
Zellweger syndrome
Zellweger syndrome, also called cerebrohepatorenal syndrome, is the most severe form of a spectrum of conditions called Zellweger spectrum disorder (ZSD) 1. Zellweger syndrome is one of a group of four related diseases called peroxisome biogenesis disorders that includes neonatal adrenoleukodystrophy, infantile Refsum disease, and rhizomelic chondroplasia punctata 2. The peroxisome biogenesis disorders are divided into two groups: Zellweger spectrum disorders and Rhizomelic Chondrodysplasia Punctua spectrum. The signs and symptoms of Zellweger syndrome typically appear during the newborn period and may include poor muscle tone (hypotonia), poor feeding, seizures, hearing loss, vision loss, distinctive facial features, and skeletal abnormalities 3. Affected children also develop life-threatening problems in other organs and tissues, such as the liver, heart, and kidneys 4. Children with Zellweger syndrome usually do not survive beyond the first year of life. Zellweger syndrome is caused by mutations in any one of at least 13 genes, termed PEX genes, required for the normal formation and function of peroxisomes; mutations in the PEX1 gene are the most common cause. It is inherited in an autosomal recessive manner 3. There is no cure for Zellweger syndrome; treatment is generally symptomatic and supportive.
Peroxisomes are cell structures that break down toxic substances and synthesize lipids (fatty acids. oils, and waxes) that are necessary for cell function. Peroxisomes are required for normal brain development and function and the formation of myelin, the whitish substance that coats nerve fibers. They are also required for normal eye, liver, kidney, and bone functions.
Zellweger spectrum disorder is comprised of three disorders that have considerable overlapping signs and symptoms and affect many parts of the body ranging from severe to mild 1. In the past before the biochemical and molecular bases of the Zellweger spectrum disorder were fully determined, Zellweger spectrum disorder includes Zellweger syndrome (the most severe form), neonatal adrenoleukodystrophy (intermediate in severity form), infantile Refsum disease (the least severe form) and more recently the very mild Zellweger syndrome disorder called Heimler syndrome 5, 6, 7. These conditions were once thought to be distinct disorders but are now considered to be part of the same condition spectrum. The original classification of Zellweger syndrome, neonatal adrenoleukodystrophy and infantile Refsum disease is less valuable now, especially since additional variant phenotypes suggestive for a disease spectrum have been identified. Instead the term “Zellweger spectrum disorder” is now used to refer to all individuals with a defect in one of the Zellweger spectrum disorder-PEX genes regardless of phenotype 1. Furthermore, some researchers prefer not to use the separate condition names but to instead refer to cases as severe, intermediate, or mild Zellweger spectrum disorder. However, for discussing prognosis and counseling patients or families the old classification (e.g., Zellweger syndrome, neonatal adrenoleukodystrophy, infantile Refsum disease, Heimler syndrome) may in some cases still be useful 1.
The incidence of Zellweger syndrome disorders is estimated to be 1 in 50,000 newborns in the United States 8. The worldwide prevalence of Zellweger syndrome disorders is estimated between 1:50,000 and 1:100,000, with reports of higher incidence of Zellweger syndrome with estimation of 1 in 12,000 in the Saguenay-Lac-St-Jean region of Quebec and much lower incidence is reported in Japan, with an estimated incidence of 1 in 500,000 births 9, 10, 11, 12.
Zellweger spectrum disorders result from dysfunctional lipid metabolism, including the over-accumulation of very long-chain fatty acids and phytanic acid, and defects of bile acids and plasmalogens–specialized lipids found in cell membranes and myelin sheaths of nerve fibers. Symptoms of these disorders include an enlarged liver; characteristic facial features such as a high forehead, underdeveloped eyebrow ridges, and wide-set eyes; and neurological abnormalities such as cognitive impairment and seizures.
Individuals with Zellweger syndrome, at the severe end of the Zellweger spectrum disorder, develop signs and symptoms of the condition during the newborn period. These infants experience weak muscle tone (hypotonia), sometimes to the point of being unable to move, and may not be able to suck or swallow, feeding problems, hearing and vision loss, and seizures. These problems are caused by the breakdown of myelin, which is the covering that protects nerves and promotes the efficient transmission of nerve impulses. The part of the brain and spinal cord that contains myelin is called white matter. Destruction of myelin (demyelination) leads to loss of white matter (leukodystrophy). Children with Zellweger syndrome also develop life-threatening problems in other organs and tissues, such as the liver, heart, and kidneys. They may have skeletal abnormalities, including a large space between the bones of the skull (fontanelles) and characteristic bone spots known as chondrodysplasia punctata that can be seen on x-ray of the patellae and the long bones. Affected individuals have distinctive facial features, including a flattened face, broad nasal bridge, and high forehead. Some babies will be born with glaucoma, retinal degeneration, and impaired hearing. Jaundice and gastrointestinal bleeding also may occur.
Children with Zellweger syndrome are significantly impaired usually having made no developmental progress and typically do not survive beyond the first year of life 1. Death is usually secondary to progressive apnea (breathing stop) or respiratory compromise from infection 1.
Children with neonatal adrenoleukodystrophy or infantile Refsum disease, which are at the less-severe end of the Zellweger spectrum disorder, have more variable features than those with Zellweger syndrome and usually do not develop signs and symptoms of the disease until late infancy or early childhood. They may have many of the features of Zellweger syndrome; however, their condition typically progresses more slowly. Children with these less-severe conditions often have weak muscle tone (hypotonia), vision problems (retinal dystrophy), sensorineural hearing loss, neurologic involvement (ataxia, polyneuropathy, and leukodystrophy), liver dysfunction, developmental delay, some degree of intellectual disability, adrenal insufficiency, and kidney oxalate stones 1. Some have osteopenia (low bone density due to a decrease in the amount of calcium and phosphorus in the bone, osteopenia can cause bones to be weak and brittle); almost all have ameleogenesis imperfecta in the secondary teeth (a disorder that affects the structure and appearance of the enamel of the teeth, causing teeth to be very small, discolored, pitted or grooved, and prone to rapid wear and breakage with early tooth decay and loss). Most people with neonatal adrenoleukodystrophy survive into childhood, and those with infantile Refsum disease may reach adulthood. In rare cases, individuals at the mildest end of the condition spectrum have developmental delay in childhood and hearing loss or vision problems beginning in adulthood and do not develop the other features of this disorder. Recently, individuals with normal intellect have been identified in the very mild end of the Zellweger syndrome disorder spectrum called Heimler syndrome 5, 6, 7.
Zellweger syndrome treatment is focused on symptomatic therapy and may include gastrostomy to provide adequate calories, hearing aids, cataract removal, glasses to correct refractive errors, supplementation of fat-soluble vitamins, and cholic acid supplementation; varices can be treated with sclerosing therapies; anti-seizure medication, early intervention services for developmental delay and intellectual disability; adrenal replacement therapy; vitamin D supplementation and consideration of bisphosphonates for osteopenia; treatment as per dentist for ameliogenesis imperfecta. Supportive treatment for kidney oxalate stones has included hydration, lithotripsy (a medical procedure that uses high frequency sound waves to break up stones in the kidney), and surgical intervention. Annual influenza and respiratory syncytial virus (RSV) vaccines should be provided 1.
Figure 1. Zellweger syndrome pictures
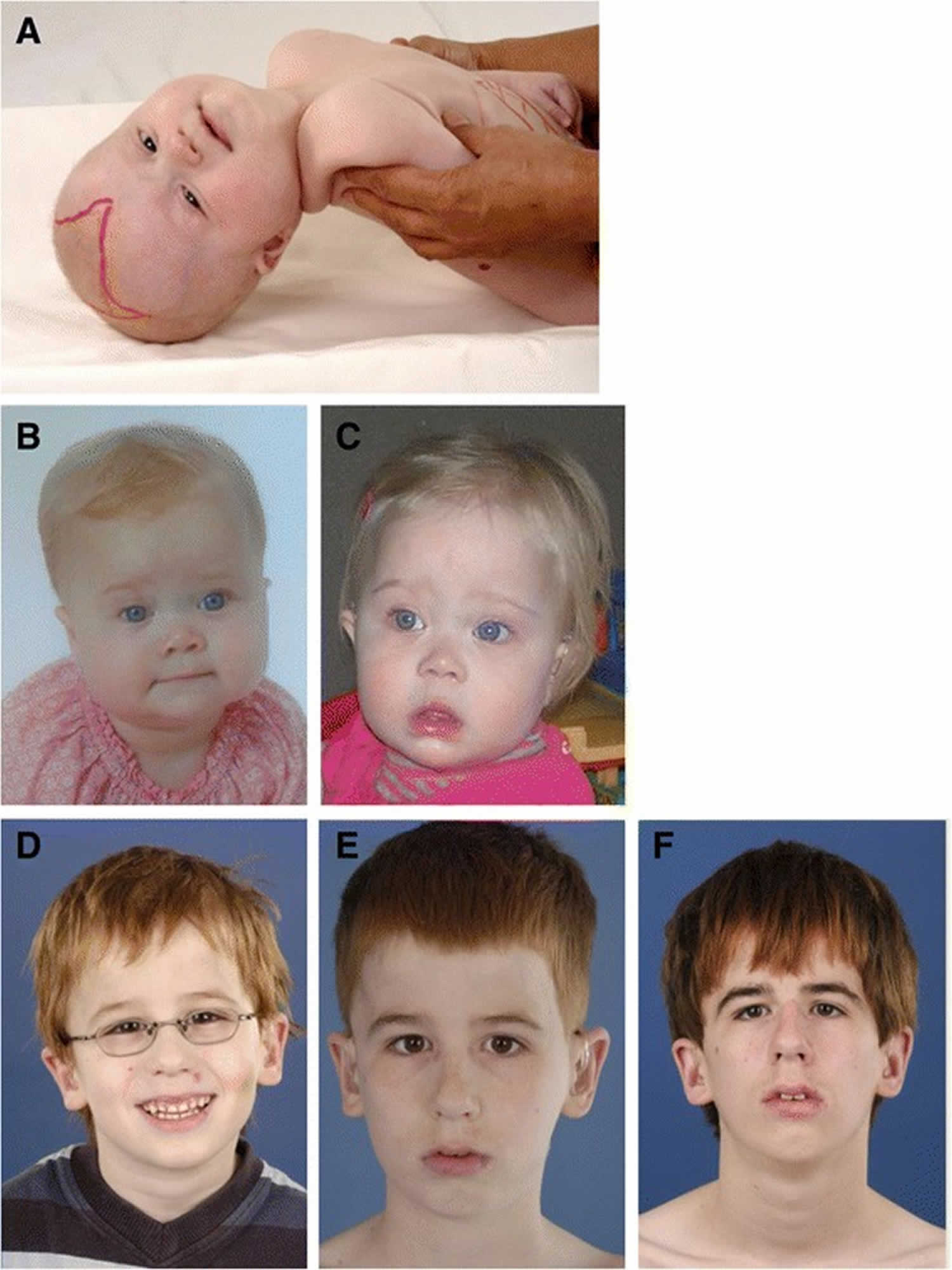
Footnotes: Craniofacial dysmorphic features in Zellweger syndrome disorder patients developing over time. (A) Photograph of a 6-month-old girl with typical craniofacial dysmorphia. Note the epicantal folds, high forehead, broad nasal bridge and hypoplastic supraorbital ridges. The anterior fontanel is drawn and enlarged. (B-C) Girl with a Zellweger syndrome disorder at the age of 9 months (B) and at the age of 1 year and two months (C). Less pronounced facial dysmorphism is present: a high forehead is seen, a broad nasal bridge, hypoplastic supraorbital ridges, anteverted nares and more subtle epicantal folds. (D-F) Photograph of a male with a Zellweger syndrome disorder at the age of 5 years (D), 10 years (E) and 15 years (F). No evident facial dysmorphic features can be recognized, although the ears seem to be slightly low-set.
