What is Alport syndrome
Alport syndrome is a rare inherited disease characterized by kidney disease, hearing loss, and eye abnormalities 1. People with Alport syndrome experience progressive loss of kidney function. Almost all affected individuals have blood in their urine (hematuria), which indicates abnormal functioning of the kidneys. Many people with Alport syndrome also develop high levels of protein in their urine (proteinuria). The kidneys become less able to function as this condition progresses, resulting in end-stage renal disease (ESRD). Alport syndrome is estimated to account for 3% of children with chronic kidney disease and 0.2% of adults with end-stage renal disease in the United States.
Alport syndrome is caused by genetic mutations that affect the type 4 collagen family of proteins. Type 4 collagen is a major part of important tissue structures called basement membranes that are present in all tissues including the kidney, inner ear, and eye.
There are three genetic types of Alport syndrome:
- X-linked Alport syndrome (XLAS) — This is the most common type. X-linked Alport syndrome (XLAS) is more severe in males than in females.
- Autosomal recessive Alport syndrome (ARAS) — Males and females have equally severe disease.
- Autosomal dominant Alport syndrome (ADAS) — This is the rarest type. Males and females have equally severe disease.
With all types of Alport syndrome the kidneys are affected. The tiny blood vessels in the glomeruli of the kidneys are damaged. The glomeruli filter blood to make urine and remove waste products from the blood.
At first, there are no symptoms. Over time, as the glomeruli are more and more damaged, kidney function is lost and waste products and fluids build up in the body. The condition can progress to end-stage renal disease (ESRD) at an early age, between adolescence and age 40. At this point, dialysis or a kidney transplant is needed.
Alport syndrome occurs in approximately 1 in 50,000 newborns. Alport syndrome is a rare disease and affects less than 200,000 people in the US. Alport syndrome is estimated to affect approximately 1 in 5,000-10,000 people in the general population in the United States, which means that approximately 30,000-60,000 people in the United States have Alport syndrome.
People with Alport syndrome frequently develop sensorineural hearing loss, which is caused by abnormalities of the inner ear, during late childhood or early adolescence. Affected individuals may also have misshapen lenses in the eyes (anterior lenticonus) and abnormal coloration of the light-sensitive tissue at the back of the eye (retina). These eye abnormalities seldom lead to vision loss.
Significant hearing loss, eye abnormalities, and progressive kidney disease are more common in males with Alport syndrome than in affected females.
Alport syndrome causes
Mutations in the COL4A3, COL4A4, and COL4A5 genes cause Alport syndrome. These genes each provide instructions for making one component of a protein called type 4 collagen. This protein plays an important role in the kidneys, specifically in structures called glomeruli. Glomeruli are clusters of specialized blood vessels that remove water and waste products from blood and create urine. Mutations in these genes result in abnormalities of the type 4 collagen in glomeruli, which prevents the kidneys from properly filtering the blood and allows blood and protein to pass into the urine. Gradual scarring of the kidneys occurs, eventually leading to kidney failure in many people with Alport syndrome.
Type 4 collagen is also an important component of inner ear structures, particularly the organ of Corti, that transform sound waves into nerve impulses for the brain. Alterations in type 4 collagen often result in abnormal inner ear function, which can lead to hearing loss. In the eye, this protein is important for maintaining the shape of the lens and the normal color of the retina. Mutations that disrupt type 4 collagen can result in misshapen lenses and an abnormally colored retina.
Alport syndrome inheritance
Alport syndrome can have different inheritance patterns. About 80 percent of cases are caused by mutations in the COL4A5 gene and are inherited in an X-linked pattern. This gene is located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the COL4A5 gene in each cell is sufficient to cause kidney failure and other severe symptoms of the disorder. In females (who have two X chromosomes), a mutation in one copy of the COL4A5 gene usually only results in hematuria, but some women experience more severe symptoms. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
In approximately 15 percent of cases, Alport syndrome results from mutations in both copies of the COL4A3 or COL4A4 gene and is inherited in an autosomal recessive pattern. The parents of an individual with the autosomal recessive form of this condition each have one copy of the mutated gene and are called carriers. Some carriers are unaffected and others develop a less severe condition called thin basement membrane nephropathy, which is characterized by hematuria.
Alport syndrome has autosomal dominant inheritance in about 5 percent of cases. People with this form of Alport syndrome have one mutation in either the COL4A3 or COL4A4 gene in each cell. It remains unclear why some individuals with one mutation in the COL4A3 or COL4A4 gene have autosomal dominant Alport syndrome and others have thin basement membrane nephropathy.
