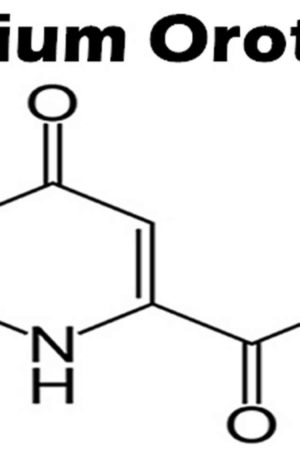

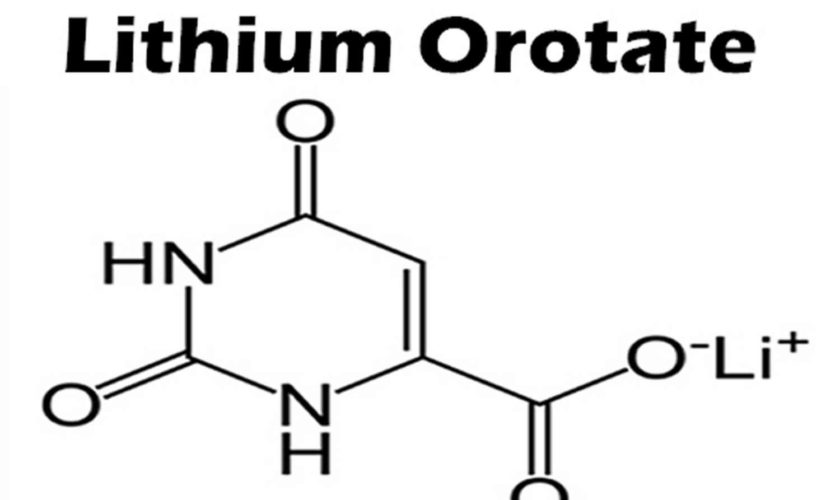
What is lithium orotate
Lithium orotate is a salt of orotic acid and lithium. Lithium orotate is widely available over the internet and marketed as a dietary supplement for conditions such as bipolar disorder, alcoholism, and aggression, although it is NOT U.S. Food and Drug Administration (FDA) approved for the treatment of any medical condition. Lithium orotate is available in some drug stores and health food stores under various brand names. Lithium orotate are advertised without regulation, and they are purchased and used without medical supervision or monitoring. The widespread availability of herbal and “dietary supplement” products from internet sources has increased the potential for poisonings. Patients who obtain Lithium orotate are subject to toxicity, drug-drug interactions, and other adverse effects.
- Proper medical diagnosis and a clear description of all possible treatment options should always be the first plan of action when treating mental disorders.
While lithium orotate is capable of providing lithium to the body, like lithium carbonate and other lithium salts, there are no systematic clinical study reviews supporting the efficacy of lithium orotate and it is only barely researched between 1973–1986 to treat certain medical conditions, such as alcoholism 1.
Animal models suggest that lithium orotate has similar pharmacokinetics, but the lithium orotate may achieve higher tissue concentrations at the same dosages than commonly prescribed for lithium carbonate and lithium citrate formulations 2. This may be secondary to lower renal clearance of the lithium orotate salt 3.
In 1973, Nieper 4 reported that lithium orotate contained 3.83 mg of elemental lithium per 100 mg and lithium carbonate contained 18.8 mg of elemental lithium per 100 mg. Nieper went on to claim that lithium did not dissolve from the orotate carrier until it passed through the blood–brain barrier 4; however, a 1976 study documented that lithium concentrations within the brains of rats were not statistically different between equivalent dosages of lithium from lithium orotate, lithium carbonate, or lithium chloride 5. While this study was conducted with rats, it directly contradicts the aforementioned assumptions made by Nieper and others 5. The pharmacokinetics of lithium orotate in human brains is poorly documented and further inquiry is needed to affirm that lithium concentrations in the brain are higher with lithium orotate. Major medical research has not been conducted on lithium orotate since the 1980s due to its patent status and the abundant availability of lithium carbonate. As previously stated, lithium intake appears to be effective even at low doses, and this may account for lithium orotates claimed effectiveness 6.
Do not use lithium without telling your doctor if you are pregnant. It could cause harm to the unborn baby. Use an effective form of birth control, and tell your doctor if you become pregnant during treatment. Early voluntary reports to international birth registries suggested an increase in cardiovascular malformations, especially for Ebstein’s anomaly, with first trimester use of Lithium. Subsequent case-control and cohort studies indicate that the increased risk for cardiac malformations is likely to be small; however, the data are insufficient to establish a drug-associated risk. There are concerns for maternal and/or neonatal Lithium toxicity during late pregnancy and the postpartum period. Published animal developmental and toxicity studies in mice and rats report an increased incidence of fetal mortality, decreased fetal weight, increased fetal skeletal abnormalities, and cleft palate (mouse fetuses only) with oral doses of Lithium that produced serum concentrations similar to the human therapeutic range. Other published animal studies report adverse effects on embryonic implantation in rats after Lithium administration. The background risk of major birth defects and miscarriage for the indicated population(s) is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
You should not breast-feed while using this medicine. Limited published data reports the presence of Lithium carbonate in human milk with breast milk levels measured at 0.12 to 0.7 mEq or 40 to 45% of maternal plasma levels. Infants exposed to Lithium during breastfeeding may have plasma levels that are 30 to 40% of maternal plasma levels. Signs and symptoms of Lithium toxicity such as hypertonia, hypothermia, cyanosis, and ECG changes have been reported in some breastfed neonates and infants. Increased prolactin levels have been measured in lactating women, but the effects on milk production are not known. Breastfeeding is not recommended with maternal Lithium use; however, if a woman chooses to breastfeed, the infant should be closely monitored for signs of Lithium toxicity. Discontinue breastfeeding if a breastfed infant develops Lithium toxicity. Consider regular monitoring of Lithium levels and thyroid function in a breastfed infant.
