
Fumarase deficiency
Fumarase deficiency also called fumaric aciduria or fumarate hydratase deficiency, is an inherited condition that primarily affects the nervous system, especially the brain 1. Affected infants may have an abnormally small head size (microcephaly), abnormal brain structure, severe developmental delay, weak muscle tone (hypotonia), and failure to gain weight and grow at the expected rate (failure to thrive). They may also experience seizures. Some people with this disorder have unusual facial features, including a prominent forehead (frontal bossing), low-set ears, a small jaw (micrognathia), widely spaced eyes (ocular hypertelorism), and a depressed nasal bridge. An enlarged liver and spleen (hepatosplenomegaly) may also be associated with this disorder, as well as an excess of red blood cells (polycythemia) or deficiency of white blood cells (leukopenia) in infancy. A variety of brain abnormalities may be detected on MRI 1. Most children with fumarase deficiency are not able to speak or walk 1. Affected individuals usually survive only a few months, but a few have lived into early adulthood 2.
Fumarase deficiency is a very rare disorder. Approximately 100 affected individuals have been reported worldwide as of 2017. Fumarase deficiency occurs in people of different ethnic backgrounds, but several people with fumarase deficiency were born in an isolated religious in the southwestern United States (around the border of northern Arizona and southern Utah) 2. The increased frequency in this area is thought to be due to a founder effect in closed, polygamist communities in which marriage between relatives is more common 3.
Fumarase deficiency is caused by mutations in the FH gene and inheritance is autosomal recessive 1. Unfortunately, there is no effective treatment at this time 1. Management aims to improve symptoms and increase quality of life.
How can I find individuals or families affected by fumarase deficiency?
You may be able to connect with individuals and/or families affected by fumarase deficiency through Climb (http://www.climb.org.uk), the National Information Centre for Metabolic Diseases. This organization works on behalf of children, young people, adults and families affected by metabolic disease. They have a Climb Family Contacts Network, which aims to help reduce the sense of isolation felt by many families affected by a rare disease, giving families the opportunity to share experiences, information and advice.
Fumarase deficiency causes
Fumarase deficiency is caused by mutations in the FH gene. This gene provides instructions for making an enzyme called fumarase (also known as fumarate hydratase). Fumarase participates in an important series of reactions known as the citric acid cycle or Krebs cycle, which allows cells to use oxygen and generate energy. Specifically, fumarase helps convert a molecule called fumarate to a molecule called malate.