Genetics counseling
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Alport syndrome symptoms
With all types of Alport syndrome the kidneys are affected. The tiny blood vessels in the glomeruli of the kidneys are damaged and cannot filter the wastes and extra fluid in your body. Many people with Alport syndrome also have hearing problems and abnormalities with their eyes.
Other signs and symptoms of Alport syndrome may include:
- Blood in the urine (hematuria), the most common and earliest sign of Alport syndrome
- Protein in the urine (proteinuria)
- High blood pressure (hypertension)
- Swelling in the legs, ankle, feet and around the eyes (called edema)
These signs and symptoms may differ, based on age, gender and inherited type of Alport syndrome.
Kidneys
Alport syndrome always affects the kidneys. The primary symptom is blood in the urine (hematuria), which is usually microscopic, meaning it can only be detected with a microscope or a urine dipstick. Sometimes children with Alport syndrome have brown, pink, or red urine (gross hematuria) for several days, associated with a cold or flu. This gross hematuria eventually stops when the child recovers and can be very frightening but is not harmful. As the disease progresses additional signs of kidney disease begin to show, such as protein in the urine (proteinuria) and high blood pressure.
Alport syndrome causes damage to the kidneys by the progressive formation of scar tissue in the normal kidney structures (glomeruli and tubules). As the kidneys filter proteins out of the blood, these molecules damage the filtering system or glomeruli because of the abnormal collagen makeup. This process is known as fibrosis and it eventually leads to kidney failure.
Symptoms of kidney problems include:
- Abnormal urine color
- Blood in the urine (which can be worsened by upper respiratory infections or exercise)
- Flank pain
- High blood pressure
- Swelling throughout the body
About 50% of males with X-linked disease require dialysis or kidney transplantation by age 25 years, and about 90% develop end-stage renal disease (ESRD) before 40 years of age. In female patients with X-linked Alport syndrome, progression to ESRD was previously thought to be rare. However, recent observations have shown that as many as 12% of female patients also develop ESRD by the age of 40 years; this rate increases to 30% by the age of 60 years and 40% by age 80 years. Among female patients, risk factors for progression to ESRD include proteinuria and hearing loss. Males and females with the autosomal recessive form (ARAS) of Alport syndrome will develop kidney failure by their teens or young adult years. People with autosomal dominant Alport syndrome (ADAS) are usually well into middle age before kidney failure develops.
Inner Ear
Hearing loss is another symptom of Alport syndrome. Hearing loss is never present at birth but becomes apparent by late childhood or early adolescence, generally before the onset of kidney failure. Some families with Alport syndrome do not experience hearing loss. It is estimated that about 80% of males with X-linked Alport syndrome (XLAS) develop hearing loss at some point in their lives, often by the time they are teenagers. In females with X-linked Alport syndrome hearing loss is less frequent and occurs later in life. Males and females with autosomal recessive Alport syndrome typically have childhood hearing loss. Patients with autosomal dominant Alport syndrome develop hearing loss at a later age.
Hearing loss usually occurs before kidney failure. Fortunately, hearing aids are usually very effective for patients with hearing loss caused by Alport syndrome. However, hearing loss is not improved by kidney transplantation.
Alport syndrome eye
Alport syndrome also leads to eye problems, including:
- Abnormal shape of the lens (anterior lenticonus), which can lead to a slow decline in vision as well as cataracts.
- Corneal erosion in which there is loss of the outer layer of the covering of the eyeball, leading to pain, itching, or redness of the eye, or blurred vision.
- Abnormal coloring of the retina, a condition called dot-and-fleck retinopathy. It doesn’t cause vision problems, but can help diagnose Alport syndrome.
- Macular hole in which there is thinning or a break in the macula. The macula is a part of the retina that makes central vision sharper and more detailed. A macular hole causes blurred or distorted central vision.
Anterior lenticonus is an abnormality in the shape of the lens of the eye and affects about 15% to 20% of patients with X-linked (XLAS) and autosomal recessive Alport syndrome (ARAS). People with anterior lenticonus may have a slow progressive deterioration of vision requiring patients to change the prescription of their glasses frequently. This condition may also lead to cataract formation.
Many people with Alport syndrome have abnormal pigment of the retina called dot-and-fleck retinopathy, but this does not result in any abnormalities of vision. This can be helpful diagnostically, though, especially in women who have no other features of Alport syndrome except hematuria (for which there are other causes).