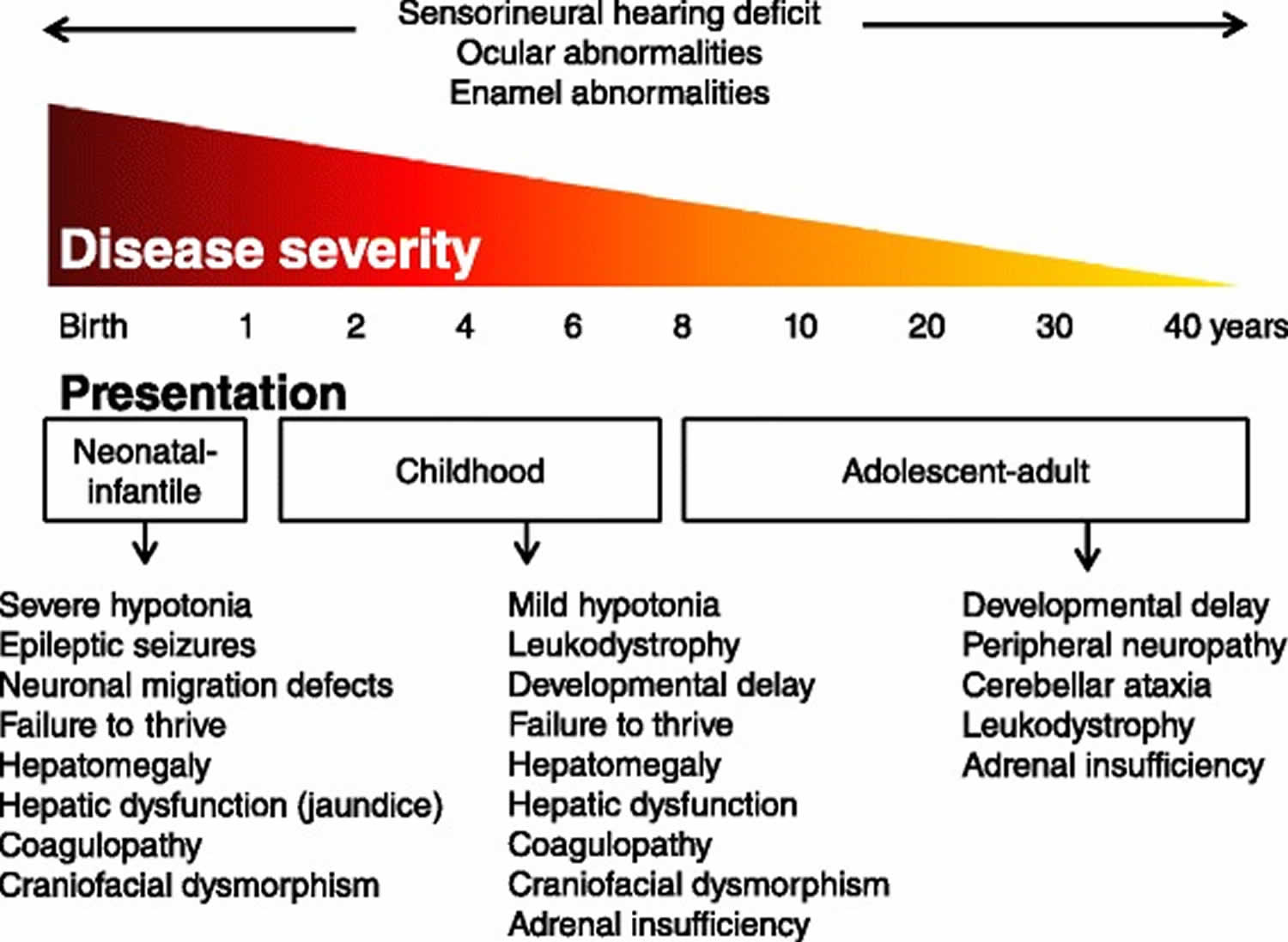
[Source 13 ]Figure 2. Zellweger syndrome disorder signs and symptoms
Zellweger syndrome cause
Mutations in at least 13 PEX genes have been found to cause Zellweger spectrum disorder (see Table 2) 13. The PEX genes provide instructions for making a group of proteins known as peroxins, which are essential for the formation and normal functioning of cell structures called peroxisomes. Peroxins assist in the formation (biogenesis) of peroxisomes by producing the membrane that separates the peroxisome from the rest of the cell and by importing enzymes into the peroxisome. Peroxisomes are sac-like organelles that contain enzymes needed to break down many different substances, including fatty acids and certain toxic compounds. Peroxisomes are also involved in many anabolic and catabolic metabolic processes, like biosynthesis of ether phospholipids and bile acids, α- and β-oxidation of fatty acids (lipids) used in digestion and in the nervous system and the detoxification of glyoxylate and reactive oxygen species. Dysfunctional peroxisomes therefore cause biochemical abnormalities in tissues, but also in readily available materials like plasma and urine 14, 15.
Mutations in the PEX genes that cause Zellweger spectrum disorder prevent peroxisomes from forming normally. Cells from Zellweger syndrome disorder patients either entirely lack functional peroxisomes, or cells can show a reduced number of functional peroxisomes or a mosaic pattern (i.e. a mixed population of cells with functional peroxisomes and cells without) 16, 17, 18. Diseases that disrupt the formation of peroxisomes, including Zellweger spectrum disorder, are called peroxisome biogenesis disorders. If the production of peroxisomes is altered, these structures cannot perform their usual functions. The signs and symptoms of Zellweger syndrome are due to the absence of functional peroxisomes within cells. Neonatal adrenoleukodystrophy and infantile Refsum disease are caused by mutations that allow some peroxisomes to form.
Mutations in the peroxisome biogenesis factor 1 (PEX1) gene are the most common cause of Zellweger spectrum disorder and are found in nearly 70 percent of affected individuals and almost 90 different mutations in PEX1 have been reported so far 19. The other genes (i.e., PEX2, PEX3, PEX5, PEX6, PEX10, PEX11, PEX12, PEX13, PEX14, PEX16, PEX19, and PEX26) associated with Zellweger spectrum disorder each account for a smaller percentage of cases of this condition.
Zellweger spectrum disorder inheritance pattern
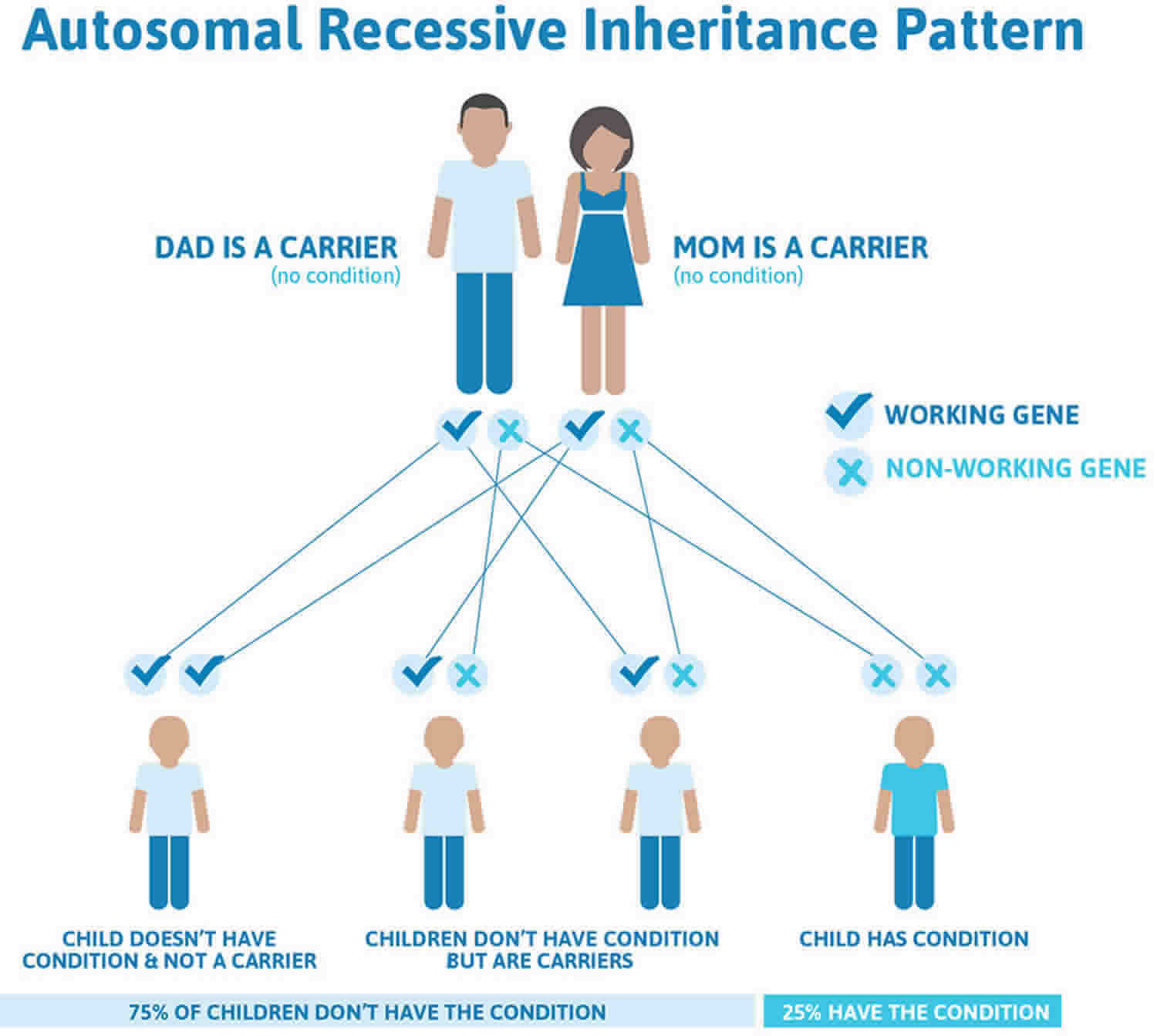
Zellweger syndrome disorder is inherited in an autosomal recessive pattern which means both copies of the gene in each cell have mutations. Recessive genetic disorders occur when an individual inherits a non-working gene from each parent. If an individual receives one working gene and one non-working gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. The risk for two carrier parents to both pass the non-working gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier, like the parents, is 50% with each pregnancy. The chance for a child to receive working genes from both parents is 25%. The risk is the same for males and females.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 3 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 3. Zellweger spectrum disorder autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Zellweger syndrome pathophysiology
Mutations in at least 13 PEX genes have been found to cause Zellweger spectrum disorder (see Table 2) 13. The PEX genes provide instructions for making a group of proteins known as peroxins, which are essential for the formation and normal functioning of cell structures called peroxisomes. Peroxins assist in the formation (biogenesis) of peroxisomes by producing the membrane that separates the peroxisome from the rest of the cell and by importing enzymes into the peroxisome. Peroxisomes are sac-like organelles that contain over 50 enzymes needed to break down many different substances, including fatty acids and certain toxic compounds. Peroxisomes are also involved in many anabolic and catabolic metabolic processes, like beta-oxidation of very-long-chain fatty acids (VLCFA), alpha oxidation of branched-chain fatty acids, catabolism of amino acids, and ethanol, biosynthesis of bile acids, steroid hormones, gluconeogenesis and plasmalogen formation which are important constituents of the cell membrane and myelin 20. Peroxisomes are also involved in the degradation of cytotoxic hydrogen peroxide and the detoxification of glyoxylate and reactive oxygen species 21. Dysfunctional peroxisomes therefore cause biochemical abnormalities in tissues, but also in readily available materials like plasma and urine 14, 15.
