Complete androgen insensitivity syndrome
Complete androgen insensitivity syndrome (CAIS) is a rare condition that occurs when the body cannot use androgens (male sex hormones) at all, thus affecting the sexual development before birth and during puberty. People with complete androgen insensitivity syndrome are genetically male (one X and one Y chromosome in each cell) but do not respond to male hormones at all. As a result, they generally have normal female external genitalia and female breasts. However, they do not have a uterus or cervix so are unable to menstruate or conceive a child (infertile). They are typically raised as females and have a female gender identity. Affected individuals have male internal sex organs (testes) that are undescended, which means they are abnormally located in the pelvis or abdomen. Undescended testes have a small chance of becoming cancerous later in life if they are not surgically removed. People with complete androgen insensitivity syndrome also have sparse or absent hair in the pubic area and under the arms. Complete androgen insensitivity syndrome affects 2 to 5 per 100,000 people who are genetically male 1. Complete androgen insensitivity syndrome is one of the most common causes of disorders of sex development (DSD) 2.
Complete androgen insensitivity syndrome is caused by changes (mutations) in the androgen receptor (AR) gene and is inherited in an X-linked manner. Treatment and gender assignment can be a very complex issue, and must be individualized with each affected person. In general, surgery may be required to remove testes that are located in unusual places and estrogen replacement therapy can be prescribed after puberty 3.
Complete androgen insensitivity syndrome causes
Mutations in the AR (androgen receptor) gene situated in the Xq11-q12 region cause androgen insensitivity syndrome 4. The AR gene provides instructions for making a protein called an androgen receptor. Androgen receptors allow cells to respond to androgens, which are hormones (such as testosterone) that direct male sexual development. Androgens and androgen receptors also have other important functions in both males and females, such as regulating hair growth and sex drive. Mutations in the AR gene prevent androgen receptors from working properly, which makes cells less responsive to androgens or prevents cells from using these hormones at all. Depending on the level of androgen insensitivity, an affected person’s sex characteristics can vary from mostly female to mostly male.
Genitalia virilization physiologically occurs between the 8th and 14th weeks of gestation and is strictly linked to androgen action and AR function 5. Specifically, testosterone is responsible for the development of the epididymis, vas deferens and seminal vesicles from the Wolffian ducts, while other male genital structures, such as the prostate, penis, and scrotum, derive from the action of dihydrotestosterone 6. On the other hand, during puberty, both adrenal and ovarian androgens favour the development of pubic and axillary hair in females, while adrenal and testicular androgens control the deepening of the voice, the enlargement of the penis and hair pattern development in males 7. Additionally, the anti-Mullerian hormone (AMH) produced by the testes causes the regression of Mullerian ducts, preventing the formation of internal feminine genitalia 6. Therefore, any type of alterations in the androgen pathway could lead to impaired virilization.
Complete androgen insensitivity syndrome inheritance pattern
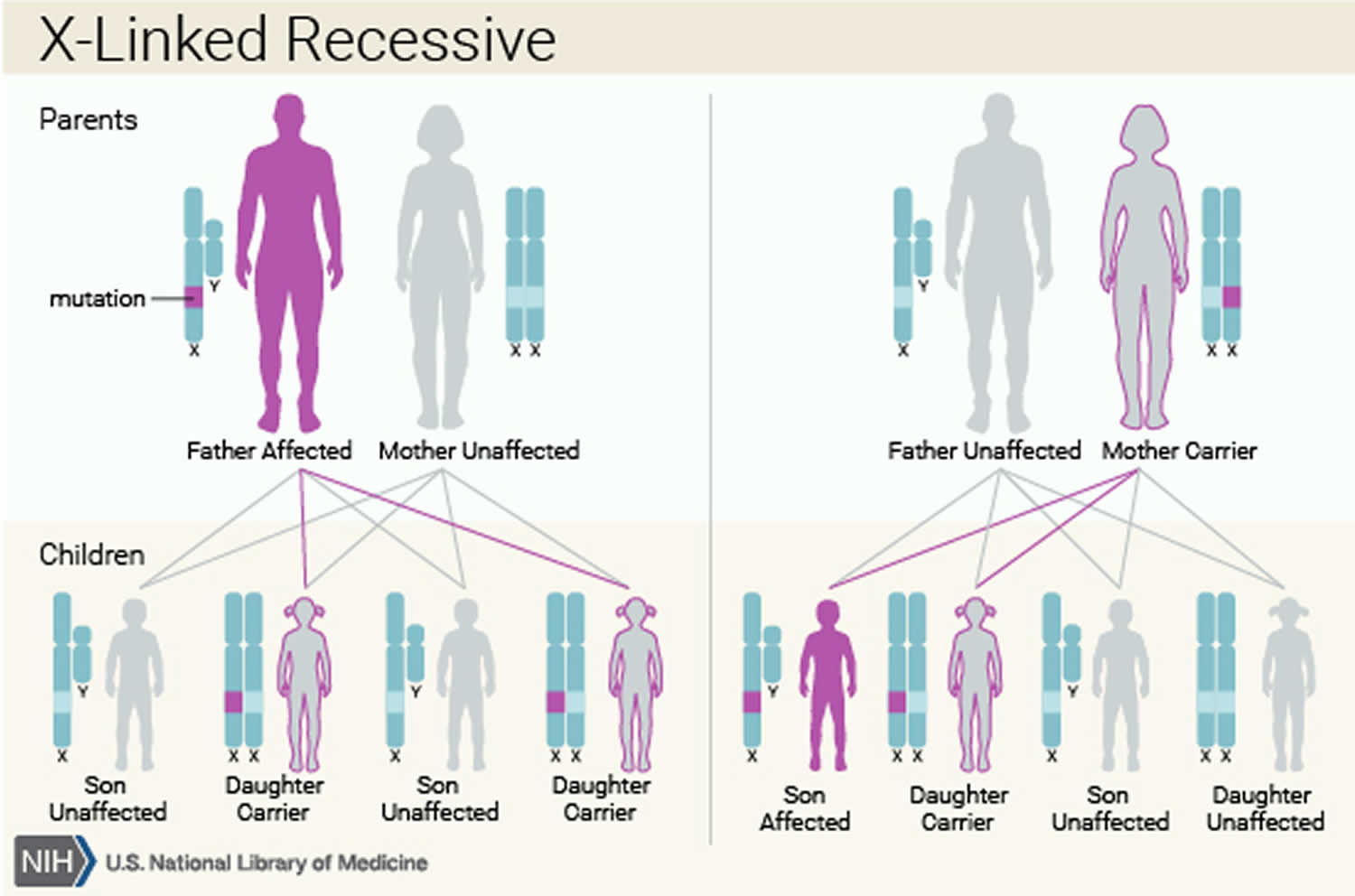
Complete androgen insensitivity syndrome is inherited in an X-linked recessive pattern. A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes in each cell. In genetic males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In genetic females (who have two X chromosomes), a mutation must be present in both copies of the gene to cause the disorder. Males are affected by X-linked recessive disorders much more frequently than females.