Recurrent corneal erosion is another eye problem that can occur in people with Alport syndrome. Individuals who suffer from this have repeated episodes of eye itchiness, and may need to take measures to protect their corneas from minor trauma such as wearing goggles when riding a bicycle.
A macular hole is another eye issue that affects only about 5% of patients. This results in a loss of vision and is an extension of the retinal thinning that occurs commonly in Alport syndrome. Affected individuals will have difficulty with central vision it will be hazy or distorted.
Figure 1. Alport syndrome eye
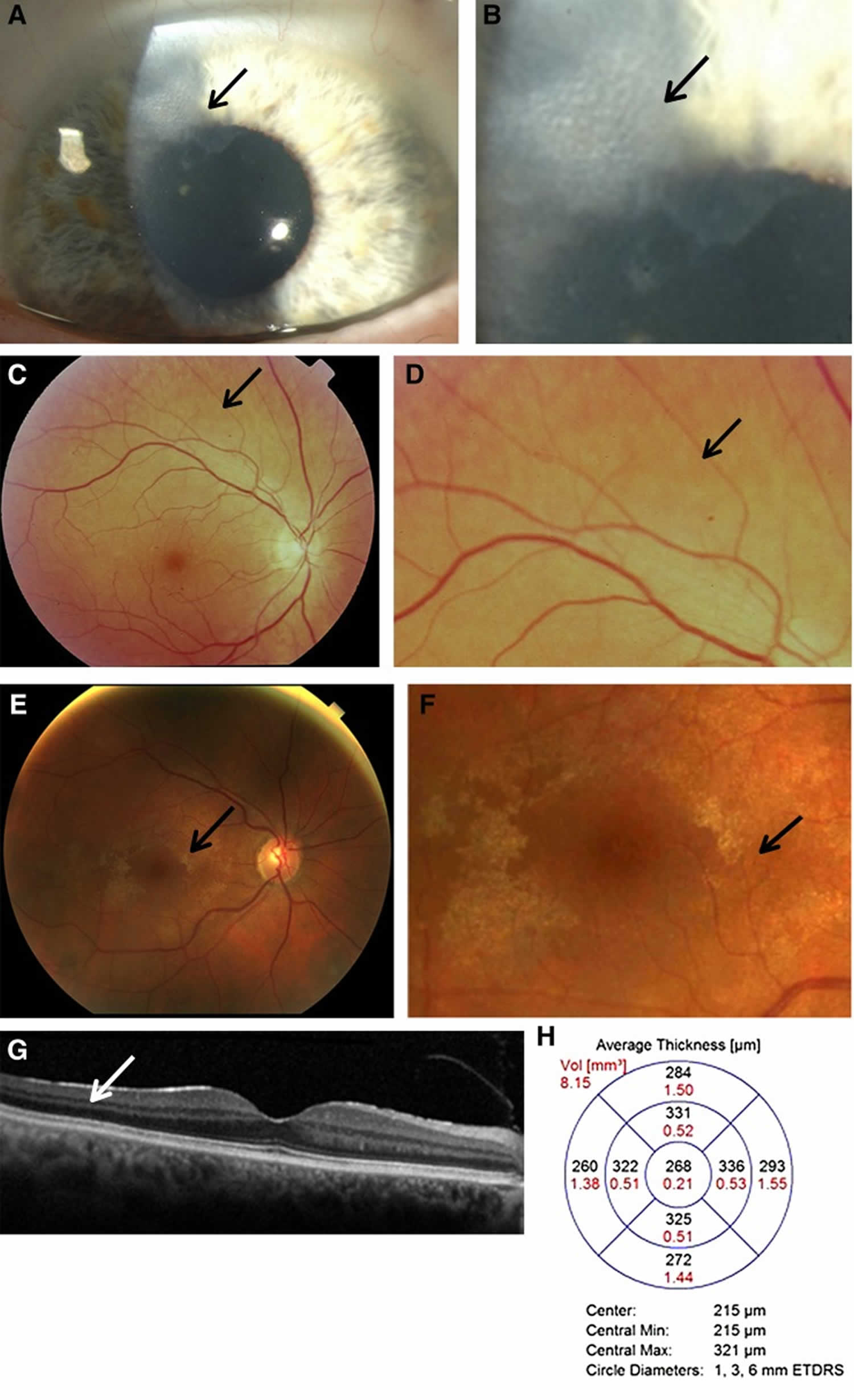
Footnote: Characteristic ocular features in women with Alport syndrome. (A) Corneal opacity (arrow) in a 60-year-old woman with X-linked Alport syndrome and an in-frame COL4A5 deletion, hearing loss, and normal renal function; and (B) higher powered view of the same abnormality. Her son had a renal transplant and had a similar corneal lesion. (C) Peripheral fleck retinopathy in a 75-year-old woman, with mild renal impairment and hearing loss; and (D) higher power view demonstrating a dappled appearance due to the coalescent fleck retinopathy (arrow). (E) Central fleck retinopathy in a 30-year-old female with a R373× in COL4A5, and normal renal function (arrow). This figure demonstrates how difficult it can be to distinguish the fleck retinopathy from the retinal sheen of youth; and (F) a higher power view of the central fleck retinopathy (arrow). Vision was not affected. (G) Optical coherence tomography demonstrating a sagittal section through the retina and subtle thinning of the temporal versus nasal quadrants, and (H) optical coherence tomography measurements demonstrating different thicknesses in the temporal versus nasal quadrants which was consistent with moderate temporal retinal thinning (7.4%)
[Source 2 ]Alport syndrome diagnosis
The diagnosis of Alport syndrome is performed using some or all of these methods:
- Medical history and physical examination (urinalysis, blood testing)
- Detailed family history and possibly urinalyses on first- and second-degree relatives
- Hearing and vision evaluation and testing
- Renal (kidney) ultrasound
- Kidney biopsy analysis
- Skin biopsy analysis
- Genetic testing.
A diagnosis of Alport syndrome is suspected based upon identification of characteristic symptoms, a detailed patient history, and a thorough clinical evaluation. The likelihood of diagnosis increases in individuals with a family history of Alport syndrome, kidney failure without known cause, early hearing loss or hematuria. A variety of specialized tests can help to confirm a suspected diagnosis.
