s common for people with dementia to experience incontinence. This can be due to urinary tract infections, constipation causing added pressure on the bladder, or medication.
A person with dementia may also simply forget to go to the toilet, or may forget where the toilet is. They may also have lost the ability to tell when they need the toilet.
How you can help
It’s important to be understanding, retain a sense of humour and remember that it’s not their fault. You may also want to try the following:
- Put a sign on the toilet door, such as a photo of the toilet.
- Keep the toilet door open and make sure that the person you care for can access it easily.
- Make sure they can remove their clothes – some people with dementia can struggle with buttons and zips.
- Look out for signs that they may need to go to the toilet, such as fidgeting and standing up and down.
- Get adaptations to the toilet if necessary – you may be able to get these through a care and support needs assessment.
If you’re still having problems with incontinence, ask your doctor to refer you to a continence advisor, who can advise on things like waterproof bedding or incontinence pads.
Helping someone with dementia with their personal hygiene
People with dementia can become anxious about certain aspects of personal hygiene and may need help with washing. For example, they may be scared of falling when getting out of the bath, or they may become disorientated in the shower.
The person you care for may not want to be left alone or they may resist washing, because they find the lack of privacy undignified and embarrassing. Try to do what’s best for them.
Poor hygiene can cause skin complaints and infections, and be a source of discomfort and low self-esteem.
How to maintain daily hygiene
To maintain daily personal hygiene, you should make sure:
- your hands are washed after you’ve used the toilet
- your genitals and anal area are washed every day
- your face is washed daily
- you’re fully bathed or showered at least twice a week
- your teeth are brushed twice a day
It is also important that you have regular dental checks. Find out more about dental treatment for people with special needs.
Help with washing and bathing
For most people, washing is a very private activity. If you are helping someone else wash or bathe, be sensitive and try to maintain their dignity. You may feel awkward and embarrassed, especially at first.
To make bathing and washing as pleasant and comfortable as possible, you might consider:
- using pleasant-smelling shampoo, bubble bath or soap
- playing music the person you care for likes and is familiar with
- if the person you’re washing is confused, explaining what’s happening as you go along
- being sensitive to their mood
Overhead showers can be frightening to some people. If you have no bath or there is a good reason for using a shower rather than a bath, use a handheld shower.
Ask the person how they would prefer to be helped and allow them as much independence as you think is safe. If they had a routine before you began caring for them, find out what it was and stick to it as much as you can. Find out which shampoo, shower gel or soap they prefer to make the experience more familiar to them.
Many people become self-conscious when undressed in front of others. Be sensitive to the situation and approach it in the way you think is most appropriate. The person you care for may feel isolated if you leave them alone. How you handle this depends on your relationship with them. Have clothes and towels with you so you don’t have to leave them alone in the bathroom if they don’t want you to.
Safety when washing or bathing
If you find it difficult to help someone move around, your local authority has a responsibility to consider your needs as a carer and the needs of the person you care for. Contact the local authority and ask for an assessment for the person you look after, as well as a carer’s assessment to help you. For advice and guidance on moving and handling, ask for an occupational therapy assessment.
If you or the person you’re looking after has limited mobility or problems balancing, make sure:
- the floor is not slippery (dry it if necessary)
- the room is a comfortable temperature
- the water is comfortably warm – older people particularly feel the cold, so bear this in mind when adjusting the temperature
- the locks are removed from the door – you or the person you care for may want privacy, but other people may need access in an emergency
Before attempting to move someone, ask yourself:
- Do they need help to move?
- Do they require help or supervision?
- Have you told them you’re moving them?
- How heavy are they?
- Are you healthy and strong enough to move them?
- Is there anyone who could help you?
- How long will it take?
- Is there enough space around you?
- Are there any obstacles in the way?
Are you wearing suitable clothing and shoes – for example, if you are on a slippery or damp surface?
If you have assessed the situation and have decided to move the person, make sure you:
- never lift above shoulder height
- make sure your feet are stable
- take a firm hold
- keep any weight close to your body
- keep your back straight and bend your knees
- lift as smoothly as possible.
Buying equipment
You may decide you need specialist moving equipment. Before you buy any equipment, get advice from a healthcare professional such as an occupational therapist or a social worker.
Try all equipment before you buy it. If you’re considering buying an expensive item, ask to use the equipment for a trial period in the home of the person you’re looking after.