About 70% of all cases of androgen insensitivity syndrome are inherited from mothers who carry an altered copy of the AR gene on one of their two X chromosomes 7. The remaining cases result from a new mutation (de novo mutations) that can occur in the mother’s egg cell before the child is conceived or during early fetal development 7.
Figure 1. Complete androgen insensitivity syndrome X-linked recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Complete androgen insensitivity syndrome signs and symptoms
Complete androgen insensitivity syndrome is characterized by feminization of the external genitalia in a 46 XY individual with unresponsiveness to androgen action and normally developed but undescended testes 8. People with complete androgen insensitivity syndrome are genetically male (one X and one Y chromosome in each cell) but do not respond to male hormones at all. As a result, they generally have normal female external genitalia and female breasts. However, they do not have a uterus or cervix so are unable to menstruate (primary amenorrhea) or conceive a child (infertile). They are typically raised as females and have a female gender identity. Affected individuals have male internal sex organs (testes) that are undescended due to complete unresponsiveness towards androgen action, which means they are abnormally located in the pelvis or abdomen 9. Undescended testes have a small chance of becoming cancerous later in life if they are not surgically removed. People with complete androgen insensitivity syndrome also have sparse or absent hair in the pubic area and under the arms. Therefore, complete androgen insensitivity syndrome should be suspected in these cases, depending on the patient’s age: in a neonate with female external genitalia when a prenatal test showed a 46XY karyotype; in a female child who presents with an inguinal hernia, which is very rare in girls, or with labial swelling containing testis; and, finally, at puberty, in females with primary amenorrhea 10.
Internal female genitalia are also absent because the abdominal testes normally produce anti-Mullerian hormone (AMH), which impedes the development of the uterus, cervix and proximal vagina 9. However, the distal part of the vagina can be observed because it is not under AMH control, but it is always shorter than normal and blind-ending 10.
In patients with complete androgen insensitivity syndrome, puberty typically appears later and has a slower advance than in the general female population. However, breasts and female adiposity can develop regularly due to the action of estradiol deriving from the peripheral aromatization of testosterone 3. In contrast, pubic and axillary hair is absent or very rare because it mostly depends on androgen action. In regard to final height, complete androgen insensitivity syndrome patients are typically taller than the healthy female population due to the presence of the Y chromosome, which intervenes on statural growth independently of hormonal status 11. The typical hormone profile is characterized by a high level of luteinizing hormone (LH) above the usual reference range, while the follicle stimulating hormone (FSH) level is usually normal, probably due to gonadal inhibin regulation 12. Moreover, the basal testosterone value results are typically within the normal male range but increased relative to the female range, while the estradiol level is normal referring it to the male range but in the lower range for females 12.
Complete androgen insensitivity syndrome diagnosis
Individuals with complete androgen insensitivity syndrome have normal female external genitalia with absence of female internal genitalia. They typically present either before puberty with masses in the inguinal canal that are subsequently identified as testes or at puberty with primary amenorrhea and sparse to absent pubic or axillary hair. Breasts and female adiposity develop normally. Sexual identity and orientation are typically female and heterosexual. The diagnosis is based on clinical presentation, laboratory tests and imaging in a female with a 46 XY karyotype and confirmed throughout AR gene analysis.
Complete androgen insensitivity syndrome diagnosis is based on the presence of female external genitalia in a 46XY individual, with normally developed but undescended testes and complete unresponsiveness of target tissues to androgens. Pelvic ultrasound or magnetic resonance imaging (MRI) could be helpful in confirming the absence of Mullerian structures, revealing the presence of a blind-ending vagina and identifying testes.
Supportive laboratory findings:
- Normal 46XY karyotype
- Evidence of normal or increased synthesis of testosterone (T) by the testes
- Evidence of normal conversion of testosterone to dihydrotestosterone (DHT)
- Evidence of normal or increased luteinizing hormone (LH) production by the pituitary gland
- In complete androgen insensitivity syndrome, but not in partial androgen insensitivity syndrome (PAIS): possible reduction in postnatal (0-3 months) surge in serum LH and serum T concentrations 13
- In the “predominantly male” phenotype:
- Less than normal decline of sex hormone-binding globulin (SHBG) in response to a standard dose of the anabolic androgen, stanozolol 14
- Higher than normal levels of anti-müllerian hormone during the first year of life or after puberty has begun
The diagnosis of androgen insensitivity syndrome is established in a 46XY proband with:
- Undermasculinization of the external genitalia, impaired spermatogenesis with otherwise normal testes, absent or rudimentary müllerian structures, evidence of normal or increased synthesis of testosterone and its normal conversion to dihydrotestosterone, and normal or increased LH production by the pituitary gland; AND/OR
- A hemizygous pathogenic variant in AR identified by molecular genetic testing.
Complete androgen insensitivity syndrome treatment
To prevent testicular malignancy, treatment of complete androgen insensitivity syndrome may include either removal of the testes after puberty when feminization is complete or prepubertal gonadectomy accompanied by estrogen replacement therapy. Because the risk of testicular cancer is low, removal of testes (gonadectomy) is increasingly controversial. In fact on one hand complete androgen insensitivity syndrome is associated with an increased risk of testicular germ cell tumor (TGCT), so testes should be removed in order to prevent testicular cancer; on the other hand, the postponement of gonadectomy until at least puberty allows spontaneous pubertal development thanks to estradiol deriving from the peripheral aromatization of testosterone produced by the retained testes. Additional treatment for complete androgen insensitivity syndrome may include vaginal dilatation to avoid painful sexual intercourse (dyspareunia).
Testicular germ cell tumor represents approximately 1–1.5% of all tumors in the general male population and is the most common malignant cancer among male subjects from 15–40 years of age 15. The occurrence of testicular germ cell tumor in adulthood could be above 22% 16, while its incidence is very low in childhood and adolescence 17.