The diagnostic approach to confirming a suspected diagnosis of Alport syndrome has been evolving over the past decade. While tissue studies (kidney or skin biopsy) are very useful tools in the evaluation of patients with hematuria, early genetic testing is becoming increasingly important. When clinical information and family history strongly suggest a diagnosis of Alport syndrome, genetic testing, using the techniques of next generation or whole exome sequencing, can confirm the diagnosis, establish the inheritance pattern and provide useful prognostic information. Genetic testing for Alport syndrome is offered by several commercial laboratories as well as some hospital laboratories, but there is wide variation in insurance coverage.
When genetic testing is unavailable or inaccessible, studies of tissue specimens (biopsies) are performed. A suspected diagnosis of X-linked Alport syndrome (XLAS) may be confirmed by skin biopsy. A specific test known as immunostaining is performed on the sample. With immunostaining, an antibody that reacts against collagen type IV alpha-5 chain proteins is added to the skin sample. This allows physicians to determine whether a specific protein is present and in what quantity. Normally, alpha-5 chains are found in skin samples, but in males with XLAS they are nearly completely absent. Alpha-3 and alpha-4 chains are not present in the skin and, therefore, skin biopsies cannot be used to diagnose autosomal recessive Alport syndrome (ARAS) or autosomal dominant Alport syndrome (ADAS).
A kidney biopsy may be also performed. A kidney biopsy can reveal characteristic changes to kidney tissue including abnormalities of the glomerular basement membrane (GBM) that can be detected by an electron microscope. Immunostaining can also be performed on a kidney biopsy sample. In addition to detecting alpha-5 chains, kidney samples can be assessed to determine whether type 4 collagen alpha-3 or alpha-4 chains are present and in what quantity.
Examination of urine samples (urinalysis) can reveal microscopic or gross amounts of blood (hematuria) in the urine. Hematuria may come and go (intermittent) in some cases, especially females with XLAS or individuals with ADAS. If kidney disease has progressed, elevated levels of protein can also be detected in urine samples.
Individuals diagnosed with Alport syndrome should undergo hearing tests that determine a person’s audible range for tones and speech (audiometry) and a complete eye (ophthalmological) exam.
In cases where a parent has a known genetic abnormality (i.e. heterozygous mothers) prenatal diagnosis or pre-implantation genetic diagnosis (PGD) may be options. Prenatal diagnosis is possible through chorionic villi sampling (CVS) or amniocentesis. During CVS, fetal tissue samples are removed and enzyme tests (assays) are performed on cultured tissue cells (fibroblasts) and/or white blood cells (leukocytes). During amniocentesis, a sample of the fluid that surrounds the developing fetus is removed and studied.