Zellweger syndrome is thus characterized by increased accumulation of very-long-chain fatty acids (VLCFA), increased C26, and C22 fatty acids in plasma, fibroblasts, and amniocytes 22. Reduced steroid biosynthesis and accumulation of very-long-chain fatty acids (VLCFA) in adrenal gland cells cause decreased levels of adrenocorticotropic hormone (ACTH) and some other steroidal hormones 23. Reduced degradation of cytotoxic hydrogen peroxide and abnormal accumulation of VLCFA causes neuronal membrane injury and demyelination 24.
Major abnormalities are present in the kidney (cortical cysts), liver (fibrotic), and brain (demyelination, centrosylvian polymicrogyria) – hence the name cerebrohepatorenal syndrome.
Zellweger syndrome symptoms
The signs and symptoms of Zellweger syndrome typically become apparent within the first few hours or days of life. Affected newborns often have poor muscle tone (hypotonia); seizures; feeding difficulties; liver cysts with liver dysfunction; vision loss; hearing loss; and distinctive facial characteristics including a flattened face, broad nasal bridge, high forehead, upslanting palpebral fissures, and epicanthal folds 3. Some people also have an abnormally small or large head size (microcephaly or macrocephaly, respectively); protruding tongue; neck skin folds; cataracts; glaucoma; and/or nystagmus 25. Many affected infants have skeletal abnormalities, which may include a large space between the bones of the skull and bone spots known as chondrodysplasia punctata that can be seen with an X-ray 4. The function of the central nervous system (brain and spinal cord) is typically severely affected 25. Children with Zellweger syndrome also develop life-threatening problems in other organs and tissues, such as the liver, heart, and kidneys 4.
80%-99% of people with Zellweger syndrome have these symptoms:
- Cognitive impairment
- Corneal opacity
- Death in infancy
- Depressed bridge of nose
- EEG abnormality
- Epicanthus (eye folds)
- Epiphyseal stippling (speckled calcifications in end part of bone)
- External ear malformation
- Failure to thrive (faltering weight)
- Feeding difficulties in infancy
- Flat facial shape
- Liver failure
- Enlarged liver
- High forehead
- Jaundice
- Profound global developmental delay
- Reduced tendon reflexes
- Respiratory insufficiency
- Severe muscular hypotonia (severely decreased muscle tone)
- Short stature
- Skeletal dysplasia
- Upslanted palpebral fissure (upward slanting of the opening between the eyelids)
- Very long chain fatty acid accumulation
- Wide anterior fontanel (wider-than-typical soft spot of skull)
- Wide nasal bridge
30%-79% of people with Zellweger syndrome have these symptoms:
- Abnormal chorioretinal morphology
- Cataract
- Clitoral hypertrophy (enlarged clitoris)
- Cryptorchidism (undescended testes)
- Flat occiput
- High palate
- Hydronephrosis
- Hypospadias
- Macrocephaly (increased size of skull)
- Intestinal malabsorption
- Microcephaly (abnormally small skull)
- Micrognathia (little lower jaw)
- Multicystic kidney dysplasia
- Nystagmus (involuntary, rapid, rhythmic eye movements)
- Optic atrophy
- Polymicrogyria (more grooves in brain)
- Posterior embryotoxon
- Premature birth
- Pyloric stenosis
- Seizures
- Sensorineural hearing impairment
- Underdeveloped supraorbital ridges (flattened bony protrusion above eyes)
- Visual impairment
5%-29% of people with Zellweger syndrome have these symptoms:
- Abnormality of coagulation (abnormal blood clotting)
- Abnormality of the tongue
- Brushfield spots
- Glaucoma
- Primary adrenal insufficiency
- Thickened nuchal skin fold (thickened skin folds of neck)
- Ventricular septal defect
Zellweger spectrum disorder should be suspected in children with the following signs and symptoms.
Zellweger spectrum disorder signs and symptoms in newborns:
- Hypotonia
- Poor feeding
- Distinctive facies
- Brain malformations
- Seizures
- Renal cysts
- Hepatomegaly, cholestasis, and hepatic dysfunction
- Bony stippling (chondrodysplasia punctata) of the patella(e) and other long bones
Zellweger spectrum disorder signs and symptoms in older infants and children:
- Developmental delays with or without hypotonia (Note: Intellect can be normal.)
- Failure to thrive
- Hearing loss
- Vision impairment
- Liver dysfunction
- Adrenal dysfunction
- Leukodystrophy
- Peripheral neuropathy and ataxia
The symptoms of Zellweger spectrum disorders vary greatly from one individual to another. The specific number and severity of symptoms present in an individual are highly variable and affected infants will not have all of the symptoms discussed below. The most severe form, Zellweger syndrome, is usually noticeable shortly after birth. Infants with Zellweger syndrome often have severe neurological deficits, progressive dysfunction of the liver and kidneys and usually develop life-threatening complications during the first year of life.
Children with neonatal adrenoleukodystrophy and infantile Refsum disease may not develop symptoms until later during infancy. Some of these children reach adolescence or adulthood although most have some degree of intellectual disability, hearing loss and vision problems. Some have profound loss of muscle tone (hypotonia or floppiness), but some learn to walk and to speak. Some children with these milder forms of Zellweger spectrum disorders do not have any craniofacial abnormalities or only very mild ones.
In extremely rare cases, affected individuals have gone undetected until adulthood. These individuals have had only mild symptoms such as adult-onset hearing loss or vision problems and/or mild developmental delays.
Many symptoms of Zellweger spectrum disorders are present at birth (congenital). Affected infants often exhibit prenatal growth failure in spite of a normal period of gestation and may also have a profound lack of muscle tone (hypotonia or floppiness). Affected infants may be limp, show little movement (lethargic) and poorly respond to environmental stimuli. Infants may be unable to suck and/or swallow leading to feeding difficulties and failure to gain weight and grow as expected (failure to thrive).
Infants also develop a variety of neurological complications including frequent seizures, poor or absent reflexes, intellectual disability, and delays in reaching developmental milestones such as sitting, crawling or walking (developmental delays). Affected infants have various brain abnormalities including defects caused by the abnormal migration of brain cells (neurons). Neurons are created in the center of the developing brain and must travel to other areas of the brain to function properly. In individuals with Zellweger spectrum disorders the neurons fail to migrate properly resulting in a variety of brain abnormalities (neuronal migration defects). Some affected infants also develop progressive degeneration of the nerve fibers (white matter) of the brain (leukodystrophy).
Infants may have distinctive facial features including a flattened appearance to the face, a high forehead, abnormally large “soft spots” (fontanelles) on the skull, broad bridge of the nose, a small nose with upturned nostrils (anteverted nares), an abnormally small jaw (micrognathia), a highly arched roof of the mouth (palate), a small chin, extra (redundant) folds of skin on the neck, and minor malformation of the outer part of the ears. The bony ridges of the eye socket may be abnormally shallow and the back of the head may be abnormally flat (flat occiput).
A variety of eye abnormalities may occur including eyes that are spaced widely apart (hypertelorism), clouding of the lenses of the eyes (cataracts) or the clear (transparent) outer layer of the eye (corneal opacities), degeneration of the nerve that carries visual images from the eye to the brain (optic atrophy), and rapid, involuntary eye movements (nystagmus). Many infants with Zellweger spectrum disorders develop degeneration of the retina, which is the thin layer of nerve cells that sense light and convert it into nerve signals, which are then relayed to the brain through the optic nerve. Glaucoma, a condition characterized by increased pressure within the eye causing a distinctive pattern of visual impairment, may also occur. The various eye abnormalities associated with Zellweger spectrum disorders can cause loss of vision to varying degrees. In addition to vision loss, infants with Zellweger spectrum disorders also experience hearing loss with onset during the first few months of life.
Some infants may have an abnormally large spleen (splenomegaly) and/or liver (hepatomegaly). The liver may also be scarred (fibrotic) and inflamed (cirrhosis), with progressive loss of function resulting in a variety of symptoms such as yellowing of the skin and whites of the eyes (jaundice). Additional findings include small cysts on the kidneys and gastrointestinal bleeding due to deficiency of a coagulation factor in the blood. Some children may develop episodes of exaggerated or uncontrolled bleeding (hemorrhaging) including bleeding within the skull (intracranial bleeding). Eventually, liver failure may occur.