According to the last published World Health Organization (WHO) classification, the majority of testicular germ cell tumor originates from noninvasive lesions, referred to as germ cell neoplasia in situ and pre-germ cell neoplasia in situ 18. The complete androgen insensitivity syndrome condition has been related to a higher incidence of testicular germ cell tumor than in the general population 19. The most common association is reported with seminoma and gonadoblastoma, although other histological forms have been found, such as choriocarcinomas, teratomas, embryonal tumours, adenomas, and Leydig and/or Sertoli cell tumors 20.
The exact incidence of cancer in patients with complete androgen insensitivity syndrome is very difficult to estimate because of the frequent change in management of this disorder over the years, particularly regarding the correct time of gonadectomy 21. Data from the literature review report a general risk of approximately 5% in androgen insensitivity syndrome disorder overall and a prevalence of <1% in complete androgen insensitivity syndrome 22. In addition, the risk of malignant progression is elevated only with increased age 23; it rarely occurs in prepubertal age (less than 1%), in contrast with other disorders of sex development, including partial androgen insensitivity syndrome 24. In the general population, germ cell neoplasia in situ advances into invasive cancer in approximately 50% of cases over five years 25, while the majority of malignant lesions described in patients with complete androgen insensitivity syndrome after puberty were pre-germ cell neoplasia in situ or germ cell neoplasia in situ, with a low likelihood of becoming invasive 26. These data suggest that malignant progression from pre-germ cell neoplasia in situ to invasive testicular germ cell tumor is very infrequent and probably takes place only in late adulthood. These findings validate the possibility of postponing a gonadectomy until after puberty 27. Even the occurrence of a bilateral inguinal hernia during childhood no longer represents an absolute indication for early gonadectomy 23.
Several studies have tried to identify factors associated with cancer development and progression. It has been suggested, for example, that there is a possible role of individual genetic susceptibility, related to one or more single nucleotide polymorphisms (SNPs) 28. Cools et al. 26 found a significantly increased genetic susceptibility to the development of invasive cancer in subjects with pre-germ cell neoplasia in situ due to the presence of specific alleles of genes related to invasive cancer. They did not find specific patterns of SNPs directly associated with pre-germ cell neoplasia in situ/germ cell neoplasia in situ/invasive cancer, but they stressed the possible role of genetic factors in cancer development together with residual androgen paracrine action and testicular cellular milieu 19. Additionally, a higher risk of malignant transformation has been associated with altered expression of the histological markers PUO5F1 and KITLG 21. POU5F1, also known as OCT3/4, represents a marker of delayed maturation of germ cells (early primordial germ cells), a condition commonly reported in a situation of insufficient hormonal action and/or defective cellular milieu, such as happens in disorders of sex development. Although an increased positivity to POU5F1 does not represent a premalignant condition ipso facto; the overexpression or the defective downregulation of POU5F1, particularly in germ cells in contact with the basal membrane, could promote the development of premalignant/malignant lesions by providing these cells with an increased survival capacity 29. On the other hand, aberrant gene expression of KITLG has only been related to pre-germ cell neoplasia in situ and not to the delayed maturation status of germ cells 30. Therefore, the ability to distinguish the delayed maturation status of germ cells from premalignant lesions could allow for early identification of suspected lesions and overdiagnosis of germ cell neoplasia in situ 26.
Furthermore, testis-specific protein, Y-linked (TSPY), could be another pivotal marker for malignant progression; indeed, it is physiologically involved in cellular proliferation 31. Normal surviving germ cells in disorders of sex development usually overexpress TSPY, whereas its expression gradually decreases simultaneously with neoplastic progression until it becomes undetectable 32.
There may be several reasons for a low trend of malignancy in retained gonads in patients with complete androgen insensitivity syndrome. First, in contrast with other disorders of sexual development with gonadal dysgenesis, testicular tissue is normally developed in complete androgen insensitivity syndrome. Second, the lack of signal coming from androgens may play a key role in modulating cellular development and differentiation. Finally, the high rate of germ cell apoptosis in complete androgen insensitivity syndrome reduces the possibility of malignant evolution 33. However, the residual paracrine actions of androgen in testicular tissue, also described in complete androgen insensitivity syndrome, could be a risk factor for cancer development, especially during and after puberty 34. It could promote neoplastic progression of germ cells and explain the increased risk of developing malignancy in adulthood [68,100]. On the other hand, some authors suggested the possible protective role of the residual androgen activity in cancer development, precisely because it allows the survival of the normal germ cell population overall 35.
Although there is a low rate of invasive cancer in complete androgen insensitivity syndrome, it is mandatory to recognize suspected lesions early. Unfortunately, both germ cell neoplasia in situ and seminomas do not usually secrete serum markers, such as β-HCG and α-fetoprotein 36; other specific serum markers are needed. For example, some microRNA clusters, such as the overexpression of miR371-3 and miR-302/367, have been associated with an invasive form of testicular germ cell tumor and with germ cell neoplasia in situ both in disorders of sex development and in the general male population 37. While these microRNAs have demonstrated promise both in the diagnosis and in the follow-up of testicular germ cell tumor, the germ cell neoplasia in situ form likely does not secrete enough microRNAs to be useful for early diagnosis 38. Currently, the real effect on the testes position still remains unclear as a promoting factor in cancer development in complete androgen insensitivity syndrome 21.
In summary, complete androgen insensitivity syndrome is a condition associated with an increased risk of cancer, although cancer results less frequently in complete androgen insensitivity syndrome compared to other disorders of sex development. The majority of tumoral lesions detected are non-invasive ones, with a low rate of progression into aggressive forms. Multiple factors seem to be involved, including individual genetic susceptibility, residual paracrine androgen effect, and testes position, and there are not any reliable serum markers to identify early lesions, though there are many suitable candidates. Nevertheless, histological, epidemiological, and prognostic features of testicular cancer in complete androgen insensitivity syndrome allow the postponing of gonadectomy until after pubertal age.
Follow-up of retained testes
Currently, about 15% of adult patients with complete androgen insensitivity syndrome decide to maintain their gonads intact, even after pubertal development 16. This is probably due to the fact that they want both to take advantage of the benefits of endogenous hormone secretion and to avoid the possible complications of the surgical procedure 19. Therefore, an effective follow-up programme is needed, in order to precociously recognize and afford the development of a testicular germ cell tumor. However, there is actually not a sufficient amount of confirmed data to guarantee safe management of these patients.