Pre-implantation genetic diagnosis can be performed on embryos created through in vitro fertilization. Pre-implantation genetic diagnosis refers to testing an embryo to determine whether it has the same genetic abnormality as the parent. Families interested such an option should seek the counsel of a certified genetics professional.
Kidney Biopsy
Alport syndrome produces unique changes in the walls of the blood vessels of the glomeruli (kidney structures) that can be detected by performing electron microscopy on the kidney biopsy material. A kidney biopsy is usually performed as an outpatient procedure at a hospital and requires minor surgery to obtain a sample of kidney tissue. Kidney biopsies can also be tested for the presence or absence of the type IV collagen alpha-3, alpha-4 and alpha-5 chains (COL4A3, COL4A4, and COL4A5 genes). This information is helpful in confirming a suspected diagnosis of Alport syndrome and can usually determine the genetic form of the disease.
Skin Biopsy
A skin biopsy can be performed when XLAS (X-linked Alport syndrome) is suspected. The type 4 collagen alpha-5 chain (COL4A5) is normally present in the skin and a biopsy of the skin can be tested for the presence or absence of this collagen chain. Approximately 80% of male patients and 60% of female patients with X-linked Alport syndrome will show abnormal staining for COL4A5 in the skin biopsy. This approach is especially useful if a kidney biopsy poses an excessive risk, such as in patients with kidney failure.
Because, the alpha-3 and alpha-4 chains are not present in the skin this test cannot be used to diagnose ARAS or ADAS.
Genetic Testing
Clinicians in many but not all parts of the world now have access to genetic testing for diagnosis of Alport syndrome through commercial laboratories or laboratories associated with medical institutions. Such testing offers high rates of diagnostic accuracy, particularly for X-linked Alport syndrome, although testing is available for autosomal recessive and autosomal dominant forms of the disease, too. Insurance coverage for genetic testing varies widely so patients are encouraged to seek the help of certified genetic counselors to determine their eligibility.
In most cases, genetic testing can confirm a diagnosis of Alport syndrome. Genetic testing is the only way to diagnose a female with no symptoms but with a family history of X-linked Alport syndrome. Genetic testing may also be useful when results of a skin or kidney biopsy are not conclusive.
In cases where a parent has a known genetic mutation, prenatal diagnosis or pre-implantation genetic diagnosis (PGD) may be options. Prenatal diagnosis is possible through chorionic villi sampling (CVS) or amniocentesis. During CVS, fetal tissue samples are removed and enzyme tests are performed on cultured tissue cells and/or white blood cells. During amniocentesis, a sample of the fluid that surrounds the developing fetus is removed and studied. Pre-implantation genetic diagnosis can be performed on embryos created through in vitro fertilization. Pre-implantation genetic diagnosis refers to testing an embryo to determine whether it has the same genetic abnormality as the parent. Families interested in such an option should seek the counsel of a certified genetics professional.
Genetics counseling
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Alport syndrome life expectancy
Alport syndrome inevitably leads to end-stage renal disease and there are no therapies known to improve outcome 3. Angiotensin-converting enzyme inhibitors can delay time to dialysis and improve life expectancy in three generations of Alport families 3. Angiotensin-converting enzyme inhibitors therapy significantly improved life expectancy beyond the median age of 55 years of the no-treatment cohort. Thus, Alport syndrome is treatable with angiotensin-converting enzyme inhibition to delay renal failure and therapy improves life expectancy in a time-dependent manner. This supports the need for early diagnosis and early nephroprotective therapy in Alport syndrome patients.
Alport syndrome prognosis
Women usually have a normal lifespan with no signs of the disease except for blood in the urine. In rare cases, women have high blood pressure, swelling, and nerve deafness as a complication of pregnancy.
Autosomic recessive Alport Syndrome has an earlier onset of renal failure than Autosomic dominant Alport Syndrome 4. As the kidneys fail, dialysis or a transplant will be needed.
In men, deafness, vision problems, and end-stage kidney disease are likely by age 50.
In the X-linked Alport syndrome (XLAS), the most common type of Alport syndrome, about 50% of males require dialysis or kidney transplantation by age 25 years, and approximately 90% develop End Stage Renal Disease (ESRD) before age 40 4. Female patients with X-linked Alport syndrome have a better prognosis with about 12% developing end-stage renal disease (ESRD) by age 40 4. By age 60, this rate increases to about 30% and by 60 years of age, the rate of ESRD approaches 40% 4. In the female population, proteinuria and hearing loss are found to be risk factors for the progression to ESRD. In comparison, the autosomal recessive form of Alport syndrome can cause kidney failure by age 20 while the autosomal dominant form of the disease typically has a delay in ESRD until middle age 5.