Minor skeletal abnormalities may also be present in Zellweger spectrum disorders including clubfoot, fingers that are fixed or stuck in a bent position and cannot extend or straighten fully (camptodactyly), and chondrodysplasia punctata, a condition characterized by the formation of small, hardened spots of calcium (stippling) on the knee cap (patella) and long bones of the arms and legs.
Certain heart defects may also occur in infants with Zellweger spectrum disorders including septal defects and patent ductus arteriosus. Septal defects are “holes” in the heart, specifically holes in the thin partition (septum) that separates the chambers of the heart. Small septal defects may close on their own; larger defects may cause various symptoms including breathing irregularities and high blood pressure. Patent ductus arteriosus is a condition in which the two large arteries of the body (aorta and pulmonary artery) remain connected by a small blood vessel (ductus arteriosus) that is supposed to close after birth.
Due to the lack of muscle tone, laryngomalacia (floppy airway) and other respiratory problems may occur in infants with Zellweger spectrum disorders. Respiratory support may entail the use of a nasal cannula for oxygen to more aggressive forms of support as the disease progresses.
In some males infants with Zellweger spectrum disorders, additional symptoms may occur including the abnormal placement of the urinary opening on the underside of the penis (hypospadias) and failure of the testes to descend into the scrotum (cryptorchidism).
Neonatal-infantile signs and symptoms
Zellweger syndrome disorder patients within this group typically present in the neonatal period with hepatic dysfunction and profound hypotonia resulting in prolonged jaundice and feeding difficulties. Epileptic seizures are usually present in these patients. Characteristic dysmorphic features can usually be found, of which the facial dysmorphic signs are most evident (Figure 1A). Sensorineural deafness and ocular abnormalities like retinopathy, cataracts and glaucoma are typical but not always recognized at first presentation. Brain magnetic resonance imaging (MRI) may show neocortical dysplasia (especially perisylvian polymicrogyria), generalized decrease in white matter volume, delayed myelination, bilaterial ventricular dilatation and germinolytic cysts 26. Neonatal onset leukodystrophy is rarely described 27. Calcific stippling (chondrodysplasia punctata) may be present, especially in the knees and hips. The neonatal-infantile presentation grossly resembles what was originally described as classic Zellweger syndrome. Prognosis is poor and survival is usually not beyond the first year of life.
Childhood signs and symptoms
These patients show a more varied symptomatology than Zellweger syndrome disorder patients with a neonatal-infantile presentation. Presentation at the outpatient clinic usually involves delayed developmental milestone achievement. Ocular abnormalities comprise retinitis pigmentosa, cataract and glaucoma, often leading to early blindness and tunnel vision 28. Sensorineural deafness is almost always present and usually discovered by auditory screening programs. Hepatomegaly and hepatic dysfunction with coagulopathy, elevated transaminases and (history of) hyperbilirubinemia are common. Some patients develop epileptic seizures. Craniofacial dysmorphic features are generally less pronounced than in the neonatal-infantile group (Figure 1 B and C). Renal calcium oxalate stones and adrenal insufficiency may develop. Early-onset progressive leukodystrophy may occur, leading to loss of acquired skills and milestones in some individuals. The progressive demyelination is diffuse and affects the cerebrum, midbrain and cerebellum with involvement of the hilus of the dentate nucleus and the peridentate white matter 26. Sequential imaging in three Zellweger syndrome disorder patients showed that the earliest abnormalities related to demyelination were consistently seen in the hilus of the dentate nucleus and superior cerebellar peduncles, chronologically followed by the cerebellar white matter, brainstem tracts, parieto-occipital white matter, splenium of the corpus callosum and eventually involvement of the whole of the cerebral white matter 29. The above described rapid progressive leukodystrophy, in combination with other symptoms described here, resemble what was originally described as neonatal adrenoleukodystrophy. A small subgroup of patients develop a relatively late-onset white matter disease, but no patients with late-onset rapid progressive white matter disease after the age of five have been reported 30. Prognosis depends on what organ systems are primarily affected (i.e. liver) and the occurrence of progressive cerebral demyelination, but life expectancy is decreased and most patients die before adolescence.
Adolescent-adult signs and symptoms
Symptoms in this group are less severe, and diagnosis can be in late child- or even adulthood 31. Eye abnormalities and a sensorineural hearing deficit are the most consistent symptoms. Craniofacial dysmorphic features can be present, but may also be completely absent (Figure 1 D-F). Developmental delay is highly variable and some patients may have normal intelligence. Daily functioning ranges from completely independent to 24 hour care. It is important to emphasize that primary adrenal insufficiency is common and is probably under diagnosed 32. In addition to some degree of developmental delay, other neurological abnormalities are usually also present: signs of peripheral neuropathy, cerebellar ataxia and pyramidal tract signs. The clinical course is usually slowly progressive, although the disease may remain stable for (many) years 33. Slowly progressive, clinically silent leukoencephalopathy is common, but MRI may be normal in other cases 26.
Zellweger syndrome diagnosis
A diagnosis of a Zellweger syndrome is usually suspected when characteristic signs and symptoms are present at birth, including the distinctive facial features. Tests that measure or detect specific substances in blood or urine samples can confirm a diagnosis of Zellweger syndrome. For example, detection of elevated levels of very long chain fatty acids (VLCFA) in the blood is the most commonly used screening test. Additional tests on blood and urine samples to find other substances associated with Zellweger spectrum disorders may be performed. An ultrasound may be used to look for cysts on the kidneys or an enlarged liver. A genetic test to find a mutation in one of the genes associated with Zellweger spectrum disorders may also be used to confirm the diagnosis 3.
Genetic testing is available for Zellweger spectrum disorders; next generation sequencing methods (sequencing millions of small fragments of DNA at the same time) are being used more frequently as a confirmatory test, and may be required for peroxisome disorders that are difficult to determine by traditional biochemical methods. Additionally, genetic determination of mutations in Zellweger spectrum disorders, in contrast to biochemical tests, will also identify carriers for Zellweger spectrum disorders, which will allow reliable genetic counseling of families and may also assist with eligibility for future clinical trials.
Methods have been developed to detect elevated levels of very long chain fatty acids in newborn screening for X-linked adrenoleukodystrophy, a related peroxisomal disorder. Legislation for X-linked adrenoleukodystrophy newborn screening has passed in many states and screening has begun in New York; continued legislative efforts are expected to expand through movements initiated by patient families and advocacy organizations to lobby their state legislatures. Recently, the Department of Health and Human Services Advisory Committee for Heritable Disorders for Newborns and Children voted to propose the addition of X-linked adrenoleukodystrophy screening in the Recommend Uniform Screening Panel. Newborn screening for X-linked adrenoleukodystrophy should increase early diagnosis of Zellweger spectrum disorders and determination of accurate incidence estimates of the disease.
Certain tests (biochemical or genetic) can be performed prenatally in the first or second trimester using chorionic villus sampling or amniocentesis. Ultrasonography, a test that uses reflected sound waves to create a picture of internal organs, may be used to detect cysts on the kidneys or an enlarged liver. Preimplantation genetic diagnosis with in vitro fertilization can also be performed when the gene mutations are known.
Figure 4. Diagnostic flow-chart for Zellweger syndrome disorders
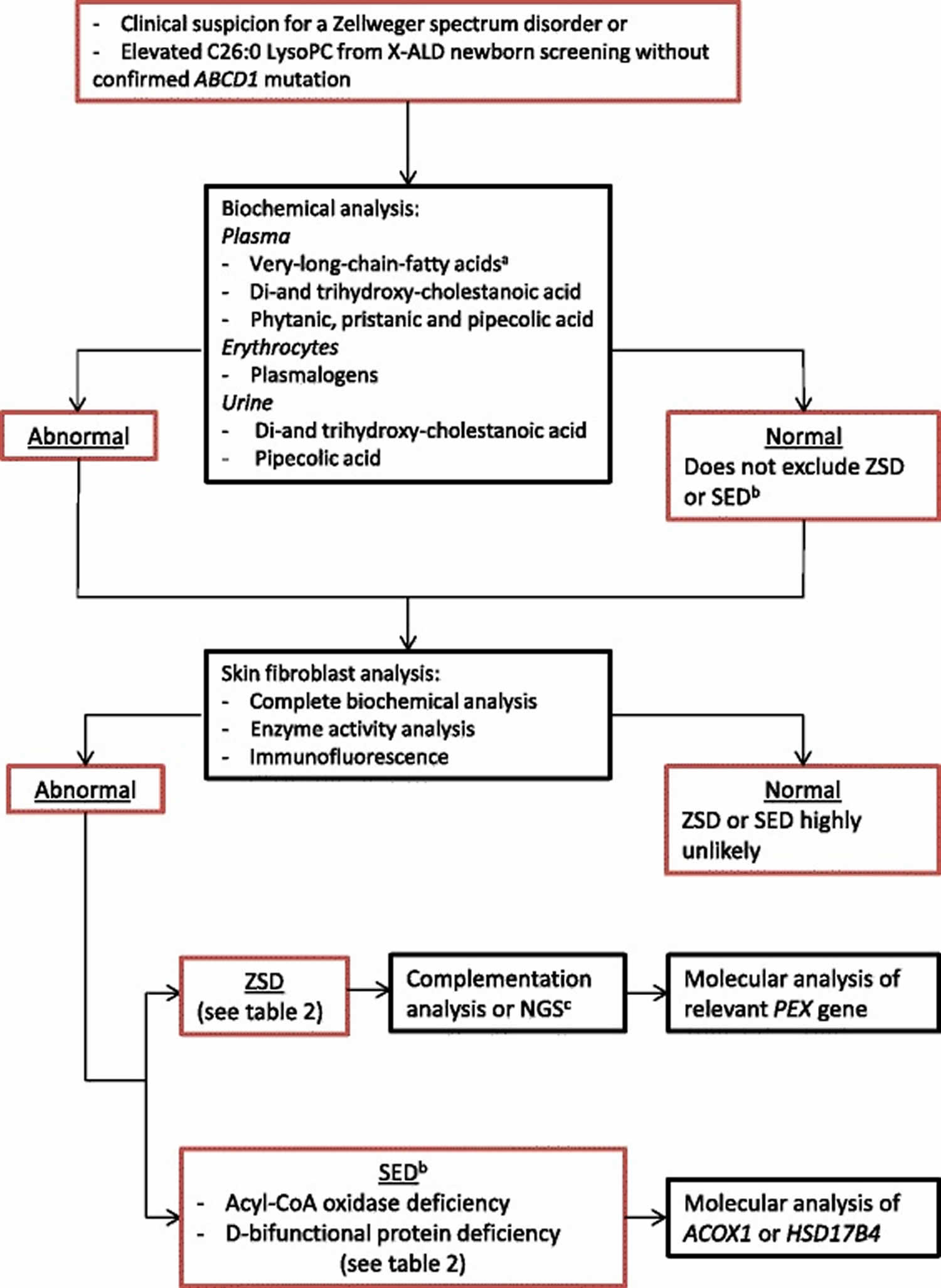
Footnotes: (a) Very long chain fatty acids (VLCFA): C26:0, C24:0/C22:0 ratio, C26:0/C22:0 ratio. (b) Single enzyme deficiency with phenotypical Zellweger syndrome disorder similarities like ACOX1 deficiency and DBP deficiency. (c) Next generation sequencing (NGS) of all PEX genes is advised when complementation analysis is not practicable
Abbreviations: X-ALD = X-linked adrenoleukodystrophy; LysoPC = lysophosphatidylcholine; ZSD = Zellweger syndrome disorder; SED = single enzyme deficiency
[Source 13 ]Table 1. Screening assays for Zellweger Spectrum Disorder
| Compound | Test | Expected Findings | Limitations of Test |
|---|---|---|---|
| C26:0 lysophosphatidylcholine 1 | Dried blood spot concentrations | ↑ C26:0-lysophosphatidylcholine concentrations | Persons with mild Zellweger syndrome disorder may not be detected. |
| Very-long-chain fatty acids (VLFCA) | Plasma concentration | ↑ plasma concentrations of C26:0 & C26:1; ↑ ratios of C24/C22 & C26/C22 2 | Non-fasting samples, hemolyzed samples, or a person on a ketogenic diet can cause false positive results. |
| Phytanic acid & pristanic acid 3 | Plasma concentration | ↑ concentrations of phytanic acid &/or pristanic acid | Branched-chain fatty acid accumulation depends on dietary intake of phytanic acid, which is minimal in formula- & breast-fed infants. Thus, phytanic & pristanic acid levels are normal in a neonate w/Zellweger syndrome disorder. |
| Plasmalogens | Erythrocyte membrane concentrations | ↓ amounts of C16 & C18 plasmalogens | Persons w/moderate-to-mild Zellweger syndrome disorder may have marginally↓-to-normal plasmalogen levels. |
| Pipecolic acid | Plasma/urine concentration | ↑ concentration of pipecolic acid in both plasma & urine | Urinary excretion of pipecolic acid is high in neonatal period but diminishes with age 4. Thus, urine should be tested in a neonate & plasma in an older child or adult. |
| Bile acids | Plasma/urine concentration | ↑ concentrations of C27 bile acid intermediates trihydroxycholestanoic acid & dihydroxycholestanoic acid | In most cases plasma testing is more sensitive than urine analysis. |
Footnotes:
1 26:0-lysophosphatidylcholine (LPC) is measured in dried blood spot test in newborn screening programs for X-linked adrenoleukodystrophy (X-ALD) in many states in the USA 34, 35. Elevated C26:0-lysophosphatidylcholine concentrations are also detected in individuals with Zellweger syndrome disorder. Clinical evaluation, molecular testing, and additional biochemical testing of newborns with elevated C26:0-lysophosphatidylcholine on dried blood spot test can help to distinguish those with peroxisomal disorders other than X-linked adrenoleukodystrophy.