Mutations in the FH gene disrupt the enzyme’s ability to help convert fumarate to malate, interfering with the function of this reaction in the citric acid cycle. Impairment of the process that generates energy for cells is particularly harmful to cells in the developing brain, and this impairment results in the signs and symptoms of fumarase deficiency.
Fumarase deficiency inheritance pattern
Fumarase deficiency is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. However, people with one mutated copy of the FH gene in each cell, including parents of individuals with fumarase deficiency, tend to develop benign tumors containing smooth muscle tissue (leiomyomas) in the skin and, in females, the uterus 4. They also have an increased risk of kidney cancer. This condition is called hereditary leiomyomatosis and renal cell cancer (HLRCC) 5.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
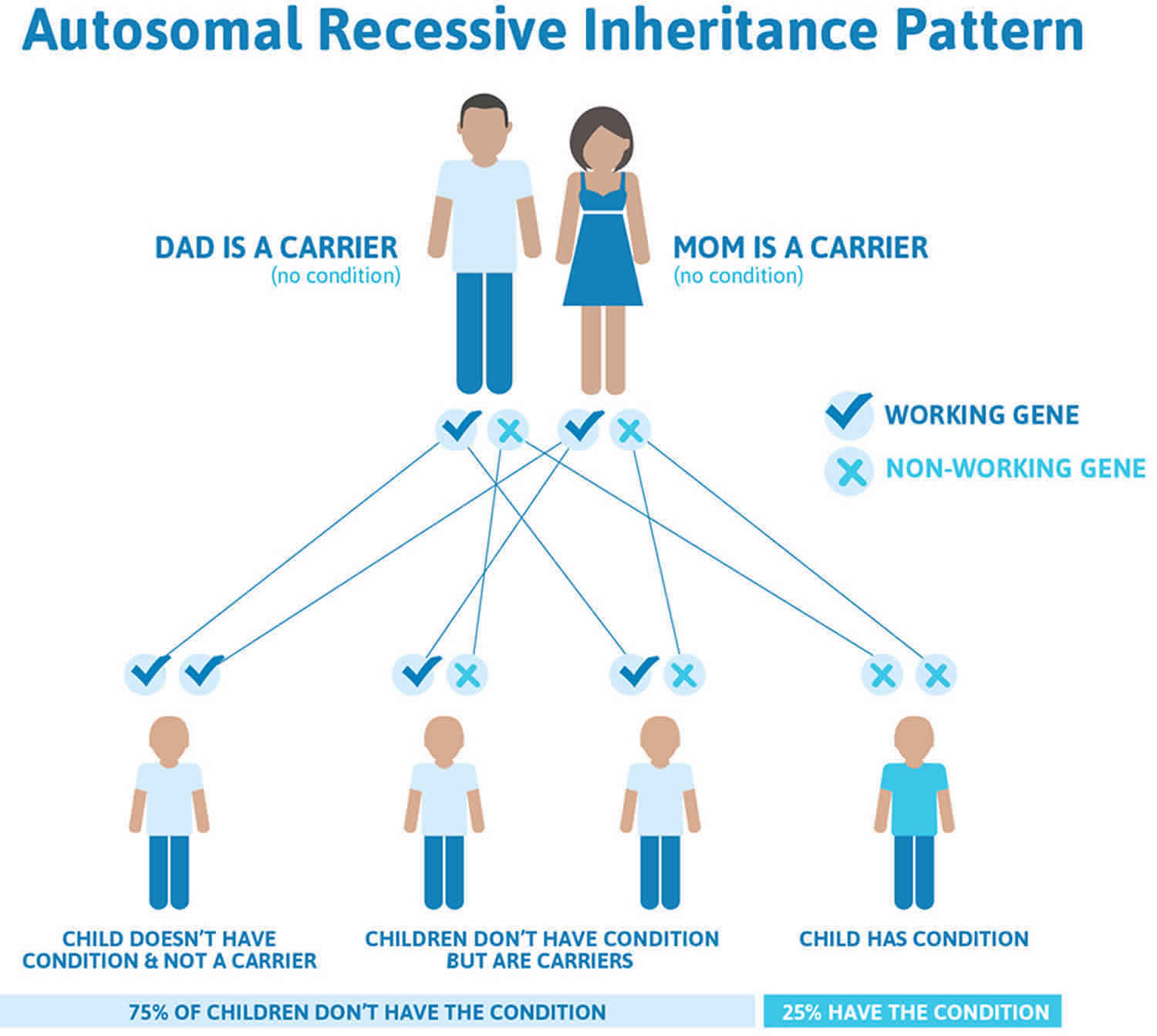
Figure 1 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 1. Fumarase deficiency autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Fumarase deficiency symptoms
Most newborns with fumarase deficiency have severe neurologic abnormalities including poor feeding, failure to thrive, and poor muscle tone (hypotonia). Early-onset infantile encephalopathy (abnormal brain structure or function), seizures, microcephaly, and severe developmental delay are also common. Most children with fumarase deficiency do not learn to walk and talk. Some also have polycythemia (an excess of red blood cells), recurrent vomiting, or pancreatitis. Distinctive facial features that may be present include a prominent forehead (frontal bossing), depressed nasal bridge, and widely-spaced eyes 1.
A variety of brain abnormalities may be identified on brain MRI. These may include cerebral atrophy (loss of neurons and their connections to each other), enlarged ventricles (cavities where cerebrospinal fluid is produced), thinning or absence (agenesis) of the corpus callosum, and an abnormally small brain stem (the part of the brain that connects to the spinal cord) 1.
Fetal manifestations. Few clinical reports comment on complications of affected pregnancies. However, polyhydramnios, oligohydramnios, intrauterine growth retardation, and premature birth (typically 33-36 weeks) are reported in approximately one-third of affected pregnancies 6. Enlarged cerebral ventricles and other brain abnormalities have been identified by fetal ultrasound 7.
Neonatal and early infantile encephalopathy. Newborns with fumarate hydratase deficiency may be symptomatic immediately following delivery or may appear normal at birth, and be discharged home from the nursery without recognized problems 8. If symptoms are not apparent at birth, affected infants show severe neurologic abnormalities within age one week to one month, including poor feeding, failure to thrive, and hypotonia. These newborns and infants manifest encephalopathy, with poor eye contact and variable degrees of depressed consciousness including lethargy, stupor, and even coma. Head and neck control may be entirely absent. Infants gain weight slowly and may require tube feedings.