Renal transplantation caused by Alport syndrome has a superior patient and graft survival than by other causes 6.
Alport syndrome treatment
The treatment of Alport syndrome is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, nephrologists, audiologists, ophthalmologists, and other healthcare professionals may need to systematically and comprehensively plan an affect child’s treatment. Genetic counseling is beneficial for affected individuals and their families. Psychosocial support for the entire family is essential as well.
The goals of treatment include monitoring and controlling the disease and treating the symptoms. Specific symptoms associated with Alport syndrome are treated by routine, accepted guidelines. For example, hearing aids may be used to treat hearing loss when appropriate. Hearing aids are usually effective in people with Alport syndrome because they do not lose the ability to distinguish one sound from another, as long as the sounds are amplified. Surgery to remove cataracts is performed when necessary.
Your doctor may recommend any of the following:
- A diet that limits salt, fluids, and potassium
- Medicines to control high blood pressure
Kidney disease is managed by:
- Taking medicines to slow kidney damage
- A diet that limits salt, fluids, and protein
Hearing loss can be managed with hearing aids. Eye problems are treated as needed. For example, an abnormal lens due to lenticonus or cataracts can be replaced.
Genetic counseling may be recommended because Alport syndrome is inherited.
Alport syndrome eye
Eye pads, topical antibiotics and pain relief can be applied to resolve corneal erosions 7.
Corneal transplantation is sometimes required to improve visual acuity for severe corneal disease.
Lens removal and intraocular lens implantation surgery are eventually required for most patients with lenticonus and cataract
No treatment is required for fleck retinopathy 7.
Macular holes do not respond well to surgery treatment 7.
Medication
The recommended medications for treatment of Alport syndrome interfere with several hormones that together make up what is known as the renin-angiotensin-aldosterone system (RAAS). The renin-angiotensin-aldosterone system (RAAS) normally plays a very important role in maintaining the body‘s fluid balance and blood pressure, helping to make sure that the kidneys get the blood flow necessary for good kidney function. The renin-angiotensin-aldosterone system (RAAS) is overactive in various chronic kidney diseases, including Alport syndrome, and has been shown to promote scarring of the kidneys.
Medications that interfere with renin-angiotensin-aldosterone system hormones protect kidney function in animals and people with chronic kidney diseases.
Medications that interfere with renin-angiotensin-aldosterone system hormones include:
- Angiotensin converting enzyme (ACE) inhibitors — these medications block the production of angiotensin 2, the active form of angiotensin,
- Angiotensin receptor blockers (ARBs) — these medications block the action of angiotensin 2,
- Aldosterone inhibitors — these medications block the action of aldosterone.
Both angiotensin converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs) have been shown to slow down the loss of kidney function in mice with Alport syndrome. People who start taking an ACE inhibitor while their kidney function is still normal are older when they develop kidney failure than Alport patients who don‘t receive ACE inhibitors or are started on ACE inhibitors after they have started to lose kidney function. ACE inhibitors, ARBs and aldosterone inhibitors all reduce elevated urine protein levels in people with Alport syndrome.
Due to the rarity of Alport syndrome, treatment trials that have been tested on a large group of patients are lacking 8. Various treatments have been reported in the medical literature as part of single case reports or small series of patients. Treatment trials would be very helpful to determine the long-term safety and effectiveness of specific medications and treatments for individuals with this disorder. Clinical guideline recommendations have been published 9, 10 that discuss the treatment of Alport syndrome, including information on identifying and treating children with a high risk of developing early-onset renal failure.
Angiotensin-converting enzyme (ACE) inhibitors have been used to treat individuals with Alport syndrome. This off-label use may not be appropriate for all affected individuals and several factors must be considered before starting the therapy such as baseline kidney function, family history, and specific symptoms present 8. ACE inhibitors may be given when elevated levels of protein are detectable in the urine (overt proteinuria) in certain cases. These drugs are blood pressure medications that prevent (inhibit) an enzyme in the body from producing angiotensin 2. Angiotensin 2 is a chemical that acts to narrow blood vessels and can raise blood pressure. ACE inhibitors in individuals with Alport syndrome have been shown to reduce proteinuria and slow the progression of kidney disease, delaying the onset of renal failure.
Some individuals do not respond to or cannot tolerate ACE inhibitors. These individuals may be treated with drugs known as angiotensin receptor blockers (ARBs). ARBs prevent angiotensin 2 from binding to the corresponding receptors on blood vessels.