2 Low plasma concentration of low-density lipoproteins (LDL, sometimes called the “bad” cholesterol because a high LDL level leads to a buildup of cholesterol in your arteries) and high-density lipoproteins (HDL, sometimes called the “good” cholesterol because HDL carries cholesterol from other parts of your body back to your liver. Your liver then removes the cholesterol from your body) can cause false negative results. In a person with low plasma concentrations of LDL and HDL without a defect in peroxisomal fatty acid metabolism, the plasma concentration of specific fatty acids (e.g., C22:0, C24:0, C26:0) are significantly lower than normal control levels. Persons with defects in peroxisomal fatty acid metabolism and very low LDL and HDL concentrations do not have significant elevations in C26:0 and C26:1, but do have modest elevations in the ratios of C24/C22 and C26/C22.
3 This analysis is usually included in very-long-chain fatty acids (VLFCA) measurement.
4 Pipecolic acid measurement is an adjunct to more definitive biomarkers such as plasma very-long-chain fatty acids (VLFCA) and red blood cell plasmalogen levels. Elevations in pipecolic acid can also occur in pyridoxine-dependent seizures 36.
[Source 1 ]Table 2. Genetic testing used in Zellweger syndrome disorder
| Gene | Percentage of Zellweger syndrome disorder attributed to pathogenic variants in gene |
|---|---|
| PEX1 | 60.5% |
| PEX6 | 14.5% |
| PEX12 | 7.6% |
| PEX26 | 4.2% |
| PEX10 | 3.4% |
| PEX2 | 3.1% |
| PEX5 | 2.0% |
| PEX13 | 1.5% |
| PEX16 | 1.1% |
| PEX3 | 0.7% |
| PEX19 | 0.6% |
| PEX14 | 0.5% |
| PEX11B | 0.1% |
Zellweger syndrome differential diagnosis
Symptoms of the following disorders can be similar to those of Zellweger spectrum disorders. Differential diagnosis of Zellweger syndrome based on the main presenting symptom includes the following 13:
- Hypotonia in newborns
- Chromosomal abnormalities (Down syndrome, Prader-Willi syndrome)
- Spinal muscular atrophy
- Hypoxic-ischemic encephalopathy
- Other peroxisomal disorders (acyl-CoA oxidase type 1 deficiency, D-bifunctional protein deficiency)
- Sensorineural hearing loss with retinitis pigmentosa
- Usher syndrome type 1,2
- Cockayne syndrome
- Alport syndrome
- Waardenburg syndrome
- Classical Refsum disease
- Bilateral cataract
- Lowe syndrome
- Galactosemia
- Congenital infections
- Rhizomelic chondrodysplasia punctata (RCDP)
- Adrenocortical insufficiency
- Adrenal hemorrhage
- X-linked adrenoleukodystrophy (X-ALD)
- Infectious adrenalitis
Rhizomelic chondrodysplasia punctata (RCDP) spectrum are a group of rare disorders that are also classified as peroxisomal biogenesis disorders. Rhizomelic chondrodysplasia punctata (RCDP) is characterized by shortening of the long bone of the arms (humerus) and legs (femur), a condition known as rhizomelia. Additional findings include distinctive facial features, the formation of small, hardened spots of calcium (stippling) on the knee cap (patella) and long bones of the arms and legs (chondrodysplasia punctata), cataracts that are present at birth or shortly thereafter, profound growth deficiency after birth, intellectual disability, and seizures. Rhizomelic chondrodysplasia punctata (RCDP) causes life-threatening complications during the first decade of life and in some cases during the newborn (neonatal) period. Milder forms of rhizomelic chondrodysplasia punctata (RCDP) have been identified in which affected individuals have less severe intellectual deficits and growth deficiency and often no rhizomelia. Many of these disorders are caused by mutations in the PEX7 gene and are inherited in an autosomal recessive pattern.
The leukodystrophies are a group of rare, progressive, metabolic, genetic disorders that can affect the brain, spinal cord and often the nerves outside the central nervous system (peripheral nerves). Each type of leukodystrophy is caused by an abnormality affecting a specific gene that results in abnormal development of one of at least 10 different chemicals that make up the white matter of the brain. The white matter is tissue composed of nerve fibers. Many of these nerve fibers are covered by a collection of fats (lipids) and proteins known as myelin. Myelin, which collectively may be referred to as the myelin sheath, protects the nerve fibers, acts as an insulator and increases the speed of transmission of nerve signals. Each type of leukodystrophy affects a different part of the myelin sheath, leading to a range of different neurological problems.
Refsum disease is a metabolic disorder characterized by the build-up of a fat (lipid) called phytanic acid in blood plasma and tissues. Individuals with Refsum disease are usually normal at birth, but between the ages of 10 and 20 years old, symptoms begin to develop starting with loss of night vision (retinitis pigmentosa), and eventually including weakness in arms and legs, or unsteadiness (cerebellar ataxia). Other common symptoms include a loss of sense of smell (anosmia), and rough, scaly skin (ichthyosis) and after many years deafness. Treatment for Refsum disease is based on limiting the intake of foods high in phytanic acid. Our bodies cannot make phytanic acid; rather, it is found in foods such as dairy, beef, lamb, and some seafood. Refsum disease is caused by a change (mutation) in the gene that makes an enzyme responsible for breaking down phytanic acid, a particular type of fatty acid which is derived by bacterial fermentation of green plants or algae. The lack of function of the enzyme (phytanoyl-CoA hydroxylase) leads to a build-up of phytanic acid in blood plasma and tissues. The disorder is inherited in an autosomal recessive manner.
X-linked adrenoleukodystrophy (X-ALD) is a rare genetic disorder that affects the white matter of the nervous system and the adrenal cortex. White matter is made up of nerve fibers called axons that relay nerve impulses from one cell to another. These nerve fibers are covered by myelin, an insulating layer or sheath that protects the nerve fibers. Myelin is made up of proteins and fats and gives white matter its white color. Without myelin, the signals between nerve cells cannot be transmitted properly, resulting in neurological symptoms. The adrenal cortex is the outer layer of cells of the adrenal glands. The adrenal glands sit atop the kidneys and produce hormones that are vital to proper health and development including cortisol and the sex hormones. Many of those affected experience serious neurological problems either during childhood or during adulthood with rather different types of disabilities. Some affected individuals also have adrenal insufficiency, which means that reduced amounts of certain hormones such as adrenaline and cortisol are produced, leading to abnormalities in blood pressure, heart rate, sexual development and reproduction. X-linked adrenoleukodystrophy (X-ALD) is an X-linked recessive disorder that is caused by variations (mutations) in the ABCD1 gene. Because it is an X-linked disorder males develop more serious complications than females, while some females will have no symptoms.
A group of disorders may result from a deficiency of a single peroxisomal enzyme. These disorders include D-bifunctional protein deficiency and pseudo-neonatal adrenoleukodystrophy (acyl-CoA oxidase deficiency). The symptoms of single enzyme peroxisomal disorders vary greatly from one individual to another. Some affect infants have severe complications similar to Zellweger syndrome; others have milder symptoms that resemble neonatal adrenoleukodystrophy or infantile Refsum disease.
Zellweger syndrome treatment
There is currently no cure or effective treatment for Zellweger syndrome. Management of Zellweger syndrome disorder is supportive and based on the signs and symptoms present in each person. For example, infants with feeding issues may require placement of a feeding tube to ensure proper intake of calories. Care is usually handled by a team of specialists that may include pediatricians, neurologists, surgeons, audiologists (specialists who assess and treat hearing problems), ophthalmologists (specialists who assess and treat vision problems), and orthopedists (specialists who assess and treat skeletal disorders) and other healthcare professionals may need to systematically and comprehensively plan an affect child’s treatment 37.