Epileptic seizures are common (40%-80%), although age of onset and seizure type are variable 9. Infantile spasms (epileptic spasms) accompanied by hypsarrhythmia on EEG have been reported 10. Seizures are often treatment resistant.
Dysmorphology. Abnormal facial features with a spectrum of specific findings have been widely reported and should be regarded as a hallmark feature of this condition (although perhaps not universal). Common features (>50% of affected individuals) include depressed nasal bridge, frontal bossing, and widely spaced eyes 9. Less frequent features (<50%) include cleft ala nasi or anteverted nares, ear anomalies, or narrow forehead 9.
Head size. Head size has been reported as abnormally small (microcephalic) in 36% of all affected individuals 9. However, in one large kindred (8 affected individuals in 1 consanguineous family), 88% (7 of 8 affected individuals) were reported to have “relative macrocephaly,” since head sizes were within the normal range, but in association with brain imaging findings of cerebral atrophy and mild communicating hydrocephalus (enlarged extra-axial CSF spaces) 11. That is, it appears that most children with fumarate hydratase deficiency have abnormally limited brain growth.
Acute metabolic derangements. Acute metabolic crises with findings such as hypoglycemia, ketosis, hyperammonemia, or acidosis are rarely observed in fumarate hydratase deficiency. One such patient is reported in detail by Allegri et al 9, with repeated episodes of hypoglycemia during the first two weeks of life, associated with metabolic acidosis, elevated lactic acidemia, and mild hyperammonemia.
Brain imaging findings. As with abnormal facial features, abnormalities of brain development are to be expected with fumarate hydratase deficiency, but significant person-to-person (or kindred-to-kindred) differences are described. Completely normal brain magnetic resonance imaging (MRI) may occur, but should make one question the diagnosis.
The most common finding is a small brain, representative of cerebral under-development. This may be described by the neuroradiologist as cerebral atrophy [73% of all cases summarized by Allegri et al 9], or ventriculomegaly [82% of all cases summarized by Allegri et al 9]. Brain volume loss (or more likely lack of brain volume development) can be accompanied by a relative decrease in CSF reabsorption, leading to a normal head size with a small brain but modestly expanded CSF compartments. In the series of Kerrigan et al 11, two such individuals were shunted for possible “hydrocephalus” leading to collapse of the CSF compartments and secondary microcephaly without clinical improvement.
Additional findings on MRI can include nonspecific white matter abnormalities, described as either delayed myelination or hypomyelination 8, deficient closure of the Sylvian opercula 8, and a small brain stem 8. Abnormalities of the corpus callosum are also reported, including thinning 8 and absence 7. Diffuse bilateral polymicrogyria of the cerebral cortex has also been reported, a universal feature in the eight affected individuals from one kindred reported by Kerrigan et al 11 but also noted in three additional unrelated individuals 12.
Other clinical features. Other findings can include neonatal polycythemia 11, recurrent vomiting with hepatosplenomegaly 9, and pancreatitis 8.
Visual disturbances and optic nerve hypoplasia were described in one family 11. Despite the congenital anomalies of the brain that occur with fumarate hydratase deficiency, birth defects involving other organ systems are uncommon.
Fumarase deficiency diagnosis
Isolated increased concentration of fumaric acid on urine organic acid analysis is highly suggestive of fumarate hydratase deficiency 1. The diagnosis is confirmed by identification of deficient fumarate hydratase enzyme activity in fibroblasts, lymphoblasts, or white blood cells and/or by molecular genetic testing of FH, the gene that encodes fumarate hydratase and the only gene in which mutation is known to cause fumarate hydratase deficiency. Fumarate hydratase enzyme activity in severely affected individuals is generally less than 10% of the control mean; however, residual fumarate hydratase enzyme activity in some affected individuals can be 11%-35% of the control mean, overlapping with that seen in some obligate heterozygotes.
Fumarase deficiency treatment
Currently there is no effective treatment for fumarase deficiency. The goal of management is to improve symptoms, prevent complications, and increase quality of life. Examples of management options include 1:
- Standard anticonvulsant medications for seizures.
- Placement of a feeding tube (if needed) for optimal nutrition and to prevent aspiration.