Is lithium orotate safe?
Lithium orotate’s safety remains in question. There have been numerous case reports of patients requiring medical attention after taking lithium orotate supplements 7, 8, 9.
Orotic acid can be mutagenic in mammalian somatic cells. It is also mutagenic for bacteria and yeast 10.
An 18-year-old woman presented to our emergency department after ingesting 18 tablets of Find Serenity Now®; each tablet contained, according to the listing, 120 mg of lithium orotate [3.83 mg of elemental lithium per 100 mg of (organic) lithium orotate compared to 18.8 mg of elemental lithium per 100 mg of (inorganic) lithium carbonate] 7. The patient complained of nausea and reported one episode of emesis (vomiting). Her examination revealed normal vital signs. The only finding was a mild tremor without rigidity. Almost 90 minutes after the ingestion, her serum lithium level was 0.31 mEq/L, a urine drug screen was negative, and an electrocardiogram (ECG) showed a normal sinus rhythm. The patient received intravenous fluids and an anti-emetic; one hour later, her repeat serum lithium level was 0.40 mEq/L. After 3 hours of observation, nausea and tremor were resolved, and she was subsequently transferred to a psychiatric hospital for further care.
Lithium’s widespread use and its narrow therapeutic index can lead to adverse effects in up to 90% of all users 11. Most toxicity is mild and includes lethargy, vomiting, ataxia, and myoclonus, but massive, acute ingestions or severe chronic toxicity can lead to coma or seizures 7. Other adverse effects include thyroid and parathyroid abnormalities, serotonergic crisis, cardiovascular abnormalities, and nephrogenic diabetes insipidus 12.
Onset and severity of symptoms vary upon the timing of ingestion and product formulation. The risk of toxicity increases with increased age, renal insufficiency, hyponatremia, volume depletion, drug-drug interactions, and comorbidities or co-ingestions 13. Significant toxicity tends to occur when levels are well above the upper therapeutic level (1.5 mEq/L); however, lithium’s variable absorption and delayed tissue concentrations make interpretation of serum levels difficult. Toxicity may also occur at lower levels, especially in the setting of chronic use 14.
Lithium orotate vs Lithium carbonate
Lithium Carbonate is a white, light, alkaline powder with molecular formula Li2CO3 and molecular weight 73.89. Lithium Carbonate is the carbonate salt of lithium, a soft alkali metal, with antimanic and hematopoietic activities. Lithium interferes with transmembrane sodium exchange in nerve cells by affecting sodium, potassium-stimulated adenosine triphosphatase (Na+, K+-ATPase); alters the release of neurotransmitters; affects cyclic adenosine monophosphate (cAMP) concentrations; and blocks inositol metabolism resulting in depletion of cellular inositol and inhibition of phospholipase C-mediated signal transduction. While lithium has no psychotropic effects in normal individuals, it has potent mood stabilizing properties in patients with bipolar disorders, mania and recurrent depression. The mechanism of action of lithium is unknown, but is thought to be mediated by its replacement of sodium ions and disruption of membrane potentials in the central nervous system. It may also act by differential effects on neurotransmitter induced depolarization of membranes or interference with phosphatidylinositol pathways. In addition, lithium stimulates granulocytopoiesis and appears to increase the level of pluripotent hematopoietic stem cells by stimulating the release of hematopoietic cytokines and/or directly acting on hematopoietic stem cells.
Lithium was approved for use in bipolar illness for the treatment of mania for more than 50 years in the United States since 1970 and it is still widely used for this indication. Manic symptoms include hyperactivity, rushed speech, poor judgment, reduced need for sleep, aggression, and anger. Lithium also helps to prevent or lessen the intensity of manic episodes. Lithium has also been used in therapy of schizophrenia, alcohol dependence, attention deficit disorder and migraine headaches. Lithium is available as capsules or tablets of 150, 300, 450 and 600 mg in generic forms as well in several brand names including Carbolith, Duralith and Eskalith. A typical maintenance dose regimen is 600 to 900 mg daily. Lithium levels are generally monitored because of the narrow therapeutic window between toxicity and effectiveness aiming for levels between 0.6 and 1.2 mEq/L in chronic situations (higher in acute). Common side effects include metallic taste, nausea, tremor, polyuria, polydipsia and weight gain. Uncommon side effects include hypothyroidism.
Concurrent administration of lithium carbonate and potassium iodide or other iodine-containing compounds may enhance hypothyroid and goitrogenic effects of either drug 15.
Lithium can cause side effects that may impair your thinking or reactions. Be careful if you drive or do anything that requires you to be awake and alert.
Call your doctor at once if you have any early signs of lithium toxicity, such as nausea, vomiting, diarrhea, drowsiness, muscle weakness, tremor, lack of coordination, blurred vision, or ringing in your ears.
Lithium is not approved for use by anyone younger than 12 years old.
Liver test abnormalities have been reported to occur in a small proportion of patients on long term therapy with lithium. These abnormalities are usually asymptomatic and transient, reversing even with continuation of medication. Instances of more marked elevations in serum aminotransferases have been reported in patients taking overdoses of lithium, but the other metabolic and systemic effects of lithium overdose generally overshadow hepatic adverse effects. Lithium has not been associated with instances of clinically apparent acute liver injury with jaundice.
Before taking lithium
You should not use lithium if you are allergic to it.