Ultrasound (US) remains the first-line evaluation for inguinal or labioscrotal gonads 39, and annual ultrasound follow-up is recommended, starting from puberty 26. Ultrasound evaluation can identify suggestive lesions, such as microlithiasis and/or irregular echogenicity of testis parenchyma, but it is not able to properly detect germ cell neoplasia in situ 40.
Magnetic resonance (MR) has to be performed in abdominal testes 39. Although it is not able to identify germ cell neoplasia in situ and/or microlithiasis [110,112], this procedure appears to be crucial for testicular germ cell tumor staging and follow-up. Nakhal et al. 41 retrospectively evaluated testicular MR images of 25 patients with complete androgen insensitivity syndrome in order to investigate the effective role of MR in early identification of suspected lesions. MR was not predictive for the diagnosis of premalignant lesions, but it detected both paratesticular cysts and Sertoli cell adenomas. Therefore, the possible role of MR in the identification of early invasive testicular germ cell tumor lesions remains debatable. Instead, Dohnert et al. 23 proposed a biannual follow-up, including both US and/or MR, along with the determination of classic serum markers (e.g., β-HCG, α-FP, LDH) and hormonal assessment (FSH, LH, testosterone and Inhibin B).
Further investigations are needed to detect how to perform the follow-up of patients with complete androgen insensitivity syndrome and unremoved testis after pubertal age. Currently, the gold standard for effective diagnosis of testicular germ cell tumor and/or its precursors still remains histological analysis at biopsy, which may result in gonadectomy 41.
Hormonal replacement therapy (HRT)
Hormonal replacement therapy is mandatory after bilateral gonadectomy in order to prevent symptoms of hypoestrogenism, inducing pubertal development if surgery has been performed before pubertal age or maintaining secondary sexual features if it has been performed later 42. Secondary therapeutic targets of hormonal replacement therapy also differ depending on the time of the gonadectomy; it allows physiological pubertal spurt development, physiological changes in body composition (fat and muscle mass distribution), achievement of bone mineral peak and maintenance of bone mineralization, and psychological and relational/sexual wellness 42.
The classic hormonal replacement therapy for complete androgen insensitivity syndrome patients is based on oestrogen therapy, but current data are not able to indicate the best daily dosage. Therefore, hormonal replacement therapy should be started at the lowest dose (i.e., oral ethinyl oestradiol 2.5–5 µg/day or 50–100 ng/kg/day) and then gradually increased to the adult dosage (i.e., oral ethinyl oestradiol 20–25 µg/day) in order to simulate physiological secretion 42. Specifically in prepubertal subjects, hormonal replacement therapy should be slowly increased every 6 months in order to complete feminization, such as breast development, changes in body composition and reaching of female body shape, in approximately two years 43. After complete breast development, therapy should be continued with a regular daily dose 43. As previously assessed, there is conflicting data about the optimal dose of oestrogen after the initial titration and, in particular, there are no specific trials conducted on complete androgen insensitivity syndrome subjects 44. Furthermore, excessive doses could lead to impaired growth development and early epiphyses closure 44. Therefore, hormonal replacement therapy could be individualized according to clinical experience and patient needs. There is also no agreement on which is the best hormone formulation. Indeed, both oral and transdermal oestrogens seem to be useful and effective 42; perhaps transdermal should be preferred to oral formulations for a more physiological delivery, an absent/lower first-pass effect, less interference with hepatic metabolism and IGF-1 serum levels, and a decreased risk of thromboembolism 45.
Furthermore, there are no consistent data on the real adverse effects of classic hormonal replacement therapy in complete androgen insensitivity syndrome. Some studies have reported a slightly increased risk of myocardial infarction, stroke and breast cancer in adult menopausal women with oral administration 45, but the absolute risk seems to be very low, and these results could be useless for young patients with complete androgen insensitivity syndrome. However, in the literature, an increased risk of osteoporosis, cardiovascular diseases, dementia or cognitive decline, and Parkinson disease has been reported in subjects with early ovarian failure if untreated with oestrogen hormonal replacement therapy 46.
The additional therapy with progesterone is not required because of the absence of a uterus in patients with complete androgen insensitivity syndrome, and there is no evidence of increased well-being with estroprogestinic therapy 44.
Despite the correct administration of classic hormonal replacement therapy, many patients with complete androgen insensitivity syndrome reported a decrease in psychological well-being and in sexual satisfaction, perhaps due to several hormonal changes after the bilateral gonadectomy 47. In a multicentre, double-blind and randomized crossover trial, the effectiveness and side effects of oestrogen versus testosterone hormonal replacement therapy were investigated in 26 patients (ranging from 18–54 years old) genetically diagnosed with complete androgen insensitivity syndrome who had undergone a bilateral gonadectomy. No significant differences were found in terms of psychological well-being, mental health and quality of life between subjects who received oestrogen and those who received testosterone. Furthermore, no signs of virilization were observed with testosterone hormonal replacement therapy, but it seemed to be better than oestradiol in improving sexual desire. Authors concluded that testosterone should be considered a valid alternative to oestrogen hormonal replacement therapy in complete androgen insensitivity syndrome 47. Future studies are needed to understand what could be the best therapeutic approach.
Interestingly, patients with complete androgen insensitivity syndrome seem to have a different hormonal status that does not follow a physiological male or female profile. In particular, postpubertal complete androgen insensitivity syndrome patients with intact gonads show increased levels of LH with normal levels of FSH and of sex hormone binding globulin (SHBG) for the female range; moreover, basal testosterone and oestradiol values, free androgen indices and androgen aromatization indices are in the normal male range 48. Increased levels of LH, despite testosterone levels, may be attributable to the role of androgen resistance in the normal negative feedback action carried out by androgens on the hypothalamus-hypophysis axis 49. Thus, Doehnert et al. 12 suggested the use of a lower dose of hormonal replacement therapy in patients with complete androgen insensitivity syndrome after gonadectomy, seeing that these patients follow neither a female nor male hormone pattern and that levels of estrogen are normally below the female range before gonadectomy. This could partially explain the reported reduced wellbeing with doses of current classic hormonal replacement therapy.
Assignment of sex
The issue of sex assignment in infancy when the child is being evaluated for ambiguous genitalia is paramount. It requires informed decision making by parents and health care personnel and should be resolved as early as possible, after a multidisciplinary evaluation has been completed.