In 2015, Cholbam (cholic acid) was approved as the first treatment for pediatric and adult patients with bile acid synthesis disorders due to single enzyme defects, and for patients with peroxisomal disorders (including Zellweger spectrum disorders). Cholbam is marketed by Retrophin, Inc.
Children with Zellweger spectrum disorders may require a feeding (gastrostomy) tube to ensure proper intake of calories. A gastrostomy tube is inserted directly into the stomach. Additional therapies that may be used to treat Zellweger spectrum disorders include hearing aids, cochlear implants, fat-soluble vitamin supplementation (particularly vitamin K to treat bleeding complications due to clotting defects), surgery to treat cataracts, and glasses to improve vision.
Anti-epileptic drugs may be used to treat seizures, but seizures may persist and be difficult to control despite such therapy.
Adrenal insufficiency occurs frequently in more intermediate forms of Zellweger spectrum disorders. It is recommended that yearly adrenal monitoring with adrenocorticotropic hormone (ACTH) and morning cortisol be performed. Treatment with adrenal replacement (Cortef) using standard dosing should be implemented if abnormal. Even if adrenal measurements appear normal, families and clinicians should be aware of the possibility of adrenal insufficiency and consider stress dosing in periods of sudden severe illness, fever, and major surgical procedures.
Progressive decreased bone mineral density has been associated with Zellweger spectrum disorders and pathologic fractures have occurred in patients. Therefore, evaluation for bone disease should be considered. Additionally, many children with Zellweger spectrum disorders have enamel abnormalities of permanent teeth and should receive appropriate dental care.
Early intervention is important in treating children with Zellweger spectrum disorders. Services that may be beneficial may include special education, physical and orthopedic therapy, special services for children with hearing, and other medical, social, and/or vocational services.
Genetic counseling may be of benefit and is often required in prenatal testing for families of affected individuals. Other treatment is symptomatic and supportive.
Table 3. Supportive therapeutic options in Zellweger spectrum disorders
| Symptom/disease | Treatment/intervention |
|---|---|
| Adrenal insufficiency | Cortisone |
| Coagulopathy | Vitamin K suppletion |
| Enamel hypoplasia | Dentist referral |
| Epilepsy | Standard antiepileptic drugs |
| Hearing impairment | Hearing aids, cochlear implant |
| High phytanic acid plasma level | Phytanic acid restricted diet |
| Hyperoxaluria | Oral citrate treatment Sufficient fluid intake |
| Insufficient calory intake | Gastrostomy |
| Low levels of fat-soluble vitamins (A, D, E) | Vitamin suppletion |
| Visual impairment | Cataract removal, glasses and ophthalmologist referral |
Table 4. Recommended surveillance for individuals with Zellweger spectrum disorders
| System/Concern | Evaluation | Frequency |
|---|---|---|
| Feeding | Measurement of growth parameters Eval of nutritional status & safety of oral intake | At each visit |
| Hearing | Audiology eval | Annually |
| Eyes | Ophthalmologic eval Visual field testing | |
| Hepatic | Coagulation factors & other synthetic liver functions (PT, PTT, AST, ALT, total & direct bilirubin) Ultrasound &/or fibroscan to evaluate liver architecture | Annually; persons w/overt hepatic dysfunction require more frequent monitoring. |
| Neurologic | Monitor those w/seizures as clinically indicated. | At each visit |
| Head MRI to evaluate for white matter changes that may explain changes in cognitive &/or motor ability | As needed | |
| Development | Monitor developmental progress & educational needs. | At each visit |
| Endocrine | Assess adrenal function (ACTH & cortisol). | By age 1 yr; annually thereafter |
| Dental | Dental exam | Every 6 mos after dental eruption |
| Renal stones | Urine oxalate-to-creatinine ratio Consider renal imaging when performing liver imaging. | Annually |
| Miscellaneous/ Other | Assess family need for social work support (e.g., palliative/respite care, home nursing, other local resources) & care coordination. | At each visit |
Docosahexaenoic acid
Docosahexaenoic acid (DHA, an omega-3 fatty acid) is a long-chain polyunsaturated fatty acid (PUFA) important for retinal and brain function 38, 39. Tetracosahexaenoic acid (C24:6omega-3) undergoes one cycle of peroxisomal beta-oxidation to be converted to docosahexaenoic acid (DHA) 40, leading to reduced levels of docosahexaenoic acid (DHA) when peroxisomes are absent. Because Zellweger syndrome disorder patients often have low levels of docosahexaenoic acid (DHA) in membranes of red blood cells, supplementation of DHA was suggested to be a possible therapy. Although some studies have claimed a beneficial effect of docosahexaenoic acid (DHA) supplementation 41, 42, a randomized double-blind placebo controlled trial showed that docosahexaenoic acid (DHA) treatment leads to increased DHA levels in plasma, but no improvement of visual function and growth could be observed 43.
Lorenzo’s oil
Lorenzo’s oil (i.e. 4:1 mix of glyceryl trioleate and glyceryl trierucate) therapy was originally developed for the single peroxisomal enzyme deficiency X-linked adrenoleukodystrophy (X-ALD), and was shown to lower very-long-chain fatty acids (VLFCA) in plasma 44, but had no effect on disease progression 45, 46. Some studies reported lowering of the VLCFA levels in plasma by Lorenzo’s oil in Zellweger syndrome
babies 47, 48. However, based on data of studies in X-linked adrenoleukodystrophy (X-ALD) individuals, there is no reason to expect that Lorenzo’s oil will be beneficial for Zellweger syndrome disorder patients at this point 13.
Cholic acid
Cholic acid is a primary 24 carbon bile acid, involved in for instance the absorption of fat-soluble vitamins. Cholic acid is formed from its precursor trihydroxycholestanoic acid (THCA) by one peroxisomal beta-oxidation cycle 13. The peroxisomal C27-bile acid intermediates dihydroxycholestanoic acid (DHCA) and trihydroxycholestanoic acid (THCA) accumulate in Zellweger syndrome disorders and are considered to be more toxic than the primary C24 bile acids due to their altered physical properties and are believed to contribute to the liver disease in Zellweger syndrome disorders (e.g. dysfunction and liver fibrosis) 49. The bile acid intermediates are only partly conjugated and are less well excreted than C24 bile acids contributing to cholestasis. Scientists hypothesize that dihydroxycholestanoic acid (DHCA) and trihydroxycholestanoic acid (THCA) cross the blood–brain barrier and cause central nerve system damage 13. Several case reports have described a beneficial effect of cholic acid in Zellweger syndrome babies, supported by reduced urinary and plasma excretion of dihydroxycholestanoic acid or trihydroxycholestanoic acid 50, 51. Clinically there was increased growth and an increase in the levels of fat-soluble vitamins. Furthermore, bile acid treatment in mice was shown to improve liver disease 52. Limitations of the studies so far, however, are the small number of treated patients and short follow-up. Current evidence is insufficient to conclude that cholic acid treatment is beneficial for patients with a Zellweger syndrome disorder 53. The Food and Drug Administration recently approved cholic acid as a safe treatment for Zellweger syndrome disorder patients in the United States 54. However, efficiency should be demonstrated in large clinical trials before this treatment can be implemented.
Plasmalogen precursors
Due to a deficiency of the first peroxisomal steps in the biosynthesis of plasmalogens 55, Zellweger syndrome disorder patients may have low levels of plasmalogens. Plasmalogens play a critical role in cell membranes and as anti-oxidants 56. It was suggested that supplementation with precursors of plasmalogens (batyl alcohol) could be beneficial for Zellweger syndrome disorder patients, as import of these alkylglycerols proceeds normally. Several case reports have described an increase in red blood cell plasmalogen levels after treatment and improvement of clinical symptoms in some patients 57, 58, 59. Although never studied systematically, ether lipid therapy could be of interest for Zellweger syndrome disorder.
Citrate
The toxic metabolite oxalate accumulates in plasma and urine from Zellweger syndrome disorder patients 40. This causes kidney calcium oxalate stones. In a large group of Dutch Zellweger syndrome disorder patients a high prevalence of 83 % of renal calcium oxalate stones was shown 60. For this reason, patients should be screened for the presence of high levels of oxalic acid in urine yearly. To prevent the formation of renal stones, patients with hyperoxaluria should start oral citrate treatment. Furthermore, sufficient fluid intake is recommended 61.
Supportive care
All Zellweger syndrome disorder patients need to be screened for adrenal insufficiency 32, epilepsy, low levels of fat-soluble vitamins, (partly) vitamin K dependent coagulopathy, high levels of phytanic acid, hearing or visual impairment and enamel hypoplasia. They should be treated according to the identified abnormalities, e.g. supplementation of cortisone, anti-epileptic drugs, vitamins and/or a phytanic restricted diet. Because supplementation of cortisone is associated with severe side effects, such as growth suppression and osteoporosis 62, only patients with a true insufficiency (i.e. altered Synacthen test) should be treated. A phytanic acid restricted diet is only necessary when levels of phytanic acid are extremely high and is not recommended when levels are moderately increased, as sufficient intake of calories is more decisive. Hearing and visual impairment should be (partly) corrected by hearing aids and glasses, with ophthalmologic and audiological evaluations yearly. Enamel hypoplasia, present in nearly all patients, should be followed-up by a dentist 63, 64. Some patients will need a gastrostomy to provide adequate intake of calories.
Zellweger syndrome prognosis
The severity and progression of Zellweger syndrome disorder is difficult to predict for individual patients 13.
The prognosis for infants with Zellweger syndrome (severe Zellweger syndrome disorder) is poor. Zellweger syndrome typically presents in the neonatal period with profound hypotonia, characteristic facies, gyral (fold in the brian cortex) malformations, seizures, inability to feed, kidney cysts, liver dysfunction, and chondrodysplasia punctata. Infants with Zellweger syndrome (severe Zellweger syndrome disorder) are significantly impaired and usually die during the first year of life, usually having made no developmental progress 20. Death is usually secondary to progressive apnea or respiratory compromise from infection. However, most infants do not survive past the first 6 months, and usually succumb to respiratory distress, gastrointestinal bleeding, or liver failure.
Intermediate or milder Zellweger syndrome disorder (previously called neonatal adrenoleukodystrophy and infantile Refsum disease) may present in the newborn period, but generally comes to attention later because of developmental delays, hearing loss, and/or visual impairment. Liver dysfunction may lead to a vitamin K-responsive blood clotting disorder. Children have also come to attention with episodes of bleeding (hemorrhage); several children have presented in the first year of life with intracranial bleeding. The intermediate or milder Zellweger syndrome disorder (neonatal adrenoleukodystrophy and infantile Refsum disease) clinical course is variable; while many children are very hypotonic, many learn to walk and talk.