Going to the toilet
Going to the toilet (toileting) is an important part of personal hygiene, regardless of whether you or the person you’re looking after is able to control their bladder and bowels (continent) or not.
Incontinence can create feelings of shame or embarrassment for both the carer and the person being cared for. Sometimes people may be in denial about their incontinence or refuse to accept help. Reassure them it’s not their fault and approach the issue in a calm, reassuring way.
Giving a bed bath
If the person you care for cannot move or has extremely limited mobility, you may need to give them a bed bath. You will need to be extra careful when moving or handling them. Specialist disposable baths are available for people who need a bath where you are put fully in the water.
Continence services
As many as one in three people have difficulty controlling their flow of urine. And while you may not have a problem controlling your bowel or bladder, a mobility problem can make successfully visiting the toilet difficult.
Continence problems can cause physical problems such as skin irritation and infection, as well as embarrassment and loss of confidence.
A continence adviser may be able to provide many small items and other equipment that can help with continence, including:
- plastic or PVC covers to protect beds
- disposable or washable continence pads
- waterproof pants
Your social services department should be able to provide small aids and adaptations for the home, including:
- hand rails
- commodes
- raised toilet seats.
Helping someone with dementia sleep well
People with dementia often experience disturbed sleep. They may wake up during the night or be restless. These problems may get worse as the illness progresses. People with dementia may also have painful illnesses such as arthritis that cause, or contribute to, sleep problems.
Some medication can cause sleepiness during the day and interfere with sleep at night. Sleeping pills can be used with care in people with dementia.
Carers’ breaks and respite care
Your carer’s assessment may identify that you need a break from caring from time to time. Equally, the person you care for may also want to have a break without you.
Replacement care is designed to replace the care that you, as a carer, would normally be giving the person you care for. It may be needed so you can look after your own health and wellbeing, and to take a break from caring. For example, it may be that regular replacement overnight care is needed so you can catch up on your own sleep.
In certain situations, respite (temporary) care may be provided by your local authority after your carer’s assessment or after the person you care for has had an assessment. You can use the directory of local carers’ services to find your nearest local carers’ center or respite service. Your local authority or local carers’ center can give you information about local support.
You might want to consider:
- homecare services – these can either be day services that give you the chance to do an activity inside or outside the home, or night services that can help you get a proper night’s sleep, or helpers coming to the home of the person you’re caring for. Different types of help can be organized, including sitting with the person you care for and keeping them company, preparing meals, and helping them to get up, washed and dressed. The care workers who come to your home can also provide social activities, such as taking the person you care for to the cinema, pub or shopping.
- residential or nursing care – this is where the person you’re looking after goes for a short stay in a residential or nursing home. If you can, visit the care or nursing home beforehand so you can see what it’s like.
- day care – this is where the person you’re looking after goes to a day center or takes part in activities away from home.
If the replacement care provided is essentially a homecare service for the person needing care and allows you to take a break, it should be considered a service provided to the cared-for person and should therefore be charged to them, not you as the carer.