Even in complete androgen insensitivity syndrome this may not always be easy. Cheikhelard et al 50 evaluated 29 individuals with complete androgen insensitivity syndrome; it was recommended that gonads be retained at least until the completion of spontaneous puberty and the possibility of virilization be evaluated before management decisions are made.
Psychological counseling and use of support groups can be of benefit 51.
Gender identity has become a topic of increasing importance due to the possibility of changes in sex assignment over time 52.
Issues of sexual orientation regardless of gender phenotype have also become increasingly important to explore and discuss 53.
Bone mineral density
In adults, monitoring of bone mineral density through DEXA (dual-emission x-ray absorptiometry) scanning 54.
Complete androgen insensitivity syndrome disorder seems to be associated with a reduced bone mineral density on dual-energy X-ray absorptiometry (DEXA) and an increased risk of osteoporosis in adulthood due to a lack of androgen function 55; bilateral gonadectomy may also play an important role 56. The early identification of bone mineral density alterations could prevent comorbidity and improve the quality of life of these subjects.
Decreased bone mineral density in complete androgen insensitivity syndrome patients with removed gonads has been widely reported in the literature 57, while bone mineral density seems to be less impaired in adult patients with intact gonads 58. Moreover, lumbar bone mineral density seems to be more affected than vertebral bone mineral density, independent of gonadal status, suggesting a different pattern of AR expression between trabecular and compact bone tissue 59. In patients who underwent gonadectomy, good adherence to HRT may play a role in bone mineral density; indeed, it has been associated with better vertebral and femoral bone mineral density levels at DEXA 60. In contrast, Danilovic et al. 11 found, at most, a slight improvement in vertebral DEXA values after two years of correct HRT administration, suggesting that other factors may be involved. Otherwise, a positive effect of HRT in bone mineral density improvement could only occur after prolonged and/or high-dose therapy (i.e., equivalent to 0.625–1.25 mg of conjugated estrogens) 61.
Some studies reported an increased fracture risk in patients with complete androgen insensitivity syndrome and removed gonads 62, but they involved only a small number of subjects, and often, there was substantial bias (i.e., reference values used for DEXA), so the data are still inconclusive; there are no consistent data about fracture rate in patients with complete androgen insensitivity syndrome with intact gonads 56. Additionally, patients with complete androgen insensitivity syndrome seem to also have a specific body composition; indeed, several animal studies have reported altered body fat mass with earlier development of obesity, an abnormal lipid profile, alterations in adipose tissue related hormones and decreased insulin sensitivity due to the resistance or absence of androgen activity 63. Dati et al. 64 have investigated body composition and metabolism assessment in middle-aged adult patients with complete androgen insensitivity syndrome, both with removed and retained testes. The body fat mass was increased and resulted in high values of total cholesterol and LDL cholesterol, and large amounts of HOMA-IR (Homeostatic Model Assessment for Insulin Resistance) were detected. Furthermore, they found an increased rate of obesity, even if the mean BMI did not differ significantly from the general female population of the same age. Interestingly, the majority of obese patients were those who retained testes. The authors suggested the importance of a regular assessment of body composition, metabolic status, and cardiovascular risk in all patients diagnosed with complete androgen insensitivity syndrome, regardless of gonadal condition. Additionally, control of BMI and regular physical exercise are recommended together with calcium and vitamin D supplementation in order to improve bone health. Bisphosphonate therapy may be indicated only in the presence of a severely reduced bone mineral density and/or fractures 3.
Complete androgen insensitivity syndrome prognosis
The medical and psychological prognosis for a woman with complete androgen insensitivity syndrome is excellent if she has appropriate support and counseling.
Complete androgen insensitivity syndrome increases the risk of testicular malignancy if the testes are not removed, with risk estimated at 3.6% at 25 years and 33% at 50 years 23, 65. Prepubertal malignancy in complete androgen insensitivity syndrome is extremely rare. The risk of germ cell tumors in partial androgen insensitivity syndrome with untreated undescended testes is significantly greater, with estimates as high as 50% 66. Not much documentation on the morbidity or mortality of these tumors specifically in individuals with androgen insensitivity syndrome is available. The tumor is considered cured without need for further therapy if it is removed while still limited to the interior of the testes capsule. The tumor is considered curable in most patients even when undetected at this early state. The use of a magnetic resonance imaging before surgery appears to be helpful in localizing and planning for removal of the gonads for malignancy risk reduction and preventing injury to other structures 67.
In contrast to medical morbidity, psychological morbidity is common. Phenotypic females who are discovered to be genetic males may have psychosocial problems. These females require sensitive psychological support. Their psychosocial problems range from identity issues to problems dealing with the gender perceptions of the outside world and the style and sensitivity (or lack thereof) they encounter within the medical system.
Most affected individuals report psychological trauma at diagnosis. Their reactions to the diagnosis frequently are compounded by their interactions with the medical care system, in which they often are treated as oddities and forced to undergo multiple examinations and interviews with students and residents for teaching purposes. Even in nonteaching situations, women with androgen insensitivity syndrome report difficulties identifying offices where physicians and staff are familiar with their condition. Many of these patients have been told that they really are not women but actually are men because of the presence of a Y chromosome and testes. These difficulties and doubts often cause shame and self-doubt, as well as anger and frustration with a medical system they had expected to take care of them. There is far-reaching lack of sexual confidence and sexual satisfaction in individuals with complete androgen insensitivity syndrome.