Lithium may be used to treat manic episodes associated with bipolar disorder; however, there is a fine line between too much and too little and ongoing monitoring is needed to prevent lithium toxicity.
Obtain serum Lithium concentration assay after 4 days, drawn 12 hours after the last oral dose. Adjust daily dosage based on serum Lithium concentration and clinical response. Fine hand tremor, polyuria and mild thirst may occur during initial therapy for the acute manic phase, and may persist throughout treatment. Transient and mild nausea and general discomfort may also appear during the first few days of Lithium administration. These adverse reactions may subside with continued treatment, concomitant administration with food, temporary reduction or cessation of dosage.
- Lithium is usually taken two to three times daily with food.
- There is a fine line between too much and too little lithium. Always take lithium exactly as directed and go to your scheduled appointments. Never take any herbal supplements or over the counter remedies without consulting your doctor or pharmacist first as many drugs may affect blood levels of lithium.
- If you miss a dose of lithium, take it as soon as you remember. If it is close to your next dose, do not double up on the dose.
- Do not crush or chew extended-release tablets; swallow whole.
- Too much caffeine may decrease the amount of lithium in your body.
- Lithium may affect your mental alertness or make you drowsy. Do not drive until you know how lithium will affect you. Avoid alcohol.
- Ensure you keep adequately hydrated while taking lithium and maintain an adequate salt intake (your doctor will discuss this requirement). The risk of side effects of lithium is increased if you are dehydrated, or if you are excessively hydrated. Excessive sweating or diarrhea may also upset the balance of lithium in the blood.
- Contact your doctor if you become ill or have an infection as your dosage of lithium may need to be altered or temporarily discontinued.
- Seek urgent medical attention if symptoms similar to diabetes (such as excessive thirst or excessive urine production), or serotonin syndrome ( occur.
- Stop lithium and contact your doctor urgently if symptoms of lithium toxicity such as diarrhea, vomiting, tremor, drowsiness, muscle weakness or confusion occur.
- Seek urgent medical advice if symptoms consistent with serotonin syndrome (such as agitation, hallucinations, fast heart rate, dizziness, flushing, nausea, diarrhea) develop.
- You will need to go for regular blood tests while you are taking lithium to ensure that the dosage is appropriate for you.
- May affect your mental and physical abilities so be careful driving or operating machinery until you know how lithium affects you.
- Do not take any other medications, including those bought over the counter, without first checking with your doctor or pharmacist that they are compatible with lithium.
Tell your doctor if you have ever had:
- an abnormal electrocardiograph or ECG (sometimes called an EKG);
- fainting spells;
- a family history of death before age 45;
- kidney disease;
- heart disease;
- a debilitating illness;
- a thyroid disorder;
- low levels of sodium in your blood; or
- if you are dehydrated.
Some medicines can interact with lithium and cause a serious condition called serotonin syndrome. Be sure your doctor knows if you also take stimulant medicine, opioid medicine, herbal products, or medicine for depression, mental illness, Parkinson’s disease, migraine headaches, serious infections, or prevention of nausea and vomiting. Ask your doctor before making any changes in how or when you take your medications.
Other drugs that will affect lithium
Tell your doctor about all your current medicines. Many drugs can interact with lithium, especially:
- a diuretic or “water pill”;
- fluoxetine (Prozac);
- metronidazole;
- potassium iodide thyroid medication;
- heart or blood pressure medication;
- seizure medicine; or
- nonsteroidal anti-inflammatory drugs – aspirin, ibuprofen (Advil, Motrin), naproxen (Aleve), celecoxib, diclofenac, indomethacin, meloxicam, and others.
This list is not complete and many other drugs may interact with lithium. This includes prescription and over-the-counter medicines, vitamins, and herbal products. Not all possible drug interactions are listed here.
How long does it take for lithium to work?
A reduction in manic symptoms should be noticed within one to three weeks. Your doctor will determine if your symptoms have improved enough to warrant lithium long-term.
Lithium is completely absorbed in the gastrointestinal tract with peak levels occurring 0.25 to 3 hours after oral administration of immediate-release preparations and two to six hours after sustained-release preparations.
Lithium dosing information
Adult Dose of Lithium for Mania
Comments:
- Dosing must be individualized according to serum levels and the response to treatment.
- Obtain serum Lithium concentrations regularly until the serum concentration and clinical condition of the patient has stabilized. Adjust daily dosage based on serum Lithium concentration and clinical response.
- Alternative extended release formulation doses are 600 mg 3 times a day (acute control) and 300 mg 3 to 4 times a day (long-term control).
Uses:
- Treatment of manic episodes of bipolar disorder
- Maintenance treatment for individuals with bipolar disorder
Acute Control:
- Titrate to serum Lithium concentrations between 0.8 and 1.2 mEq/L.
- Usual dose: 1800 mg/day
- Extended release formulations: 900 mg orally in the morning and at nighttime
- Regular release formulations: 600 mg orally 3 times a day, in the morning, afternoon, and nighttime
Long-term Control:
- Titrate to serum Lithium concentrations between 0.8 and 1 mEq/L
- Maintenance dose: 900 to 1200 mg/day
- Extended release formulations: 600 mg orally in the morning and at nighttime
- Regular release formulations: 300 mg orally 3 to 4 times a day
Adult Dose of Lithium for Bipolar Disorder
Comments:
- Dosing must be individualized according to serum levels and the response to treatment.
- Alternative extended release formulation doses are 600 mg 3 times a day (acute control) and 300 mg 3 to 4 times a day (long-term control).