References- National Health Service of UK. Symptoms of dementia. http://www.nhs.uk/Conditions/dementia-guide/Pages/symptoms-of-dementia.aspx
- U.S. National Library of Medicine. MedlinePlus. Dementia. https://medlineplus.gov/dementia.html
- National Institutes of Health. National Institute on Aging. What Is Dementia ? https://www.nia.nih.gov/health/what-dementia
- National Institutes of Health. National Institute on Aging. What Is Dementia ? https://www.nia.nih.gov/health/what-dementia
- National Institutes of Health. National Institute on Aging. Do Memory Problems Always Mean Alzheimer’s Disease ? https://www.nia.nih.gov/health/do-memory-problems-always-mean-alzheimers-disease
- U.S. National Library of Medicine. MedlinePlus. Mild Cognitive Impairment. https://medlineplus.gov/mildcognitiveimpairment.html
- Corbo, I., & Casagrande, M. (2022). Higher-Level Executive Functions in Healthy Elderly and Mild Cognitive Impairment: A Systematic Review. Journal of clinical medicine, 11(5), 1204. https://doi.org/10.3390/jcm11051204
- Petersen R. C. (2016). Mild Cognitive Impairment. Continuum (Minneapolis, Minn.), 22(2 Dementia), 404–418. https://doi.org/10.1212/CON.0000000000000313
- National Institutes of Health. National Institute on Aging. What Is Mild Cognitive Impairment ? https://www.nia.nih.gov/health/what-mild-cognitive-impairment
- Simon SS, Yokomizo JE, Bottino CM. Cognitive intervention in amnestic Mild Cognitive Impairment: a systematic review. Neurosci Biobehav Rev. 2012;36:1163–78. https://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0048106/
- Gates NJ, Sachdev PS, Fiatarone Singh MA, Valenzuela M. Cognitive and memory training in adults at risk of dementia: a systematic review. BMC Geriatr. 2011;25;11:55. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3191477/
- Miller DI, Taler V, Davidson PS, Messier C. Measuring the impact of exercise on cognitive aging: methodological issues. Neurobiol Aging. 2012;33:622.e29–43. https://www.ncbi.nlm.nih.gov/pubmed/21514694
- McDonnell MN, Smith AE, Mackintosh SF. Aerobic exercise to improve cognitive function in adults with neurological disorders: a systematic review. Arch Phys Med Rehabil. 2011;92:1044–52. https://www.ncbi.nlm.nih.gov/pubmed/21704783
- Lautenschlager NT, Cox K, Kurz AF. Physical activity and mild cognitive impairment and Alzheimer’s disease. Curr Neurol Neurosci Rep. 2010;10:352–58. https://www.ncbi.nlm.nih.gov/pubmed/20556547
- Alzheimer’s Australia. What is dementia ? https://www.fightdementia.org.au/about-dementia/what-is-dementia
- National Institutes of Health. National Institute on Aging. Types of Dementia. https://www.nia.nih.gov/health/what-dementia
- National Institutes of Health. National Institute on Aging. Diagnosing Dementia. https://www.nia.nih.gov/health/diagnosing-dementia
- National Health Services of U.K. Tests for diagnosing dementia. http://www.nhs.uk/Conditions/dementia-guide/Pages/dementia-diagnosis-tests.aspx
- Oxford Medical Education. Mini-mental state examination (MMSE). http://www.oxfordmedicaleducation.com/geriatrics/mini-mental-state-examination-mmse/
- National Institutes of Health. National Institute on Aging. What Is Alzheimer’s Disease ? https://www.nia.nih.gov/health/what-alzheimers-disease
- U.S. National Library of Medicine. MedlinePlus. Alzheimer’s Disease. https://medlineplus.gov/alzheimersdisease.html
- Purves D, Augustine GJ, Fitzpatrick D, et al., editors. Neuroscience. 2nd edition. Sunderland (MA): Sinauer Associates; 2001. Neuroglial Cells. Available from: https://www.ncbi.nlm.nih.gov/books/NBK10869/
- National Institutes of Health. National Institute on Aging. What Happens to the Brain in Alzheimer’s Disease ? https://www.nia.nih.gov/health/what-happens-brain-alzheimers-disease
- National Institutes of Health. National Institute on Aging. What Causes Alzheimer’s Disease ? https://www.nia.nih.gov/health/what-causes-alzheimers-disease
- National Institutes of Health. National Institute on Aging. How Is Alzheimer’s Disease Diagnosed ? https://www.nia.nih.gov/health/how-alzheimers-disease-diagnosed
- National Institutes of Health. National Institute on Aging. How Is Alzheimer’s Disease Treated ? https://www.nia.nih.gov/health/how-alzheimers-disease-treated