References- Androgen insensitivity syndrome. https://ghr.nlm.nih.gov/condition/androgen-insensitivity-syndrome
- Hughes I.A., Davies J.D., Bunch T.I., Pasterski V., Mastroyannopoulou K., Macdougall J. Androgen insensitivity syndrome. Lancet. 2012;380:1419–1428. doi: 10.1016/S0140-6736(12)60071-3
- Gottlieb B, Trifiro MA. Androgen Insensitivity Syndrome. 1999 Mar 24 [Updated 2017 May 11]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1429
- Lanciotti L, Cofini M, Leonardi A, Bertozzi M, Penta L, Esposito S. Different Clinical Presentations and Management in Complete Androgen Insensitivity Syndrome (CAIS). Int J Environ Res Public Health. 2019;16(7):1268. Published 2019 Apr 9. doi:10.3390/ijerph16071268 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6480640
- Mendonca B.B., Domenice S., Arnhold I.J.P., Costa E.M.F. 46, XY disorders of sex development (DSD) Clin. Endocrinol. 2009;70:173–187. doi: 10.1111/j.1365-2265.2008.03392.x
- Lanciotti L., Cofini M., Leonardi A., Penta L., Esposito S. Up-To-Date Review About Minipuberty and Overview on Hypothalamic-Pituitary-Gonadal Axis Activation in Fetal and Neonatal Life. Front. Endocrinol. 2018;9:410. doi: 10.3389/fendo.2018.00410
- Oakes M.B., Eyvazzadeh A.D., Quint E., Smith Y.R. Complete Androgen Insensitivity Syndrome-A Review. J. Pediatr. Adolesc. Gynecol. 2008;21:305–310. doi: 10.1016/j.jpag.2007.09.006
- Petroli R.J., Hiort O., Struve D., Gesing J.K., Soardi F.C., Spínola-Castro A.M., Melo K., Prado Arnhold I.J., Maciel-Guerra A.T., Guerra-Junior G., et al. Functional impact of novel androgen receptor mutations on the clinical manifestation of androgen insensitivity syndrome. Sex. Dev. 2018;11:238–247. doi: 10.1159/000484882
- Gulía C., Baldassarra S., Zangari A., Briganti V., Gigli S., Gaffi M., Signore F., Vallone C., Nucciotti R., Costantini F.M., et al. Androgen insensitivity syndrome. Eur. Rev. Med. Pharmacol. Sci. 2018;22:3873–3887. doi: 10.26355/eurrev_201806_15272
- Batista R.L., Costa E.M.F., Rodrigues A.D., Gomes N.L., Faria J.A., Jr., Nishi M.Y., Arnhold I.J.P., Domenice S., Mendonca B.B. Androgen insensitivity syndrome: A review. Arch. Endocrinol. Metab. 2018;62:227–235. doi: 10.20945/2359-3997000000031
- Danilovic D.L.S., Correa P.H.S., Costa E.M.F., Melo K.F.S., Mendonca B.B., Arnhold I.J.P. Height and bone mineral density in androgen insensitivity syndrome with mutations in the androgen receptor gene. Osteoporos. Int. 2007;18:369–374. doi: 10.1007/s00198-006-0243-6
- Doehnert U., Bertelloni S., Werner R., Dati E., Hiort O. Characteristic features of reproductive hormone profiles in late adolescent and adult females with complete androgen insensitivity syndrome. Sex. Dev. 2015;9:69–74. doi: 10.1159/000371464
- Bouvattier C, Carel JC, Lecointre C, David A, Sultan C, Bertrand AM, Morel Y, Chaussain JL. Postnatal changes of T, LH, and FSH in 46,XY infants with mutations in the AR gene. J Clin Endocrinol Metab. 2002;87:29–32.
- Sinnecker GH, Hiort O, Nitsche EM, Holterhus PM, Kruse K. Functional assessment and clinical classification of androgen sensitivity in patients with mutations of the androgen receptor gene. German Collaborative Intersex Study Group. Eur J Pediatr. 1997;156:7–14.
- Parkin D.M., Whelan S.L., Ferlay J., Teppo L., Thomas D.B. Cancer Incidence in Five Continents. Volume 8 IARC Scientific Publications; Lyon, France: 2002.
- Deans R., Creighton S.M., Liao L.M., Conway G.S. Timing of gonadectomy in adult women with complete androgen insensitivity syndrome (CAIS): Patient preferences and clinical evidence. Clin. Endocrinol. 2012;76:894–898. doi: 10.1111/j.1365-2265.2012.04330.x
- Hurt W.G., Bodurtha J.N., McCall J.B., Ali M.M. Seminoma in pubertal patient with androgen insensitivity syndrome. Am. J. Obstet. Gynecol. 1989;161:530–531. doi: 10.1016/0002-9378(89)90350-5
- Moch H., Cubilla A.L., Humphrey P.A., Reuter V.E., Ulbright T.M. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs—Part A: Renal, Penile, and Testicular Tumours. Eur. Urol. 2016;70:93–105. doi: 10.1016/j.eururo.2016.02.029
- Cools M., Wolffenbuttel K.P., Hersmus R., Mendonca B.B., Kaprová J., Drop S.L.S., Stoop H., Gillis A.J.M., Oosterhuis J.W., Costa E.M.F., et al. Malignant testicular germ cell tumors in postpubertal individuals with androgen insensitivity: Prevalence, pathology and relevance of single nucleotide polymorphism-based susceptibility profiling. Hum. Reprod. 2017;32:2561–2573. doi: 10.1093/humrep/dex300
- Kravarusic D., Seguier-Lipszyc E., Feigin E., Nimri R., Nagelberg N., Freud E. Androgen insensitivity syndrome: Risk of malignancy and timing of surgery in a paediatric and adolescent population. Afr. J. Paediatr. Surg. 2011;8:194–198. doi: 10.4103/0189-6725.86061
- Kaprova-Pleskacova J., Stoop H., Brüggenwirth H., Cools M., Wolffenbuttel K.P., Drop S.L., Snajderova M., Lebl J., Oosterhuis J.W., Looijenga L.H. Complete androgen insensitivity syndrome: Factors influencing gonadal histology including germ cell pathology. Mod. Pathol. 2014;27:721–730. doi: 10.1038/modpathol.2013.193
- Cools M., Drop S.L.S., Wolffenbuttel K.P., Oosterhuis J.W., Looijenga L.H.J. Germ Cell Tumors in the Intersex Gonad: Old Paths, New Directions, Moving Frontiers. Endocr. Rev. 2006;27:468–484. doi: 10.1210/er.2006-0005