Uses:
- Treatment of manic episodes of bipolar disorder
- Maintenance treatment for individuals with bipolar disorder
Acute Control:
- Usual dose: 1800 mg/day
- Extended release formulations: 900 mg orally in the morning and at nighttime
- Regular release formulations: 600 mg orally 3 times a day, in the morning, afternoon, and nighttime
Long-term Control:
- Maintenance dose: 900 to 1200 mg/day
- Extended release formulations: 600 mg orally in the morning and at nighttime
- Regular release formulations: 300 mg orally 3 to 4 times a day
Pediatric 12 years and older dose of Lithium for Mania
Comments:
- Dosing must be individualized according to serum levels and the response to treatment.
- Alternative extended release formulation doses are 600 mg 3 times a day (acute control) and 300 mg 3 to 4 times a day (long-term control).
- Maintenance therapy reduces the frequency of manic episodes and diminishes the intensity of the episodes.
Uses:
- Treatment of manic episodes of bipolar disorder
- Maintenance treatment for individuals with bipolar disorder
12 years and older acute control:
Usual dose: 1800 mg/day
- Extended release formulations: 900 mg orally in the morning and at nighttime
- Regular release formulations: 600 mg orally 3 times a day, in the morning, afternoon, and nighttime
Long-term Control:
- Maintenance dose: 900 to 1200 mg/day
- Extended release formulations: 600 mg orally in the morning and at nighttime
- Regular release formulations: 300 mg orally 3 to 4 times a day
Pediatric 12 years and older dose of Lithium for Bipolar Disorder
Comments:
- Dosing must be individualized according to serum levels and the response to treatment.
- Alternative extended release formulation doses are 600 mg 3 times a day (acute control) and 300 mg 3 to 4 times a day (long-term control).
- Maintenance therapy reduces the frequency of manic episodes and diminishes the intensity of the episodes.
Uses:
- Treatment of manic episodes of bipolar disorder
- Maintenance treatment for individuals with bipolar disorder
12 years and older acute control:
- Usual dose: 1800 mg/day
- Extended release formulations: 900 mg orally in the morning and at nighttime
- Regular release formulations: 600 mg orally 3 times a day, in the morning, afternoon, and nighttime
Long-term Control:
- Maintenance dose: 900 to 1200 mg/day
- Extended release formulations: 600 mg orally in the morning and at nighttime
- Regular release formulations: 300 mg orally 3 to 4 times a day
Lithium side effects
Get emergency medical help if you have signs of an allergic reaction to lithium: hives; difficulty breathing; swelling of your face, lips, tongue, or throat.
If you are between the ages of 18 and 60, take no other medication or have no other medical conditions, side effects you are more likely to experience include:
- Fine hand tremor, frequent urination, and mild thirst commonly occur during lithium initiation. Sometimes these effects may persist throughout treatment.
- Nausea during initiation is common but usually subsides with continued administration.
- Diarrhea, vomiting, drowsiness, muscular weakness, loss of appetite and coordination difficulties may be an early sign of lithium toxicity. Dizziness, blurred vision, ringing in the ears and excessive production of dilute urine may occur with higher (toxic) lithium levels. Seek urgent medical advice.
- Lithium may also cause irregular heartbeat, drying and thinning of hair, alopecia, dry mouth, weight gain, itchiness, and other side effects. Long-term use may lead to hypothyroidism or other thyroid problems.
- Dosing may be difficult because there is not much of a margin between an adequate dose of lithium and a toxic dose.
- Monitoring is required, particularly during therapy initiation but also long-term.
- Not suitable for people with significant renal or cardiovascular disease, in those who are frail, dehydrated, taking diuretics or with low levels of sodium. Not recommended for children aged less than 12.
- Full effects of lithium in pregnancy have not been fully determined so advice is to avoid lithium, particularly in the first trimester.
- May interact with several other medications including diuretics (water pills), NSAIDs and ACE inhibitors.
- Interaction or overdosage may cause serotonin syndrome (symptoms include mental status changes [such as agitation, hallucinations, coma, delirium]), fast heart rate, dizziness, flushing, muscle tremor or rigidity and stomach symptoms (including nausea, vomiting, and diarrhea).
Notes: In general, seniors or children, people with certain medical conditions (such as liver or kidney problems, heart disease, diabetes, seizures) or people who take other medications are more at risk of developing a wider range of side effects.
Call your doctor at once if you have:
- a light-headed feeling, like you might pass out;
- irregular heartbeats, shortness of breath;
- fever, increased thirst or urination;
- weakness, dizziness or spinning sensation;
- confusion, memory problems, hallucinations;
- uncontrolled muscle movements, slurred speech;
- loss of bowel or bladder control;
- a seizure (blackout or convulsions);
- dehydration symptoms – feeling very thirsty or hot, being unable to urinate, heavy sweating, or hot and dry skin; or
- increased pressure inside the skull – severe headaches, ringing in your ears, dizziness, nausea, vision problems, pain behind your eyes.
Seek medical attention right away if you have symptoms of serotonin syndrome, such as: agitation, hallucinations, fever, sweating, shivering, fast heart rate, muscle stiffness, twitching, loss of coordination, nausea, vomiting, or diarrhea.
Common lithium side effects may include:
- drowsiness;
- tremors in your hands;
- dry mouth, increased thirst or urination;
- nausea, vomiting, loss of appetite, stomach pain;
- changes in your skin or hair;
- cold feeling or discoloration in your fingers or toes;
- feeling uneasy; or
- impotence, loss of interest in sex.
This is not a complete list of side effects and others may occur. Call your doctor for medical advice about side effects.