- National Institutes of Health. National Institute on Aging. Noticing Memory Problems ? What to Do Next. https://www.nia.nih.gov/health/noticing-memory-problems-what-do-next
- National Institutes of Health. National Institute on Aging. Cognitive Health and Older Adults. https://www.nia.nih.gov/health/cognitive-health-and-older-adults
- Erickson KI, Voss MW, Prakash RS, et al. Exercise training increases size of hippocampus and improves memory. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(7):3017-3022. doi:10.1073/pnas.1015950108. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3041121/
- Taylor, J. P., McKeith, I. G., Burn, D. J., Boeve, B. F., Weintraub, D., Bamford, C., Allan, L. M., Thomas, A. J., & O’Brien, J. T. (2020). New evidence on the management of Lewy body dementia. The Lancet. Neurology, 19(2), 157–169. https://doi.org/10.1016/S1474-4422(19)30153-X
- Lewy body dementia. https://www.mayoclinic.org/diseases-conditions/lewy-body-dementia/symptoms-causes/syc-20352025
- Diagnosing and Managing Lewy Body Dementia. A Comprehensive Guide for Healthcare Professionals. https://www.lbda.org/wp-content/uploads/2011/02/3737-lbda-physicians-book-22dec17.pdf
- McKeith, I. G., Boeve, B. F., Dickson, D. W., Halliday, G., Taylor, J. P., Weintraub, D., Aarsland, D., Galvin, J., Attems, J., Ballard, C. G., Bayston, A., Beach, T. G., Blanc, F., Bohnen, N., Bonanni, L., Bras, J., Brundin, P., Burn, D., Chen-Plotkin, A., Duda, J. E., … Kosaka, K. (2017). Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology, 89(1), 88–100. https://doi.org/10.1212/WNL.0000000000004058
- Barker WW, Luis CA, Kashuba A, Luis M, Harwood DG, Loewenstein D, Waters C, Jimison P, Shepherd E, Sevush S, Graff-Radford N, Newland D, Todd M, Miller B, Gold M, Heilman K, Doty L, Goodman I, Robinson B, Pearl G, Dickson D, Duara R. Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis Assoc Disord. 2002 Oct-Dec;16(4):203-12. doi: 10.1097/00002093-200210000-00001
- Haider A, Spurling BC, Sánchez-Manso JC. Lewy Body Dementia. [Updated 2021 Jul 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482441
- Osterberg, V. R., Spinelli, K. J., Weston, L. J., Luk, K. C., Woltjer, R. L., & Unni, V. K. (2015). Progressive aggregation of alpha-synuclein and selective degeneration of lewy inclusion-bearing neurons in a mouse model of parkinsonism. Cell reports, 10(8), 1252–1260. https://doi.org/10.1016/j.celrep.2015.01.060
- Savica, R., Grossardt, B. R., Bower, J. H., Boeve, B. F., Ahlskog, J. E., & Rocca, W. A. (2013). Incidence of dementia with Lewy bodies and Parkinson disease dementia. JAMA neurology, 70(11), 1396–1402. https://doi.org/10.1001/jamaneurol.2013.3579
- Kim CY, Alcalay RN. Genetic Forms of Parkinson’s Disease. Semin Neurol. 2017 Apr;37(2):135-146. doi: 10.1055/s-0037-1601567
- Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Gomez Tortosa E, del Ser T, Muñoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004 Feb;55(2):164-73. doi: 10.1002/ana.10795
- Bras, J., Guerreiro, R., Darwent, L., Parkkinen, L., Ansorge, O., Escott-Price, V., Hernandez, D. G., Nalls, M. A., Clark, L. N., Honig, L. S., Marder, K., Van Der Flier, W. M., Lemstra, A., Scheltens, P., Rogaeva, E., St George-Hyslop, P., Londos, E., Zetterberg, H., Ortega-Cubero, S., Pastor, P., … Hardy, J. (2014). Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Human molecular genetics, 23(23), 6139–6146. https://doi.org/10.1093/hmg/ddu334
- Mata, Ignacio F et al. “APOE, MAPT, and SNCA genes and cognitive performance in Parkinson disease.” JAMA neurology vol. 71,11 (2014): 1405-12. https://doi.org/10.1001/jamaneurol.2014.1455
- Nalls, M. A., Duran, R., Lopez, G., Kurzawa-Akanbi, M., McKeith, I. G., Chinnery, P. F., Morris, C. M., Theuns, J., Crosiers, D., Cras, P., Engelborghs, S., De Deyn, P. P., Van Broeckhoven, C., Mann, D. M., Snowden, J., Pickering-Brown, S., Halliwell, N., Davidson, Y., Gibbons, L., Harris, J., … Sidransky, E. (2013). A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA neurology, 70(6), 727–735. https://doi.org/10.1001/jamaneurol.2013.1925