In the medical literature, ACE inhibitor therapy or ARB therapy is recommended in certain individuals with Alport syndrome who show overt proteinuria. These therapies may also be considered in affected individuals who have small amounts of albumin in the urine (microalbuminuria), but have not yet developed overt proteinuria. Albumin is a marker for kidney disease because the kidney may leak small amounts of albumin when damaged.
Angiotensin converting enzyme (ACE) inhibitors that have been used to treat Alport syndrome patients include, but are not limited to:
- Benazepril
- Cilazapril
- Enalapril (Vasotec)
- Fosinopril (Monopril)
- Lisinopril (Zestril, Prinivil)
- Perinopril
- Ramapril
- Quinapril (Accupril)
- Trandolapril
Angiotensin receptor blockers (ARBs) that have been used to treat Alport syndrome patients include, but are not limited to:
- Candesartan (Atacand)
- Epresartan
- Irbesartan
- Losartan (Cozaar)
- Telmisartan
- Valsartan
Spironolactone is medication used for aldosterone inhibition.
Side Effects
Both ACE inhibitors and ARBs can cause lightheadedness, especially when a person stands up quickly. Sometimes these medications need to be stopped or their doses lowered because of persistent lightheadedness or fainting, but this is unusual. ACE inhibitors and ARBs should not be taken by females who are pregnant or who can become pregnant because they can injure a developing fetus. ACE inhibitors, ARBs and aldosterone inhibitors can cause elevated blood potassium levels, but this is not a common problem in people who have normal kidney function.
A common side effect of ACE inhibitors is a cough, which may take up to a month to subside, and if one ACE inhibitor causes cough it is likely that the others will too. Coughing occurs less often with ARBs which may be used instead of an ACE inhibitor. The most serious, but rare, side effects are allergic reactions, a decrease in white blood cells, and swelling of tissues (angioedema).
Monitoring
In addition to taking ACE inhibitors and/or ARBs to control proteinuria (protein in the urine), Alport syndrome patients need to control their blood pressure and be monitored regularly. Monitoring tests include urine and blood chemistry testing. As a general recommendation, Alport syndrome patients without any kidney function problems should be monitored yearly, patients with moderate kidney function problems should be monitored every 6 months, and those with advanced kidney failure should be monitored every 1 to 3 months.
The Clinical Practice recommendations 11 advise testing of urine protein levels in children with Alport syndrome, starting at 1 year of age and then at least annually. The test is called the urine protein“creatinine ratio. Children with urine protein“creatinine ratios above 0.2 should be treated with an ACE inhibitor with the goal of reducing the protein“creatinine ratio as much as possible. This may require a gradual increase in the dose of the medication, and may be limited by side effects such as lightheadedness.
If a child is receiving the maximum dose of the ACE inhibitor and still has high urine protein levels, the Clinical Practice recommendations 11 suggest starting either an ARB or an aldosterone inhibitor (the choice is up to the child‘s doctor). Some nephrologists may prefer to use an ARB initially and add an ACE inhibitor if necessary to suppress proteinuria.
End Stage Renal Disease (ESRD)
Although treatment may slow the progression of kidney disease in Alport syndrome, there is no cure for the disorder and no treatment that can completely stop kidney decline. The rate of progression of kidney decline in individuals with Alport syndrome is highly variable. In most affected individuals, kidney function eventually deteriorates to the point where dialysis or a kidney transplant is required.
The options for a patient with End Stage Renal Disease (ESRD) are dialysis or a kidney transplant. Kidney transplantation has a very high success rate in people with Alport syndrome. Because Alport syndrome is a familial condition, related kidney donors must be carefully evaluated for this disease. Under most circumstances, any family member with a mutation in one of the type 4 collagen chain genes should not be a kidney donor.
Dialysis is a procedure in which a machine is used to perform some of the functions of the kidney — filtering waste products from the bloodstream, helping to control blood pressure, and helping to maintain proper levels of essential chemicals such as potassium. End-stage renal disease is not reversible so individuals will require lifelong dialysis treatment or a kidney transplant.