Nervous system
Frequency not reported: Abnormal reflex convulsions, acute dystonia, ataxia, benign intracranial hypertension, blackout spells, choreoathetotic movements, cerebellar syndrome, clonic movements of whole limbs, coarse tremor of the extremities and lower jaw, cogwheel rigidity, coma, convulsions, diffuse slowing of EEG, dizziness, downbeat nystagmus, drowsiness, dysarthria, dysgeusia/taste distortion, encephalopathy, encephalopathic syndrome, epileptiform seizures, extrapyramidal syndrome, fine hand tremor, giddiness, headache, hyperactive deep tendon reflexes, hypertonicity, impaired consciousness, lack of coordination, lethargy, metallic/salty taste, myoclonus, nystagmus, peripheral sensorimotor neuropathy, poor memory, potentiation and disorganization of EEG background rhythm, pseudotumor cerebri (increased intracranial pressure and papilledema), psychomotor retardation, seizures, serotonin syndrome, slowed intellectual functioning, slurred speech/speech disorder, somnolence, startle response, stupor, tendency to sleep, tongue movements, transient electroencephalogram (EEG), tremor, vertigo, widening of EEG frequency spectrum
- Drowsiness and lack of coordination may be early signs of lithium toxicity, and may occur at lithium levels below 2 mEq/L.
- Ataxia and giddiness occurred at levels above 2 mEq/L.
- Fine hand tremor may occur during initial therapy for the acute manic phase, and may persist during therapy.
- The development of transient EEG changes, headache, dysgeusia/taste distortion, and metallic taste were unrelated to dosage.
- Peripheral neuropathy may occur in patients on long-term treatment, but is usually reversible after discontinuation of therapy.
Cardiovascular
The development of transient ECG changes, chest tightness, and edematous swelling of ankles/wrists were unrelated to dosage.
Painful discoloration of the fingers/toes and coldness of extremities (resembling Raynaud’s syndrome) occurred within one day of initiation; the patient recovered after discontinuation. The exact mechanism for this side effect is unknown.
Frequency not reported: Atrioventricular block, bradycardia, cardiac arrhythmia, cardiomyopathy, chest tightness, conduction disturbance, ECG changes, edema, hypotension, inversion of T-waves, isoelectricity of ECG, peripheral circulatory collapse, peripheral edema/edematous swelling of ankles or wrists, peripheral vasculopathy, QT prolongation, Raynaud’s phenomena/syndrome, reversible flattening of ECG, sinus node dysfunction with severe bradycardia and/or sinoatrial block (may result in syncope), transient ECG changes, unmasking of Brugada syndrome, ventricular tachyarrhythmia
Gastrointestinal
Frequency not reported: Abdominal pain/discomfort, constipation, dental caries, diarrhea, dry mouth, excessive salivation, flatulence, gastritis, incontinence of feces, indigestion, nausea/transient and mild nausea, salivary gland swelling, swollen lips, vomiting
- Diarrhea and vomiting may be early signs of lithium toxicity, and may occur at lithium levels below 2 mEq/L.
- Transient and mild nausea may occur within the first few days of therapy.
- The development of metallic/salty taste, dental caries, and swollen lips were unrelated to dosage.
Dermatologic
Frequency not reported: Acne/acneform eruptions, alopecia, anesthesia of skin, chronic folliculitis/folliculitis, cutaneous ulcers, drying and thinning of hair, generalized pruritus with/without rash, papular skin disorders, pruritus, psoriasis onset/exacerbation, urticaria, xerosis cutis
The development of generalized pruritus with/without rash and cutaneous ulcers were unrelated to dosage.
Endocrine
Frequency not reported: Diffuse nontoxic goiter with/without hypothyroidism, euthyroid goiter, hyperparathyroidism, hyperthyroidism, hypothyroidism (including myxedema), iodine 131 uptake increased, lower T3 and T4 levels, thyrotoxicosis
Hyperthyroidism has been rarely reported, and may persist after discontinuation of treatment.
Hyperparathyroidism may persist after discontinuation of treatment.
The development of diffuse nontoxic goiter with/without hypothyroidism and hyperparathyroidism were unrelated to dosage.
Musculoskeletal
Muscular weakness develops early in lithium toxicity, and may occur at lithium levels below 2 mEq/L.
Muscle hyperirritability includes fasciculations, twitching, clonic movements of whole limbs.
The development of swollen/painful joints and polyarthralgia were unrelated to dosage.
Frequency not reported: Arthralgia/polyarthralgia, muscle hyperirritability, muscular weakness, myalgia, myasthenia gravis, myoclony, rhabdomyolysis, swollen/painful joints, twitching
Renal
Frequency not reported: Decreased creatinine clearance, glycosuria, histological renal changes with interstitial fibrosis, lithium-induced chronic kidney disease, microcysts, nephrogenic diabetes insipidus, nephrotic syndrome, oliguria, renal dysfunction
Diabetes insipidus may persist after discontinuation of treatment.
Histological renal changes with interstitial fibrosis occurred in patients on prolonged treatment, and was usually reversible upon discontinuation. Long-term treatment may cause permanent kidney changes and impairment of renal function; high serum concentrations and/or acute lithium toxicity may worsen these changes.
Metabolic
Frequency not reported: Anorexia, dehydration, excessive weight gain, hypercalcemia, hypermagnesemia, hyponatremia, polydipsia, thirst/mild thirst, transient hyperglycemia/hyperglycemia, weight loss
The development of transient hyperglycemia, hypercalcemia, and excessive weight gain were unrelated to dosage.
Other side effects
Tinnitus occurred at levels above 2 mEq/L.
Mild thirst may occur during initial therapy for the acute manic phase, and may persist during therapy; in some cases, thirst resembled diabetes insipidus. The development of thirst was unrelated to dosage.