- Davis, M. Y., Johnson, C. O., Leverenz, J. B., Weintraub, D., Trojanowski, J. Q., Chen-Plotkin, A., Van Deerlin, V. M., Quinn, J. F., Chung, K. A., Peterson-Hiller, A. L., Rosenthal, L. S., Dawson, T. M., Albert, M. S., Goldman, J. G., Stebbins, G. T., Bernard, B., Wszolek, Z. K., Ross, O. A., Dickson, D. W., Eidelberg, D., … Zabetian, C. P. (2016). Association of GBA Mutations and the E326K Polymorphism With Motor and Cognitive Progression in Parkinson Disease. JAMA neurology, 73(10), 1217–1224. https://doi.org/10.1001/jamaneurol.2016.2245
- Boeve BF, Silber MH, Saper CB, Ferman TJ, Dickson DW, Parisi JE, Benarroch EE, Ahlskog JE, Smith GE, Caselli RC, Tippman-Peikert M, Olson EJ, Lin SC, Young T, Wszolek Z, Schenck CH, Mahowald MW, Castillo PR, Del Tredici K, Braak H. Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain. 2007 Nov;130(Pt 11):2770-88. doi: 10.1093/brain/awm056
- Jellinger KA, Wenning GK, Seppi K. Predictors of survival in dementia with lewy bodies and Parkinson dementia. Neurodegener Dis. 2007;4(6):428-30. doi: 10.1159/000107703
- Litvan, I., Goldman, J. G., Tröster, A. I., Schmand, B. A., Weintraub, D., Petersen, R. C., Mollenhauer, B., Adler, C. H., Marder, K., Williams-Gray, C. H., Aarsland, D., Kulisevsky, J., Rodriguez-Oroz, M. C., Burn, D. J., Barker, R. A., & Emre, M. (2012). Diagnostic criteria for mild cognitive impairment in Parkinson’s disease: Movement Disorder Society Task Force guidelines. Movement disorders : official journal of the Movement Disorder Society, 27(3), 349–356. https://doi.org/10.1002/mds.24893
- Akhter, F., Persaud, A., Zaokari, Y., Zhao, Z., & Zhu, D. (2021). Vascular Dementia and Underlying Sex Differences. Frontiers in aging neuroscience, 13, 720715. https://doi.org/10.3389/fnagi.2021.720715
- Vijayan, M., & Reddy, P. H. (2016). Stroke, Vascular Dementia, and Alzheimer’s Disease: Molecular Links. Journal of Alzheimer’s disease : JAD, 54(2), 427–443. https://doi.org/10.3233/JAD-160527
- Song, J., Lee, W. T., Park, K. A., & Lee, J. E. (2014). Association between risk factors for vascular dementia and adiponectin. BioMed research international, 2014, 261672. https://doi.org/10.1155/2014/261672
- What Is Vascular Dementia? https://www.alzheimers.gov/alzheimers-dementias/vascular-dementia
- Multi-infarct dementia. A cause of mental deterioration in the elderly. Hachinski VC, Lassen NA, Marshall J. Lancet. 1974 Jul 27; 2(7874):207-10. https://www.ncbi.nlm.nih.gov/pubmed/4135618/
- U.S. National Library of Medicine. MedlinePlus. Multi-infarct dementia. https://wwwqa.nlm.nih.gov/medlineplus/275/ency/article/000746.htm
- U.S. National Library of Medicine. Genetics Home Reference. cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. https://ghr.nlm.nih.gov/condition/cerebral-autosomal-dominant-arteriopathy-with-subcortical-infarcts-and-leukoencephalopathy
- The National Institute of Neurological Disorders and Stroke. Binswanger’s disease. https://www.ninds.nih.gov/Disorders/All-Disorders/Binswangers-Disease-Information-Page
- Strub RL. Vascular Dementia. The Ochsner Journal. 2003;5(1):40-43. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3314418/
- Sattel H., Forstl H., Biedert S. Senile dementia of Alzheimer type and multi-infarct dementia investigated by transcranial Doppler sonography. Dementia. 1996;7:41–46. https://www.ncbi.nlm.nih.gov/pubmed/8788081
- Mulkey M. Understanding Frontotemporal Disease Progression and Management Strategies. Nurs Clin North Am. 2019 Sep;54(3):437-448. doi: 10.1016/j.cnur.2019.04.011
- Bang, J., Spina, S., & Miller, B. L. (2015). Frontotemporal dementia. Lancet (London, England), 386(10004), 1672–1682. https://doi.org/10.1016/S0140-6736(15)00461-4
- Vieira, R. T., Caixeta, L., Machado, S., Silva, A. C., Nardi, A. E., Arias-Carrión, O., & Carta, M. G. (2013). Epidemiology of early-onset dementia: a review of the literature. Clinical practice and epidemiology in mental health : CP & EMH, 9, 88–95. https://doi.org/10.2174/1745017901309010088
- Khan I, De Jesus O. Frontotemporal Lobe Dementia. [Updated 2021 Aug 30]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK559286
- Pick A. Über die Beziehungen der senilen Hirnatrophie zur Aphasie. Prager Med Wochenschr. 1892;17:165–67.