- Special needs services for support with motor, language, and social development.
The ketogenic diet is usually considered to be contraindicated for treating epilepsy associated with fumarate hydratase deficiency or other enzymatic defects within the Krebs tricarboxylic acid cycle 1.
Fumarase deficiency life expectancy
Many children with fumarase deficiency die during early childhood, but some are less severely affected, have mild cognitive impairment, and survive beyond this time 13.
The clinical outcome for individuals with fumarate hydratase deficiency is not favorable. Many such individuals do not survive infancy, or may die of secondary complications (e.g., respiratory failure) during the first decade of life 10. Many children are unable to feed successfully, with failure to gain weight and increased risk for aspiration. Accordingly, feedings administered through gastrostomies may be required.
Over time, severely affected children (usually nonverbal and nonambulatory) develop evidence of spasticity, and consequently are at risk for contractures and orthopedic deformities, including scoliosis. Extrapyramidal motor features, including athetosis and dystonic posturing, can also be observed. Epileptic seizures often become more frequent and less responsive to treatment efforts. Seizures may occur daily in some individuals.
However, less severely affected children, who may be ambulatory and capable of engaging in special needs school programs (despite the presence of bilateral polymicrogyria), are also recognized 12. Consequently, counseling families with children with fumarate hydratase deficiency should include recognition of the range of severity.
References- Ewbank C, Kerrigan JF, Aleck K. Fumarate Hydratase Deficiency. 2006 Jul 5 [Updated 2013 Apr 4]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1506
- Fumarase deficiency. https://ghr.nlm.nih.gov/condition/fumarase-deficiency
- Allegri G. et al. Fumaric aciduria: an overview and the first Brazilian case report. J Inherit Metab Dis. August 2010; 33(4):411-419.
- Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S, Roylance RR, Olpin S, Bevan S, Barker K, Hearle N, Houlston RS, Kiuru M, Lehtonen R, Karhu A, Vilkki S, Laiho P, Eklund C, Vierimaa O, Aittomäki K, Hietala M, Sistonen P, Paetau A, Salovaara R, Herva R, Launonen V, Aaltonen LA., Multiple Leiomyoma Consortium. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30:406–10.
- Maradin M, Fumić K, Hansikova H, Tesarova M, Wenchich L, Dorner S, Sarnavka V, Zeman J, Barić I. Fumaric aciduria: mild phenotype in a 8-year-old girl with novel mutations. J Inherit Metab Dis. 2006;29:683.
- Saini AG, Singhi P. Infantile metabolic encephalopathy due to fumarase deficiency. J Child Neurol. 2013;28:535–7.
- Coughlin EM, Christensen E, Kunz PL, Krishnamoorthy KS, Walker V, Dennis NR, Chalmers RA, Elpeleg ON, Whelan D, Pollitt RJ, Ramesh V, Mandell R, Shih VE. Molecular analysis and prenatal diagnosis of human fumarase deficiency. Mol Genet Metab. 1998;63:254–62.
- Phillips TM, Gibson JB, Ellison DA. Fumarate Hydratase Deficiency in Monozygotic Twins. Pediatr Neurol. 2006;35:150–3.
- Allegri G, Fernandes MJ, Scalco FB, Correia P, Simoni RE, Llerena JC Jr, de Oliveira ML. Fumaric aciduria: an overview and the first Brazilian case report. J Inherit Metab Dis. 2010;33:411–9.
- Loeffen J, Smeets R, Voit T, Hoffmann G, Smeitink J. Fumarase deficiency presenting with periventricular cysts. J Inherit Metab Dis. 2005;28:799–800.
- Kerrigan JF, Aleck KA, Tarby TJ, Bird CR, Heidenreich RA. Fumaric aciduria: clinical and imaging features. Ann Neurol. 2000;47:583–8.
- Ottolenghi C, Hubert L, Allanore Y, Brassier A, Altuzarra C, Mellot-Draznieks C, Bekri S, Goldenberg A, Veyrieres S, Boddaert N, Barbier V, Valayannopoulos V, Slama A, Chrétien D, Ricquier D, Marret S, Frebourg T, Rabier D, Munnich A, de Keyzer Y, Toulhoat H, de Lonlay P. Clinical and biochemical heterogeneity associated with fumarase deficiency. Hum Mutat. 2011;32:1046–52.
- Fumaric aciduria. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=24





{kind=link}