General discomfort may also appear within the first few days of therapy.
The development of fever was unrelated to dosage.
Frequency not reported: Fall, fasciculations, fatigue, feeling dazed, fever, general discomfort, lithium toxicity, tinnitus
Genitourinary
Frequency not reported: Albuminuria, impotence/sexual dysfunction, incontinence of urine, large output of dilute urine, lithium-induced polyuria/polyuria
At levels above 2 mEq/L, patients excreted a large output of dilute urine.
Polyuria may occur during initial therapy for the acute manic phase, and may persist during therapy; in some cases, polyuria resembled diabetes insipidus. The development of polyuria was unrelated to dosage.
The development of albuminuria was unrelated to dosage.
Psychiatric
Frequency not reported: Confusion, delirium, hallucinations, restlessness, tics, worsening of organic brain syndromes
The worsening of organic brain syndromes was unrelated to dosage.
Hypersensitivity
Frequency not reported: Allergic rashes, angioedema
Ocular
Frequency not reported: Blindness, blurred vision, enlargement of the blind spot, exophthalmos, optic atrophy, transient scotomata/scotoma, visual field constriction
Blurred vision occurred at levels above 2 mEq/L.
Oncologic
Frequency not reported: Collecting duct renal carcinoma, oncocytoma
Collecting duct renal carcinoma occurred in patients on long-term therapy.
Hematologic
Frequency not reported: Leukocytosis
The development of leukocytosis was unrelated to dosage.
Lithium-Induced Polyuria
Chronic Lithium treatment may be associated with diminution of renal concentrating ability, occasionally presenting as nephrogenic diabetes insipidus, with polyuria and polydipsia. The concentrating defect and natriuretic effect characteristic of this condition may develop within weeks of Lithium initiation. Lithium can also cause renal tubular acidosis, resulting in hyperchloremic metabolic acidosis. Such patients should be carefully managed to avoid dehydration with resulting Lithium retention and toxicity. This condition is usually reversible when Lithium is discontinued, although for patients treated with long-term Lithium, nephrogenic diabetes insipidus may be only partly reversible upon discontinuation of Lithium. Amiloride may be considered as a therapeutic agent for Lithium-induced nephrogenic diabetes insipidus.
Hyponatremia
Lithium can cause hyponatremia by decreasing sodium reabsorption by the renal tubules, leading to sodium depletion. Therefore, it is essential for patients receiving Lithium treatment to maintain a normal diet, including salt, and an adequate fluid intake (2500 to 3000 mL) at least during the initial stabilization period. Decreased tolerance to Lithium has also been reported to ensue from protracted sweating or diarrhea and, if such occur, supplemental fluid and salt should be administered under careful medical supervision and Lithium intake reduced or suspended until the condition is resolved. In addition, concomitant infection with elevated temperatures may also necessitate a temporary reduction or cessation of medication.
Symptoms are also more severe with faster-onset hyponatremia. Mild hyponatremia (i.e., serum Na > 120 mEq/L) can be asymptomatic. Below this threshold, clinical signs are usually present, consisting mainly of changes in mental status, such as altered personality, lethargy, and confusion. For more severe hyponatremia (serum Na < 115 mEq/L), stupor, neuromuscular hyperexcitability, hyperreflexia, seizures, coma, and death can result. During treatment of hyponatremia, serum sodium should not be elevated by more than 10 to 12 meq/L in 24 hours, or 18 meq/L in 48 hours. In the case of severe hyponatremia where severe neurologic symptoms are present, a faster infusion rate to correct serum sodium concentration may be needed. Patients rapidly treated or with serum sodium <120mEq/L are more at risk of developing osmotic demyelination syndrome (previously called central pontine myelinolysis). Occurrence is more common among patients with alcoholism, undernutrition, or other chronic debilitating illness. Common signs include flaccid paralysis, dysarthria. In severe cases with extended lesions patients may develop a locked-in syndrome (generalized motor paralysis). Damage often is permanent. If neurologic symptoms start to develop during treatment of hyponatremia, serum sodium correction should be suspended to mitigate the development of permanent neurologic damage.
Lithium-Induced Chronic Kidney Disease
The predominant form of chronic renal disease associated with long-term Lithium treatment is a chronic tubulointerstitial nephropathy. The biopsy findings in patients with Lithium induced chronic tubulointerstitial nephropathy include tubular atrophy, interstitial fibrosis, sclerotic glomeruli, tubular dilation, and nephron atrophy with cyst formation. The relationship between renal function and morphologic changes and their association with Lithium treatment has not been established. Chronic tubulointerstitial nephropathy patients might present with nephrotic proteinuria (>3.0g/dL), worsening renal insufficiency and/or nephrogenic diabetes insipidus. Postmarketing cases consistent with nephrotic syndrome in patients with or without chronic tubulointerstitial nephropathy have also been reported. The biopsy findings in patients with nephrotic syndrome include minimal change disease and focal segmental glomerulosclerosis. The discontinuation of Lithium in patients with nephrotic syndrome has resulted in remission of nephrotic syndrome.
Kidney function should be assessed prior to and during Lithium treatment. Routine urinalysis and other tests may be used to evaluate tubular function (e.g., urine specific gravity or osmolality following a period of water deprivation, or 24-hour urine volume) and glomerular function (e.g., serum creatinine, creatinine clearance, or proteinuria). During Lithium treatment, progressive or sudden changes in renal function, even within the normal range, indicate the need for re evaluation of treatment.