- Döhnert U., Wünsch L., Hiort O. Gonadectomy in Complete Androgen Insensitivity Syndrome: Why and When? Sex. Dev. 2017;11:171–174. doi: 10.1159/000478082
- Herman M., Wernicke G.A., Yan W., Nori D., Parashar B. Pure seminoma in the setting of androgen insensitivity syndrome treated with surgical resection and para-aortic radiation: A case report and review of literature. J. Cancer Res. Ther. 2010;6:318–320. doi: 10.4103/0973-1482.73337
- Dieckmann K.P., Skakkebaek N.E. Carcinoma in situ of the testis: Review of biological and clinical features. Int. J. Cancer. 1999;83:815–822. doi: 10.1002/(SICI)1097-0215(19991210)83:6<815::AID-IJC21>3.0.CO;2-Z
- Cools M., Looijenga L. Update on the Pathophysiology and Risk Factors for the Development of Malignant Testicular Germ Cell Tumors in Complete Androgen Insensitivity Syndrome. Sex. Dev. 2017;11:175–181. doi: 10.1159/000477921
- Lee P.A., Nordenström A., Houk C.P., Ahmed S.F., Auchus R., Baratz A., Baratz Dalke K., Liao L.M., Lin-Su K., Looijenga L.H., et al. Global Disorders of Sex Development Update since 2006: Perceptions, Approach and Care. Horm. Res. Paediatr. 2016;85:158–180. doi: 10.1159/000442975
- Chung C.C., Kanetsky P.A., Wang Z., Hildebrandt M.A., Koster R., Skotheim R.I., Kratz C.P., Turnbull C., Cortessis V.K., Bakken A.C., et al. Meta-analysis identifies four new loci associated with testicular germ cell tumor. Nat. Genet. 2013;45:680–685. doi: 10.1038/ng.2634
- Cools M., van Aerde K., Kersemaekers A.-M., Boter M., Drop S.L., Wolffenbuttel K.P., Steyerberg E.W., Oosterhuis J.W., Looijenga L.H. Morphological and immunohistochemical differences between gonadal maturation delay and early germ cell neoplasia in patients with undervirilization syndromes. J. Clin. Endocrinol. Metab. 2005;90:5295–5303. doi: 10.1210/jc.2005-0139
- Stoop H., Honecker F., van de Geijn G., Gillis A.J., Cools M.C., de Boer M., Bokemeyer C., Wolffenbuttel K.P., Drop S.L., de Krijger R.R., et al. Stem cell factor as a novel diagnostic marker for early malignant germ cells. J. Pathol. 2008;216:43–54. doi: 10.1002/path.2378
- Lau Y.-F.C., Lau H.W., Kömüves L.G. Expression pattern of a gonadoblastoma candidate gene suggests a role of the Y chromosome in prostate cancer. Cytogenet. Genome Res. 2003;101:250–260. doi: 10.1159/000074345
- Kersemaekers A.-M.F., Honecker F., Stoop H., Cools M., Molier M., Wolffenbuttel K., Bokemeyer C., Li Y., Lau Y.F., Oosterhuis J.W., et al. Identification of germ cells at risk for neoplastic transformation in gonadoblastoma. Hum. Pathol. 2005;36:512–521. doi: 10.1016/j.humpath.2005.02.016
- Van der Zwan Y.G., Biermann K., Wolffenbuttel K.P., Cools M., Looijenga L.H.J. Gonadal maldevelopment as risk factor for germ cell cancer: Towards a clinical decision model. Eur. Urol. 2015;67:692–701. doi: 10.1016/j.eururo.2014.07.011
- Rey R.A. Mini-puberty and true puberty: Differences in testicular function. Ann. Endocrinol. 2014;75:58–63. doi: 10.1016/j.ando.2014.03.001
- O’Shaughnessy P.J. Hormonal control of germ cell development and spermatogenesis. Semin. Cell Dev. Biol. 2014;29:55–65. doi: 10.1016/j.semcdb.2014.02.010
- Patel V., Casey R.K., Gomez-Lobo V. Timing of Gonadectomy in Patients with Complete Androgen Insensitivity Syndrome-Current Recommendations and Future Directions. J. Pediatr. Adolesc. Gynecol. 2016;29:320–325. doi: 10.1016/j.jpag.2015.03.011
- Rijlaarsdam M.A., van Agthoven T., Gillis A.J.M., Patel S., Hayashibara K., Lee K.Y., Looijenga L.H. Identification of known and novel germ cell cancer-specific (embryonic) miRs in serum by high-throughput profiling. Andrology. 2015;3:85–91. doi: 10.1111/andr.298
- Van Agthoven T., Looijenga L.H.J. Accurate primary germ cell cancer diagnosis using serum based microRNA detection (ampTSmiR test) Oncotarget. 2017;8:58037–58049. doi: 10.18632/oncotarget.10867
- Kim W., Rosen M.A., Langer J.E., Banner M.P., Siegelman E.S., Ramchandani P. US MR imaging correlation in pathologic conditions of the scrotum. Radiographics. 2007;27:1239–1253. doi: 10.1148/rg.275065172
- Elzinga-Tinke J.E., Sirre M.E., Looijenga L.H.J., van Casteren N., Wildhagen M.F., Dohle G.R. The predictive value of testicular ultrasound abnormalities for carcinoma in situ of the testis in men at risk for testicular cancer. Int. J. Androl. 2010;33:597–603. doi: 10.1111/j.1365-2605.2009.00997.x
- Nakhal R.S., Hall-Craggs M., Freeman A., Kirkham A., Conway G.S., Arora R., Woodhouse C.R., Wood D.N., Creighton S.M. Evaluation of Retained Testes in Adolescent Girls and Women with Complete Androgen Insensitivity Syndrome. Radiology. 2013;268:153–160. doi: 10.1148/radiol.13121068
- Bertelloni S., Dati E., Baroncelli G.I. Disorders of sex development: Hormonal management in adolescence. Gynecol. Endocrinol. 2008;24:339–346. doi: 10.1080/09513590802055708
- Arnhold I.J.P., Melo K., Costa E.M.F., Danilovic D., Inacio M., Domenice S., Mendonca B.B. Advances in Experimental Medicine and Biology. Springer; New York, NY, USA: 2011. 46,XY Disorders of Sex Development (46,XY DSD) due to Androgen Receptor Defects: Androgen Insensitivity Syndrome.