- Alzheimer A. Über eigenartige Krankheitsfälle der späteren Alters. Z Gesamte Neurol Psychiatr. 1911;4:356–85.
- Mesulam MM. Primary progressive aphasia. Ann Neurol. 2001 Apr;49(4):425-32.
- Clinical and neuropathological criteria for frontotemporal dementia. The Lund and Manchester Groups. (1994). Journal of neurology, neurosurgery, and psychiatry, 57(4), 416–418. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1072868/pdf/jnnpsyc00034-0016.pdf
- Olney, N. T., Spina, S., & Miller, B. L. (2017). Frontotemporal Dementia. Neurologic clinics, 35(2), 339–374. https://doi.org/10.1016/j.ncl.2017.01.008
- Frontotemporal dementia. https://rarediseases.org/gard-rare-disease/frontotemporal-dementia
- Rohrer, J D et al. “The heritability and genetics of frontotemporal lobar degeneration.” Neurology vol. 73,18 (2009): 1451-6. https://doi.org/10.1212/WNL.0b013e3181bf997a
- Shpilyukova YA, Fedotova EY, Illarioshkin SN. [Genetic Diversity in Frontotemporal Dementia]. Mol Biol (Mosk). 2020 Jan-Feb;54(1):17-28. Russian. doi: 10.31857/S0026898420010139
- Borroni, B., Benussi, A., Premi, E., Alberici, A., Marcello, E., Gardoni, F., Di Luca, M., & Padovani, A. (2018). Biological, Neuroimaging, and Neurophysiological Markers in Frontotemporal Dementia: Three Faces of the Same Coin. Journal of Alzheimer’s disease : JAD, 62(3), 1113–1123. https://doi.org/10.3233/JAD-170584
- Bretag-Norris R, Gallur L, Flynn P. Heterogeneity in the psychiatric presentation of behavioural variant frontotemporal dementia (bvFTD). Australas Psychiatry. 2019 Oct;27(5):491-495. doi: 10.1177/1039856219860031
- Coyle-Gilchrist, I. T., Dick, K. M., Patterson, K., Vázquez Rodríquez, P., Wehmann, E., Wilcox, A., Lansdall, C. J., Dawson, K. E., Wiggins, J., Mead, S., Brayne, C., & Rowe, J. B. (2016). Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology, 86(18), 1736–1743. https://doi.org/10.1212/WNL.0000000000002638
- Knopman, D. S., & Roberts, R. O. (2011). Estimating the number of persons with frontotemporal lobar degeneration in the US population. Journal of molecular neuroscience : MN, 45(3), 330–335. https://doi.org/10.1007/s12031-011-9538-y
- The Association for Frontotemporal Degeneration. Genetics of FTD. https://www.theaftd.org/wp-content/uploads/2009/03/Final-FTD-Genetics-Brochure-with-Cover-8.2.2012.pdf
- McCarter SJ, St Louis EK, Boeve BF. Sleep Disturbances in Frontotemporal Dementia. Curr Neurol Neurosci Rep. 2016 Sep;16(9):85. doi: 10.1007/s11910-016-0680-3
- Burrell JR, Hodges JR. Falls in frontotemporal dementia and related syndromes. Handb Clin Neurol. 2018;159:195-203. doi: 10.1016/B978-0-444-63916-5.00012-4
- Aiello M, Silani V, Rumiati RI. You stole my food! Eating alterations in frontotemporal dementia. Neurocase. 2016 Aug;22(4):400-9. doi: 10.1080/13554794.2016.1197952
- Alzheimer’s Australia. HIV associated dementia. https://www.fightdementia.org.au/about-dementia/types-of-dementia/aids-related-dementia
- Creutzfeldt-Jakob Disease Fact Sheet. National Institute of Neurological Disorders and Stroke (NINDS). February 2, 2016; https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Creutzfeldt-Jakob-Disease-Fact-Sheet
- National Institutes of Health. Genetic and Rare Diseases Information Center. Creutzfeldt-Jakob disease. https://rarediseases.info.nih.gov/diseases/6956/creutzfeldt-jakob-disease
- CJD Fact Sheet. Creutzfeld-Jakob Disease Foundation. http://www.cjdfoundation.org/webfm_send/76.
- National Institutes of Neurological Disorders and Stroke. Huntington’s disease. https://www.ninds.nih.gov/Disorders/All-Disorders/Huntingtons-Disease-Information-Page




{kind=link}