Encephalopathic Syndrome
An encephalopathic syndrome, characterized by weakness, lethargy, fever, tremulousness and confusion, extrapyramidal symptoms, leukocytosis, elevated serum enzymes, BUN (blood urea nitrogen) and fasting blood glucose, has occurred in patients treated with Lithium and an antipsychotic. In some instances, the syndrome was followed by irreversible brain damage. Because of a possible causal relationship between these events and the concomitant administration of Lithium and antipsychotics, patients receiving such combined treatment should be monitored closely for early evidence of neurological toxicity and treatment discontinued promptly if such signs appear. This encephalopathic syndrome may be similar to or the same as neuroleptic malignant syndrome.
Serotonin Syndrome
Lithium can precipitate serotonin syndrome, a potentially life-threatening condition. The risk is increased with concomitant use of other serotonergic drugs (including selective serotonin reuptake inhibitors, serotonin and norepinephrine reuptake inhibitors, triptans, tricyclic antidepressants, fentanyl, tramadol, tryptophan, buspirone, and St. John’s Wort) and with drugs that impair metabolism of serotonin, i.e., MAOIs (monoamine oxidase inhibitors).
Serotonin syndrome signs and symptoms may include mental status changes (e.g., agitation, hallucinations, delirium, and coma), autonomic instability (e.g., tachycardia, labile blood pressure, dizziness, diaphoresis, flushing, hyperthermia), neuromuscular symptoms (e.g., tremor, rigidity, myoclonus, hyperreflexia, incoordination), seizures, and gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea).
Monitor all patients taking Lithium for the emergence of serotonin syndrome. Discontinue treatment with Lithium and any concomitant serotonergic agents immediately if the above symptoms occur, and initiate supportive symptomatic treatment. If concomitant use of Lithium with other serotonergic drugs is clinically warranted, inform patients of the increased risk for serotonin syndrome and monitor for symptoms.
Hypothyroidism or Hyperthyroidism
Lithium is concentrated within the thyroid and can inhibit thyroid synthesis and release which can lead to hypothyroidism. Where hypothyroidism exists, careful monitoring of thyroid function during Lithium stabilization and maintenance allows for correction of changing thyroid parameters, if any. Where hypothyroidism occurs during Lithium stabilization and maintenance supplemental thyroid treatment may be used. Paradoxically, some cases of hyperthyroidism have been reported including Grave’s disease, toxic multinodular goiter and silent thyroiditis.
Monitor thyroid function before the initiation of treatment, at three months and every six to twelve months while treatment is ongoing. If serum thyroid tests warrant concern, monitoring should occur more frequently.
Hypercalcemia and Hyperparathyroidism
Long-term Lithium treatment is associated with persistent hyperparathyroidism and hypercalcemia. When clinical manifestations of hypercalcemia are present, Lithium withdrawal and change to another mood stabilizer may be necessary. Hypercalcemia may not resolve upon discontinuation of Lithium, and may require surgical intervention. Lithium-induced cases of hyperparathyroidism are more often multiglandular compared to standard cases. False hypercalcemia due to plasma volume depletion resulting from nephrogenic diabetes insipidus should be excluded in individuals with mildly increased serum calcium. Monitor serum calcium concentrations regularly.
Unmasking of Brugada Syndrome
There have been postmarketing reports of a possible association between treatment with Lithium and the unmasking of Brugada Syndrome. Brugada Syndrome is a disorder characterized by abnormal electrocardiographic (ECG) findings and a risk of sudden death. Lithium should be avoided in patients with Brugada Syndrome or those suspected of having Brugada Syndrome. Consultation with a cardiologist is recommended if: (1) treatment with Lithium is under consideration for patients suspected of having Brugada Syndrome or patients who have risk factors for Brugada Syndrome, e.g., unexplained syncope, a family history of Brugada Syndrome, or a family history of sudden unexplained death before the age of 45 years, (2) patients who develop unexplained syncope or palpitations after starting Lithium treatment.
Pseudotumor Cerebri
Cases of pseudotumor cerebri (increased intracranial pressure and papilledema) have been reported with Lithium use. If undetected, this condition may result in enlargement of the blind spot, constriction of visual fields and eventual blindness due to optic atrophy. Consider discontinuing Lithium if this syndrome occurs.
Symptoms of lithium overdose
The toxic concentrations for Lithium (≥1.5 mEq/L) are close to the therapeutic range (0.8 to 1.2mEq/L). Some patients abnormally sensitive to Lithium may exhibit toxic signs at serum concentrations that are considered within the therapeutic range.
To reduce the risk of acute Lithium toxicity during treatment initiation, facilities for prompt and accurate serum Lithium determinations should be available before initiating treatment.
Lithium may take up to 24 hours to distribute into brain tissue, so occurrence of acute toxicity symptoms may be delayed.
Diarrhea, vomiting, drowsiness, muscular weakness and lack of coordination may be early signs of Lithium toxicity, and can occur at Lithium concentrations below 2.0 mEq/L. At higher concentrations, giddiness, ataxia, blurred vision, tinnitus and a large output of dilute urine may be seen. Serum Lithium concentrations above 3.0 mEq/L may produce a complex clinical picture involving multiple organs and organ systems, coma, and eventually death. Serum Lithium concentrations should not be permitted to exceed 2.0 mEq/L.
Neurological signs of Lithium toxicity range from mild neurological adverse reactions such as fine tremor, lightheadedness, and weakness; to moderate manifestations like apathy, drowsiness, hyperreflexia, muscle twitching, and slurred speech; and severe manifestations such as clonus, confusion, seizure, coma and death. Cardiac manifestations involve electrocardiographic changes, such as prolonged QT interval, ST and T-wave changes and myocarditis. Renal manifestations include urine concentrating defect, nephrogenic diabetes insipidus, and renal failure. Respiratory manifestations include dyspnea, aspiration pneumonia, and respiratory failure. Gastrointestinal manifestations include nausea, vomiting, and bloating.