- Warne G.L., Grover S., Zajac J.D. Hormonal therapies for individuals with intersex conditions: Protocol for use. Treat. Endocrinol. 2005;4:19–29. doi: 10.2165/00024677-200504010-00003
- Kopper N.W., Gudeman J., Thompson D.J. Transdermal hormone therapy in postmenopausal women: A review of metabolic effects and drug delivery technologies. Drug Des. Dev. Ther. 2009;2:193–202. doi: 10.2147/DDDT.S4146
- Schenck-Gustafsson K., Brincat M., Erel C.T., Gambacciani M., Lambrinoudaki I., Moen M.H., Tremollieres F., Vujovic S., Rozenberg S., Rees M. EMAS position statement: Managing the menopause in the context of coronary heart disease. Maturitas. 2011;68:94–97. doi: 10.1016/j.maturitas.2010.10.005
- Birnbaum W., Marshall L., Werner R., Kulle A., Holterhus P.M., Rall K., Köhler B., Richter-Unruh A., Hartmann M.F., Wudy S.A., et al. Oestrogen versus androgen in hormone-replacement therapy for complete androgen insensitivity syndrome: A multicentre, randomised, double-dummy, double-blind crossover trial. Lancet Diabetes Endocrinol. 2018;8587:1–10. doi: 10.1016/S2213-8587(18)30197-9
- Ahmed S.F., Cheng A., Hughes I.A. Assessment of the gonadotrophin-gonadal axis in androgen insensitivity syndrome. Arch. Dis. Child. 1999;80:324–329. doi: 10.1136/adc.80.4.324
- Hughes I., Werner R., Bunch T., Hiort O. Androgen Insensitivity Syndrome. Semin. Reprod. Med. 2012;30:432–442. doi: 10.1016/S0140-6736(12)60071-3
- Cheikhelard A, Morel Y, Thibaud E, Lotat-Jacob S, Jaubert F, Ploak M, Nihoul-Fekete C. Long-term follow up and comparison between genotype and phenotype in 29 cases of complete androgen insensitivity. J Urol. 2008;180:1496–501.
- Cull ML, Simmonds M. Importance of support groups for intersex (disorders of sexual development) patients, families and the medical profession. Sex Dev. 2010;4:310–2.
- Kolesinska Z, Ahmed AF, Niedziela M, Bryce J, Molinska-Gur M, Rodie M, Jiang J, Sinnott RO, Hughes IA, Darendeliler F, Hiort O, van der Zwan Y, Cools M, Guran T, Holterhus PM, Bertelloni S, Lisa L, Arlt W, Krone N, Ellaithi M, Balsamo A, Mazen I, Nordenstrom A, Lachlan K, Alkhawari M, Chatelain P, Weintrob N. Changes over time in sex assignments for disorders of sex development. Pediatrics. 2014;134:e710–5
- Brunner F, Fliegner M, Krupp K, Rall K, Brucker S, Richter-Appelt H. Gender role, gender identity and sexual orientation in CAIS (“XY-Women”) compared with subfertile and infertile 46,XX,women. J Sex Res. 2016;53:109–24.
- Oakes MB, Eyvazzadeh AD, Quint E, Smith YR. Complete androgen insensitivity syndrome – a review. J Pediatr Adolesc Gynecol. 2008;21:305–10.
- Bertelloni S., Baroncelli G.I., Mora S. Bone Health in Disorders of Sex Differentiation. Sex. Dev. 2010;4:270–284. doi: 10.1159/000315961
- Bertelloni S., Meriggiola M.C., Dati E., Balsamo A., Baroncelli G.I. Bone Mineral Density in Women Living with Complete Androgen Insensitivity Syndrome and Intact Testes or Removed Gonads. Sex. Dev. 2017;11:182–189. doi: 10.1159/000477599
- Taes Y., Lapauw B., Vandewalle S., Zmierczak H., Goemaere S., Vanderschueren D., Kaufman J.M., T’Sjoen G. Estrogen-specific action on bone geometry and volumetric bone density: Longitudinal observations in an adult with complete androgen insensitivity. Bone. 2009;45:392–397. doi: 10.1016/j.bone.2009.04.198
- Sobel V., Schwartz B., Zhu Y.-S., Cordero J.J., Imperato-McGinley J. Bone Mineral Density in the Complete Androgen Insensitivity and 5α-Reductase-2 Deficiency Syndromes. J. Clin. Endocrinol. Metab. 2006;91:3017–3023. doi: 10.1210/jc.2005-2809
- Sinnesael M., Claessens F., Laurent M., Dubois V., Boonen S., Deboel L., Vanderschueren D. Androgen receptor (AR) in osteocytes is important for the maintenance of male skeletal integrity: Evidence from targeted AR disruption in mouse osteocytes. J. Bone Miner. Res. 2012;27:2535–2543. doi: 10.1002/jbmr.1713
- Han T.S., Goswami D., Trikudanathan S., Creighton S.M., Conway G.S. Comparison of bone mineral density and body proportions between women with complete androgen insensitivity syndrome and women with gonadal dysgenesis. Eur. J. Endocrinol. 2008;159:179–185. doi: 10.1530/EJE-08-0166
- Birnbaum W., Bertelloni S. Sex Hormone Replacement in Disorders of Sex Development. Endocr. Dev. 2014;27:149–159. doi: 10.1159/000363640
- Marcus R., Leary D., Schneider D.L., Shane E., Favus M., Quigley C.A. The contribution of testosterone to skeletal development and maintenance: Lessons from the androgen insensitivity syndrome. J. Clin. Endocrinol. Metab. 2000;85:1032–1037. doi: 10.1210/jcem.85.3.6428
- Yu I.-C., Lin H.-Y., Liu N.-C., Wang R.S., Sparks J.D., Yeh S., Chang C. Hyperleptinemia without Obesity in Male Mice Lacking Androgen Receptor in Adipose Tissue. Endocrinology. 2008;149:2361–2368. doi: 10.1210/en.2007-0516
- Dati E., Baroncelli G.I., Mora S., Russo G., Baldinotti F., Parrini D., Erba P., Simi P., Bertelloni S. Body Composition and Metabolic Profile in Women with Complete Androgen Insensitivity Syndrome. Sex. Dev. 2009;3:188–193. doi: 10.1159/000228719
- Liu A.X., Shi H.Y., Cai Z.J., Liu A., Zhang D., Huang H.F., Jin H.M. Increased risk of gonadal malignancy and prophylactic gonadectomy: A study of 102 phenotypic female patients with y chromosome or Y-derived sequences. Hum. Reprod. 2014;29:1413–1419. doi: 10.1093/humrep/deu109
- Kathrins M, Kolon TF. Malignancy in disorders of sex development. Transl Androl Urol. 2016 Oct. 5 (5):794-798.
- Khan S, Mannel L, Koopman CL, Chimpiri R, Hansen KR, Craig LB. The use of MRI in the pre-surgical evaluation of patients with androgen insensitivity syndrome. J Pediatr Adolesc Gynecol. 2014 Feb. 27(1):e17-20.





{kind=link}