Intermediate or milder Zellweger syndrome disorder (neonatal adrenoleukodystrophy and infantile Refsum disease) is a progressive disorder with hearing and vision worsening with time. Some individuals may develop progressive degeneration of brain and spinal cord myelin, a leukodystrophy, which may lead to loss of previously acquired skills and ultimately death.
Children who survive the first year and who have a non-progressive course have a 77% probability of reaching school age 65. Some have normal intellect. They are at risk for adrenal insufficiency over time. Typically, they also have ameliogenesis imperfecta of the secondary teeth.
The remaining milder individuals can reach adulthood without progression or with long periods of stabilization. When progression occurs, it is mainly related to peripheral neuropathy and pyramidal signs, while cognition remains stable 33.
Zellweger syndrome life expectancy
Children with Zellweger syndrome (severe Zellweger syndrome disorder) usually do not survive beyond the first year of life 66, 1. Most infants do not survive past the first 6 months, and usually succumb to respiratory distress, gastrointestinal bleeding, or liver failure.
Intermediate or milder Zellweger syndrome disorder may present in the newborn period, but generally comes to attention later because of developmental delays, hearing loss, and/or visual impairment. Liver dysfunction may lead to a vitamin K-responsive blood clotting disorder. Children have also come to attention with episodes of bleeding (hemorrhage); several children have presented in the first year of life with intracranial bleeding. The intermediate or milder Zellweger syndrome disorder clinical course is variable; while many children are very hypotonic, many learn to walk and talk.
Intermediate or milder Zellweger syndrome disorder is a progressive disorder with hearing and vision worsening with time. Some individuals may develop progressive degeneration of brain and spinal cord myelin, a leukodystrophy, which may lead to loss of previously acquired skills and ultimately death.
Children who survive the first year and who have a non-progressive course have a 77% probability of reaching school age 65. Some have normal intellect. They are at risk for adrenal insufficiency over time. Typically, they also have ameliogenesis imperfecta of the secondary teeth.
The remaining milder individuals can reach adulthood without progression or with long periods of stabilization. When progression occurs, it is mainly related to peripheral neuropathy and pyramidal signs, while cognition remains stable 33.
References- Steinberg SJ, Raymond GV, Braverman NE, et al. Zellweger Spectrum Disorder. 2003 Dec 12 [Updated 2020 Oct 29]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1448
- Lee PR, Raymond GV. Child neurology: Zellweger syndrome. Neurology. 2013 May 14;80(20):e207-10. doi: 10.1212/WNL.0b013e3182929f8e
- Steinberg SJ, Raymond GV, Braverman NE, et al. Zellweger Spectrum Disorder. 2003 Dec 12 [Updated 2017 Dec 21]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1448
- Zellweger syndrome. https://ghr.nlm.nih.gov/condition/zellweger-spectrum-disorder
- Ratbi I, Falkenberg KD, Sommen M, Al-Sheqaih N, Guaoua S, Vandeweyer G, Urquhart JE, Chandler KE, Williams SG, Roberts NA, El Alloussi M, Black GC, Ferdinandusse S, Ramdi H, Heimler A, Fryer A, Lynch SA, Cooper N, Ong KR, Smith CE, Inglehearn CF, Mighell AJ, Elcock C, Poulter JA, Tischkowitz M, Davies SJ, Sefiani A, Mironov AA, Newman WG, Waterham HR, Van Camp G. Heimler Syndrome Is Caused by Hypomorphic Mutations in the Peroxisome-Biogenesis Genes PEX1 and PEX6. Am J Hum Genet. 2015 Oct 1;97(4):535-45. doi: 10.1016/j.ajhg.2015.08.011
- Ratbi I, Jaouad IC, Elorch H, Al-Sheqaih N, Elalloussi M, Lyahyai J, Berraho A, Newman WG, Sefiani A. Severe early onset retinitis pigmentosa in a Moroccan patient with Heimler syndrome due to novel homozygous mutation of PEX1 gene. Eur J Med Genet. 2016 Oct;59(10):507-11. doi: 10.1016/j.ejmg.2016.09.004
- Smith CE, Poulter JA, Levin AV, Capasso JE, Price S, Ben-Yosef T, Sharony R, Newman WG, Shore RC, Brookes SJ, Mighell AJ, Inglehearn CF. Spectrum of PEX1 and PEX6 variants in Heimler syndrome. Eur J Hum Genet. 2016 Nov;24(11):1565-1571. doi: 10.1038/ejhg.2016.62
- Gould S, Raymond G, Valle D: The Peroxisome biogenesis disorders. In The Metabolic and Molecular Bases of Inherited Disease. 8th edition. New York, NY: McGraw-Hill; 2001:3181–3218.
- Levesque S, Morin C, Guay S-P, Villeneuve J, Marquis P, Yik WY, Jiralerspong S, Bouchard L, Steinberg S, Hacia JG, Dewar K, Braverman NE. A founder mutation in the PEX6 gene is responsible for increased incidence of Zellweger syndrome in a French Canadian population. BMC Med Genet. 2012;13:72. doi: 10.1186/1471-2350-13-72
- Shimozawa N, Nagase T, Takemoto Y, Ohura T, Suzuki Y, Kondo N. Genetic heterogeneity of peroxisome biogenesis disorders among Japanese patients: evidence for a founder haplotype for the most common PEX10 gene mutation. Am J Med Genet A. 2003;120A:40–43. doi: 10.1002/ajmg.a.20030
- Child Neurology: Zellweger syndrome. Paul R. Lee, Gerald V. Raymond. Neurology May 2013, 80 (20) e207-e210; DOI: 10.1212/WNL.0b013e3182929f8e https://n.neurology.org/content/80/20/e207
- Braverman NE, Raymond GV, Rizzo WB, Moser AB, Wilkinson ME, Stone EM, et al. Peroxisome biogenesis disorders in the Zellweger spectrum: an overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol Genet Metab. 2016;117:313–321. doi: 10.1016/j.ymgme.2015.12.009
- Klouwer FC, Berendse K, Ferdinandusse S, Wanders RJ, Engelen M, Poll-The BT. Zellweger spectrum disorders: clinical overview and management approach. Orphanet J Rare Dis. 2015 Dec 1;10:151. doi: 10.1186/s13023-015-0368-9
- Braverman NE, D’Agostino MD, Maclean GE. Peroxisome biogenesis disorders: Biological, clinical and pathophysiological perspectives. Dev Disabil Res Rev. 2013;17:187–196. doi: 10.1002/ddrr.1113
- Wanders RJA, Waterham HR. Peroxisomal disorders I: biochemistry and genetics of peroxisome biogenesis disorders. Clin Genet. 2005;67:107–133. doi: 10.1111/j.1399-0004.2004.00329.x
- Waterham HR, Ebberink MS. Genetics and molecular basis of human peroxisome biogenesis disorders. Biochim Biophys Acta. 2012 Sep;1822(9):1430-41. doi: 10.1016/j.bbadis.2012.04.006
- Pineda M, Girós M, Roels F, Espeel M, Ruiz M, Moser A, Moser HW, Wanders RJ, Pavia C, Conill J, Aracil A, Amat L, Pampols T. Diagnosis and follow-up of a case of peroxisomal disorder with peroxisomal mosaicism. J Child Neurol. 1999;14:434–439. doi: 10.1177/088307389901400705
- Gootjes J, Schmohl F, Mooijer PAW, Dekker C, Mandel H, Topcu M, Huemer M, Von Schütz M, Marquardt T, Smeitink JA, Waterham HR, Wanders RJA. Identification of the molecular defect in patients with peroxisomal mosaicism using a novel method involving culturing of cells at 40 degrees C: implications for other inborn errors of metabolism. Hum Mutat. 2004;24:130–139. doi: 10.1002/humu.20062
- Ebberink MS, Mooijer PAW, Gootjes J, Koster J, Wanders RJA, Waterham HR. Genetic classification and mutational spectrum of more than 600 patients with a Zellweger syndrome spectrum disorder. Hum Mutat. 2011;32:59–69. doi: 10.1002/humu.21388
- Elumalai V, Pasrija D. Zellweger Syndrome. [Updated 2023 Feb 15]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560676
- Roth KS. Peroxisomal disease–common ground for pediatrician, cell biologist, biochemist, pathologist, and neurologist. Clin Pediatr (Phila). 1999 Feb;38(2):73-5. doi: 10.1177/000992289903800202
- Moser, A.B., Kreiter, N., Bezman, L., Lu, S.-E., Raymond, G.V., Naidu, S. and Moser, H.W. (1999), Plasma very long chain fatty acids in 3,000 peroxisome disease patients and 29,000 controls. Ann Neurol., 45: 100-110. https://doi.org/10.1002/1531-8249(199901)45:1<100::AID-ART16>3.0.CO;2-U
- Knazek RA, Rizzo WB, Schulman JD, Dave JR. Membrane microviscosity is increased in the erythrocytes of patients with adrenoleukodystrophy and adrenomyeloneuropathy. J Clin Invest. 1983 Jul;72(1):245-8. doi: 10.1172/jci110963
- Powers JM, Moser HW. Peroxisomal disorders: genotype, phenotype, major neuropathologic lesions, and pathogenesis. Brain Pathol. 1998 Jan;8(1):101-20. doi: 10.1111/j.1750-3639.1998.tb00139.x
- Zellweger syndrome. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=912
- Poll-The BT, Gärtner J. Clinical diagnosis, biochemical findings and MRI spectrum of peroxisomal disorders. Biochim Biophys Acta. 2012 Sep;1822(9):1421-9. doi: 10.1016/j.bbadis.2012.03.011
- Poll-The BT, Gootjes J, Duran M, De Klerk JBC, Wenniger Prick LJM De B, Admiraal RJC, Waterham HR, Wanders RJA, Barth PG. Peroxisome biogenesis disorders with prolonged survival: phenotypic expression in a cohort of 31 patients. Am J Med Genet A. 2004;126A:333–338. doi: 10.1002/ajmg.a.20664
- Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis. 2006;1:40. doi: 10.1186/1750-1172-1-40
- Van der Knaap MS, Wassmer E, Wolf NI, Ferreira P, Topçu M, Wanders RJA, Waterham HR, Ferdinandusse S. MRI as diagnostic tool in early-onset peroxisomal disorders. Neurology. 2012;78:1304–1308. doi: 10.1212/WNL.0b013e31825182dc
- Barth PG, Gootjes J, Bode H, Vreken P, Majoie CBLM, Wanders RJA. Late onset white matter disease in peroxisome biogenesis disorder. Neurology. 2001;57:1949–1955. doi: 10.1212/WNL.57.11.1949
- Moser AB, Rasmussen M, Naidu S, Watkins PA, McGuinness M, Hajra AK, Chen G, Raymond G, Liu A, Gordon D. Phenotype of patients with peroxisomal disorders subdivided into sixteen complementation groups. J Pediatr. 1995;127:13–22. doi: 10.1016/S0022-3476(95)70250-4
- Berendse K, Engelen M, Linthorst GE, van Trotsenburg ASP, Poll-The BT. High prevalence of primary adrenal insufficiency in Zellweger spectrum disorders. Orphanet J Rare Dis. 2014;9:133. doi: 10.1186/s13023-014-0133-5
- Berendse K, Engelen M, Ferdinandusse S, Majoie CB, Waterham HR, Vaz FM, Koelman JH, Barth PG, Wanders RJ, Poll-The BT. Zellweger spectrum disorders: clinical manifestations in patients surviving into adulthood. J Inherit Metab Dis. 2016 Jan;39(1):93-106. doi: 10.1007/s10545-015-9880-2
- Moser AB, Jones RO, Hubbard WC, Tortorelli S, Orsini JJ, Caggana M, Vogel BH, Raymond GV. Newborn Screening for X-Linked Adrenoleukodystrophy. Int J Neonatal Screen. 2016 Dec;2(4):15. doi: 10.3390/ijns2040015
- Vogel BH, Bradley SE, Adams DJ, D’Aco K, Erbe RW, Fong C, Iglesias A, Kronn D, Levy P, Morrissey M, Orsini J, Parton P, Pellegrino J, Saavedra-Matiz CA, Shur N, Wasserstein M, Raymond GV, Caggana M. Newborn screening for X-linked adrenoleukodystrophy in New York State: diagnostic protocol, surveillance protocol and treatment guidelines. Mol Genet Metab. 2015 Apr;114(4):599-603. doi: 10.1016/j.ymgme.2015.02.002
- Plecko, B., Stöckler-Ipsiroglu, S., Paschke, E., Erwa, W., Struys, E.A. and Jakobs, C. (2000), Pipecolic acid elevation in plasma and cerebrospinal fluid of two patients with pyridoxine-dependent epilepsy. Ann Neurol., 48: 121-125. https://doi.org/10.1002/1531-8249(200007)48:1<121::AID-ANA20>3.0.CO;2-V
- Zellweger Syndrome Information Page. https://www.ninds.nih.gov/Disorders/All-Disorders/Zellweger-Syndrome-Information-Page
- Horrocks LA, Yeo YK. Health benefits of docosahexaenoic acid (DHA) Pharmacol Res. 1999;40:211–225. doi: 10.1006/phrs.1999.0495
- Birch EE, Hoffman DR, Uauy R, Birch DG, Prestidge C. Visual acuity and the essentiality of docosahexaenoic acid and arachidonic acid in the diet of term infants. Pediatr Res. 1998;44:201–209. doi: 10.1203/00006450-199808000-00011
- Wanders RJA, Waterham HR. Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem. 2006;75:295–332. doi: 10.1146/annurev.biochem.74.082803.133329
- Noguer MT, Martinez M. Visual follow-up in peroxisomal-disorder patients treated with docosahexaenoic Acid ethyl ester. Invest Ophthalmol Vis Sci. 2010;51:2277–2285. doi: 10.1167/iovs.09-4020
- Martínez M, Vázquez E, García-Silva MT, Manzanares J, Bertran JM, Castelló F, Mougan I. Therapeutic effects of docosahexaenoic acid ethyl ester in patients with generalized peroxisomal disorders. Am J Clin Nutr. 2000 Jan;71(1 Suppl):376S-85S. doi: 10.1093/ajcn/71.1.376s
- Paker M, Sunness JS, Brereton NH, Speedie LJ, Albanna L, Dharmaraj S, Mosera B, Jones RO, Raymond GV. Docosahexaenoic acid therapy in peroxisomal diseases: results of a double-blind, randomized trial. Neurology. 2010;75:826–830. doi: 10.1212/WNL.0b013e3181f07061
- Moser AB, Borel J, Odone A, Naidu S, Cornblath D, Sanders DBMH. A new dietary therapy for adrenoleukodystrophy: biochemical and preliminary clinical results in 36 patients. Ann Neurol. 1987;21:240–249. doi: 10.1002/ana.410210305
- Van Geel BM, Assies J, Haverkort EB, Koelman JHTM, Verbeeten B, Wanders RJA, Barth PG. Progression of abnormalities in adrenomyeloneuropathy and neurologically asymptomatic X-linked adrenoleukodystrophy despite treatment with “Lorenzo’s oil.” J Neurol Neurosurg Psychiatry. 1999;67:290–299. doi: 10.1136/jnnp.67.3.290
- Aubourg P, Adamsbaum C, Lavallard-Rousseau MC, Rocchiccioli F, Cartier N, Jambaqué I, Jakobezak C, Lemaitre A, Boureau F, Wolf C, et al. A two-year trial of oleic and erucic acids (“Lorenzo’s oil”) as treatment for adrenomyeloneuropathy. N Engl J Med. 1993 Sep 9;329(11):745-52. doi: 10.1056/NEJM199309093291101
- Tanaka K, Shimizu T, Ohtsuka Y, Yamashiro Y, Oshida K. Early dietary treatments with Lorenzo’s oil and docosahexaenoic acid for neurological development in a case with Zellweger syndrome. Brain Dev. 2007;29:586–589. doi: 10.1016/j.braindev.2007.02.005
- Arai Y, Kitamura Y, Hayashi M, Oshida K, Shimizu T, Yamashiro Y. Effect of dietary Lorenzo’s oil and docosahexaenoic acid treatment for Zellweger syndrome. Congenit Anom (Kyoto) 2008;48:180–182. doi: 10.1111/j.1741-4520.2008.00201.x
- Ferdinandusse S, Denis S, Dacremont G, Wanders RJA. Toxicity of peroxisomal C27-bile acid intermediates. Mol Genet Metab. 2009;96:121–128. doi: 10.1016/j.ymgme.2008.11.165
- Setchell KDR, Bragetti P, Zimmer-Nechemias L, Daugherty C, Pelli MA, Vaccaro R, Gentili G, Distrutti E, Dozzini G, Morelli A, Clerici C. Oral bile acid treatment and the patient with zellweger syndrome. Hepatology. 1992;15:198–207. doi: 10.1002/hep.1840150206
- Maeda K, Kimura A, Yamato Y, Nittono H, Takei H, Sato T, Mitsubuchi H, Murai T, Kurosawa T. Oral bile Acid treatment in two Japanese patients with Zellweger syndrome. J Pediatr Gastroenterol Nutr. 2002 Aug;35(2):227-30. doi: 10.1097/00005176-200208000-00025
- Keane MH, Overmars H, Wikander TM, Ferdinandusse S, Duran M, Wanders RJ A, Faust PL. Bile acid treatment alters hepatic disease and bile acid transport in peroxisome-deficient PEX2 Zellweger mice. Hepatology. 2007;45:982–997. doi: 10.1002/hep.21532
- Keane MH, Overmars H, Wikander TM, Ferdinandusse S, Duran M, Wanders RJ, Faust PL. Bile acid treatment alters hepatic disease and bile acid transport in peroxisome-deficient PEX2 Zellweger mice. Hepatology. 2007 Apr;45(4):982-97. doi: 10.1002/hep.21532
- Drug Trials Snapshot: Cholbam (peroxisomal disorders). https://www.fda.gov/drugs/drug-approvals-and-databases/drug-trials-snapshot-cholbam-peroxisomal-disorders
- De Vet EC, van den Bosch H. Alkyl-dihydroxyacetonephosphate synthase. Cell Biochem Biophys. 2000;32:117–121. doi: 10.1385/CBB:32:1-3:117
- Braverman NE, Moser AB. Functions of plasmalogen lipids in health and disease. Biochim Biophys Acta. 2012 Sep;1822(9):1442-52. doi: 10.1016/j.bbadis.2012.05.008
- Holmes RD, Wilson GN, Hajra A. Oral ether lipid therapy in patients with peroxisomal disorders. J Inherit Metab Dis. 1987;10:239–241. doi: 10.1007/BF01811415
- Das AK, Holmes RD, Wilson GN, Hajra AK. Dietary ether lipid incorporation into tissue plasmalogens of humans and rodents. Lipids. 1992;27:401–405. doi: 10.1007/BF02536379
- Wilson GN, Holmes RG, Custer J, Lipkowitz JL, Stover J, Datta N, Hajra A. Zellweger syndrome: diagnostic assays, syndrome delineation, and potential therapy. Am J Med Genet. 1986;24:69–82. doi: 10.1002/ajmg.1320240109
- Van Woerden CS, Groothoff JW, Wijburg FA, Duran M, Wanders RJA, Barth PG, Poll-The BT High incidence of hyperoxaluria in generalized peroxisomal disorders. Mol Genet Metab. 2006;88:346–350. doi: 10.1016/j.ymgme.2006.03.004
- Leumann E, Hoppe B, Neuhaus T, Blau N. Efficacy of oral citrate administration in primary hyperoxaluria. Nephrol Dial Transplant. 1995;10(Suppl 8):14–16. doi: 10.1093/ndt/10.supp8.14
- Buchman AL. Side effects of corticosteroid therapy. J Clin Gastroenterol. 2001;33:289–294. doi: 10.1097/00004836-200110000-00006
- Lertsirivorakul J, Wongswadiwat M, Treesuwan P. Oral manifestations and dental management of a child with Zellweger syndrome. Spec Care Dent. 2012;34:46–50. doi: 10.1111/scd.12003
- Acharya BS, Ritwik P, Velasquez GM, Fenton SJ. Medical-dental findings and management of a child with infantile Refsum disease: a case report. Spec Care Dentist. 2012;32:112–117. doi: 10.1111/j.1754-4505.2012.00248.x
- Poll-The, B.T., Gootjes, J., Duran, M., de Klerk, J.B.C., Maillette de Buy Wenniger-Prick, L.J., Admiraal, R.J.C., Waterham, H.R., Wanders, R.J.A. and Barth, P.G. (2004), Peroxisome biogenesis disorders with prolonged survival: Phenotypic expression in a cohort of 31 patients. Am. J. Med. Genet., 126A: 333-338. https://doi.org/10.1002/ajmg.a.20664
- Zellweger syndrome. https://rarediseases.info.nih.gov/diseases/7917/zellweger-syndrome





{kind=link}