No specific antidote for Lithium poisoning is known. Early symptoms of Lithium toxicity can usually be treated by reduction or cessation of Lithium, before restarting treatment at a lower dose 24 to 48 hours later.
- blurred vision
- clumsiness or unsteadiness
- convulsions (seizures)
- diarrhea
- drowsiness
- increase in the amount of urine
- lack of coordination
- loss of appetite
- muscle weakness
- nausea or vomiting
- ringing in the ears
- slurred speech
- trembling (severe)
The risk of acute toxicity is increased with a recent onset of concurrent illness or with the concomitant administration of drugs which increase Lithium serum concentrations by pharmacokinetic interactions. Additional risk factors for acute Lithium toxicity include acute ingestion, age-related decline in renal function, volume depletion and/or changes in electrolyte concentrations, especially sodium and potassium. Dose requirements during the acute manic phase are higher to maintain therapeutic serum concentrations and decrease when manic symptoms subside. The risk of Lithium toxicity is very high in patients with significant renal or cardiovascular disease, severe debilitation or dehydration, or sodium depletion, and for patients receiving prescribed medications that may affect kidney function, such as angiotensin converting enzyme inhibitors (ACE inhibitors), diuretics (loops and thiazides) and NSAIDs. For these patients, consider starting with lower doses and titrating slowly while frequently monitoring serum Lithium concentrations and signs of Lithium toxicity.
In severe cases of Lithium poisoning, the first and foremost goal of treatment consists of elimination of this ion from the patient. Administration of gastric lavage should be performed, but use of activated charcoal is not recommended as it does not significantly absorb Lithium ions. Hemodialysis is the treatment of choice as it is an effective and rapid means of removing Lithium in patients with severe toxicity. As an alternative option, urea, mannitol and aminophylline can induce a significant increase in Lithium excretion. Appropriate supportive care for the patient should be undertaken. In particular, patients with impaired consciousness should have their oral airway protected and it is critical to correct any volume depletion or electrolyte imbalance. Specifically, patients should be monitored to prevent hypernatremia while receiving normal saline and careful regulation of kidney function is of utmost importance.
Serum Lithium concentrations should be closely monitored as there may be a rebound in serum Lithium concentrations as a result of delayed diffusion from the body tissues. Likewise, during the late recovery phase, Lithium should be re- administered with caution taking into account the possible release of significant Lithium stores in body tissues.
- Lithium orotate in the treatment of alcoholism and related conditions. Alcohol. 1986 Mar-Apr;3(2):97-100. https://www.ncbi.nlm.nih.gov/pubmed/3718672[↩]
- Smith DF, Schou M. Kidney function and lithium concentrations of rats given an injection of lithium orotate or lithium carbonate. J Pharm Pharmacol. 1979;31(3):61–63.[↩]
- Kling MA, Manowitz P, Pollack IW. Rat brain and serum lithium concentrations after acute injections of lithium carbonate and orotate. J Pharm Pharmacol. 1978;30(6):368–370.[↩]
- The clinical applications of lithium orotate. A two years study. Agressologie. 1973;14(6):407-11. https://www.ncbi.nlm.nih.gov/pubmed/4607169[↩][↩]
- Smith DF. Lithium orotate, carbonate and chloride: pharmacokinetics, polyuria in rats. British Journal of Pharmacology. 1976;56(4):399-402. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1666891/pdf/brjpharm00512-0006.pdf[↩][↩]
- Low dosage lithium augmentation in venlafaxine resistant depression: an open-label study. Psychiatriki. 2012 Apr-Jun;23(2):143-8. https://www.ncbi.nlm.nih.gov/pubmed/22796912[↩]
- Pauzé DK, Brooks DE. Lithium toxicity from an internet dietary supplement. Journal of Medical Toxicology. 2007;3(2):61-62. doi:10.1007/BF03160910. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3550087/pdf/13181_2009_Article_BF03160910.pdf[↩][↩][↩]
- Possible dangers of a “nutritional supplement” lithium orotate. Ann Clin Psychiatry. 2013 Feb;25(1):71. https://www.ncbi.nlm.nih.gov/pubmed/23376874[↩]
- Kwan, D.; Beyene, J.; Shah, P. S. (1 November 2009). “Adverse Consequences of Internet Purchase of Pharmacologic Agents or Dietary Supplements”. Journal of Pharmacy Technology. 25 (6): 355–360. https://doi.org/10.1177/875512250902500602[↩]
- Orotic Acid, anhydrous MSDS. http://www.sciencelab.com/msds.php?msdsId=9926339[↩]
- Chen KP, Shen W, Lu ML. Implication of serum concentration monitoring in patients with lithium intoxication. Psychiatry and Clinical Neurosciences. 2004;58:25–29.[↩]
- Oakley P, Whyte I, Carter G. Lithium toxicity: An iatrogenic problem in susceptible individuals. Australian and New Zealand J of Psychiatry. 2001;35:833.[↩]
- Okusa MD, Crystal LJT. Clinical manifestations and management of acute lithium intoxication. American J of Med. 1994;97:383–388.[↩]
- Astruc B, Petit P, Abbar M. Overdose with sustainedrelease lithium preparations. EUR Psychiatry. 1999;14:172–174.[↩]
- Evaluations of Drug Interactions. 2nd ed. and supplements. Washington, DC: American Pharmaceutical Assn., 1976, 1978., p. 140[↩]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}