A kidney transplant is preferred for individuals with Alport syndrome over dialysis and has generally been associated with excellent outcomes in treating affected individuals. Some individuals with Alport syndrome will require a kidney transplant in adolescence or the teen-age years, while others may not require a transplant until they are in their 40s or 50s. Most females with XLAS and some individuals will ADAS syndrome never require a transplant. If a kidney transplant is indicated, great care must be taken in selecting living related kidney donors to ensure that affected individuals are not chosen. Alport syndrome does not recur in kidney transplants. However about 3-5% of transplanted Alport patients make antibodies to the normal collagen 4 proteins in the transplanted kidney, causing severe inflammation called post-transplant anti-GBM (Glomerular Basement Membrane) nephritis. It is an aggressive nephritis that often leads to failure of the transplant, so it is fortunate that it is an unusual problem. Most (not all) of patients who develop anti-GBM nephritis after kidney transplant have an underlying diagnosis of Alport syndrome.
The majority of anti-GBM cases occur in males with X-linked Alport syndrome who reach end-stage renal disease before the age of 40 and who have sensorineural deafness. The risk of post-transplant anti-GBM nephritis is very low in females with X-linked Alport syndrome, patients with detectable collagen IV alpha-5 chains in their skin or in their own kidneys, and in people who reach end-stage renal disease after age 40. Post-transplant anti-GBM nephritis has occurred in women with Autosomal Recessive Alport syndrome. It could also occur in a male with Autosomal Recessive Alport syndrome. It has not been reported in anyone with Autosomal Dominant Alport syndrome.
The diagnosis of post-transplant anti-GBM nephritis is made by transplant biopsy. Alport patients can be screened for antibodies to GBM in the blood, but a negative test does not rule out post-transplant anti-GBM nephritis. About 75% of cases of post-transplant anti-GBM nephritis occur in the first year after transplant.
Signs of post-transplant anti-GBM nephritis include hematuria, proteinuria and rising serum creatinine. Alport patients who develop these abnormalities after transplant should be evaluated for post-transplant anti-GBM nephritis which includes a biopsy of the transplant.
Other Considerations
Patients with Alport syndrome should avoid drugs that are nephro-toxic or harmful to the kidneys. This includes over the counter medicines such as non-steroidal anti-inflammatory drugs (NSAIDs) containing aspirin, ibuprofen and naproxen, as well as some decongestants. Patients should speak with their Nephrologist to get guidance on medicines that should be avoided.
Hearing and vision should also be monitored every one to two years beginning in children, particularly boys, at 7 to 8 years of age and continued regularly. Hearing aids should be prescribed as needed.
Maintaining a healthy lifestyle and a balanced diet is also beneficial. Discussion of nutritional considerations, such as sodium reduction and moderating protein consumption, should be discussed with your doctor.
References- Alport syndrome. https://ghr.nlm.nih.gov/condition/alport-syndrome
- Alport Syndrome in Women and Girls. Clin J Am Soc Nephrol. 2016 Sep 7; 11(9): 1713–1720. doi: 10.2215/CJN.00580116 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5012472/
- Early angiotensin-converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy. Gross, Oliver et al. Kidney International , Volume 81 , Issue 5 , 494 – 501 https://www.kidney-international.org/article/S0085-2538(15)55330-1/pdf
- Watson S, Bush JS. Alport Syndrome. [Updated 2018 Dec 15]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK470419
- Fallerini C, Baldassarri M, Trevisson E, Morbidoni V, La Manna A, Lazzarin R, Pasini A, Barbano G, Pinciaroli AR, Garosi G, Frullanti E, Pinto AM, Mencarelli MA, Mari F, Renieri A, Ariani F. Alport syndrome: impact of digenic inheritance in patients management. Clin. Genet. 2017 Jul;92(1):34-44.
- Zhang Y, Ding J.Renal, auricular and ocular outcomes of Alport syndrome and their current management. Pediatr Nephrol. 2017.
- Alport syndrome. http://eyewiki.org/ALPORT_SYNDROME
- Alport syndrome. https://rarediseases.org/rare-diseases/alport-syndrome
- Kashtan CE, Ding J, Gregory M, et al. Clinical practice recommendations for the treatment of Alport syndrome: a statement of the Alport Syndrome Recommendation Collaborative. Pediatr Nephrol. 2013;28:5-11. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3505543
- Savige J, Gregory M, Gross O, et al. Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy. J Am Soc Nephrol. 2013;24:364-375. http://www.ncbi.nlm.nih.gov/pubmed/23349312
- Clinical practice recommendations for the treatment of Alport syndrome: a statement of the Alport Syndrome Research Collaborative. Kashtan, C.E., Ding, J., Gregory, M. et al. Pediatr Nephrol (2013) 28: 5. https://doi.org/10.1007/s00467-012-2138-4





{kind=link}