Hemoglobin H disease
Hemoglobin H disease also known as Alpha thalassemia intermedia that is caused by deletion of three alpha-globin genes (only one normal alpha gene has been inherited) 1. Hemoglobin H disease is named for the abnormal hemoglobin H (created by the remaining beta globin) that destroys red blood cells. Alpha-thalassemia is an inherited blood disorder that reduces the body’s production of hemoglobin. Affected people have anemia, which can cause pale skin, weakness, fatigue, and more serious complications. Two types of alpha-thalassemia can cause health problems: the more severe type is known as hemoglobin Bart syndrome (Alpha Thalassemia Major or Hemoglobin Bart’s Hydrops Fetalis); the milder form is called hemoglobin H disease (HbH disease). Hemoglobin Bart syndrome may be characterized by hydrops fetalis; severe anemia; hepatosplenomegaly; heart defects; and abnormalities of the urinary system or genitalia. Most babies with Hemoglobin Bart syndrome are stillborn or die soon after birth. Hemoglobin H disease is characterized by microcytic hypochromic hemolytic anemia which may cause mild to moderate anemia; hepatosplenomegaly; mild jaundice; and sometimes thalassemia-like bone changes 2. Individuals with hemoglobin H disease may also develop gallstones and experience acute episodes of hemolysis in response to oxidant drugs and infections. One of these most common non-deletion subtypes of hemoglobin H is called Hemoglobin H-Constant Spring. Hemoglobin H-Constant Spring is more severe than hemoglobin H disease. Individuals with Hemoglobin H-Constant Spring tend to have a more severe anemia and suffer more frequently from enlargement of the spleen and viral infections. Hemoglobin H disease tends to be more severe in patients with the non-deletion-type likely due to interference with the transcription of the normal alpha chain gene by the abnormal one 1. No treatment is effective for hemoglobin Bart syndrome. For hemoglobin H disease, occasional red blood cell transfusions may be needed 2.
Alpha thalassemia traits are thought to be protective against malaria, and in populations with high incidences of malaria, the trait can be found in up to 90% of the population. Hemoglobin H disease is similar and found mostly in warm climates. The populations with the highest incidences are found in Southeast Asia, the Mediterranean, and the Middle East. Hemoglobin Constant Spring is the most common form of non-deletion alpha thalassemia. One percent to 2% of individuals living in northeastern Thailand, 5% to 8% of individuals in southern China, and one-quarter of women in an ethnic minority population in Vietnam are found to have Hemoglobin-H Constant Spring 3.
What is hemoglobin?
Hemoglobin (Hb or Hgb) is made of iron-containing portion (heme) and proteins (globin chains). The globin protein consists of chains of amino acids, the “building blocks” of proteins. One portion of hemoglobin called heme is the molecule with iron at the center (see Figure 1). Another portion is made of up four protein chains called globins. Depending on their structure, the globin chains are designated as alpha (α), beta (ß), delta (δ), and gamma (γ). Each of the four globin chains holds a heme group containing one iron atom. Hemoglobin (Hb) is the iron-containing protein found in all red blood cells (RBCs) that gives red blood cells their characteristic red color and it carries oxygen (O2) throughout the body. Hemoglobin enables red blood cells to bind to oxygen in the lungs and carry oxygen to tissues and organs throughout your body. Hemoglobin also helps transport a small portion of carbon dioxide (CO2), a product of cell metabolism, from tissues and organs to the lungs, where it is exhaled 4. Under normal circumstances, adult blood contains 4 types of hemoglobin: oxygenated hemoglobin or oxyhemoglobin, hemoglobin that contains no oxygen (reduced hemoglobin) or deoxyhemoglobin, hemoglobin transporting carbon dioxide or carboxyhemoglobin, and oxidized hemoglobin or methemoglobin. The last 2 are usually found in small amounts in blood.
Not all hemoglobin is the same. Different types of hemoglobin are classified according to the type of globin chains they contain. The type of globin chains present is important in hemoglobin’s ability to transport oxygen. There are three main types of normal hemoglobin found in adults: Hemoglobin A, hemoglobin A2, and hemoglobin F are the types of globin molecule combinations, namely alpha (α), beta (ß), delta (δ), and gamma (γ), determine the type of hemoglobin. All of the normal hemoglobin is a combination of alpha and non-alpha chains. The gene for alpha globin is located on chromosome 16. Hemoglobin A is composed of one pair of alpha-globin chains and one pair of beta-globin chains. Hemoglobin A (HbA), composed of both alpha and beta-globin chains, is the type of hemoglobin that typically makes up 95% to 98% of adult hemoglobin. Hemoglobin A2 (HbA2) is a pair of alpha chains and a pair of delta chains. Hemoglobin A2 (HbA2) makes 1% to 3% of adult hemoglobin. Hemoglobin F (HbF) is comprised of two alpha and two gamma chains. Hemoglobin F (HbF) makes up for the majority of neonatal hemoglobin, but in normal adults is 2% to 3% of the total hemoglobin. The percentages fluctuate based on age, genetics, medications, and underlying conditions 5.
Normal hemoglobin types include:
- Hemoglobin A (Hb A) – this is the predominant type of hemoglobin (Hb) in adults, makes up about 95%-98% of hemoglobin found in adults; Hb A contains two alpha (α) protein chains and two beta (ß) protein chains.
- Hemoglobin A2 (Hb A2) – makes up about 2-3.5% of hemoglobin (Hb) found in adults; it has two alpha (α) and two delta (δ) protein chains.
- Hemoglobin F (Hb F, fetal hemoglobin) – makes up to 1%-2% of hemoglobin (Hb) found in adults; it has two alpha (α) and two gamma (γ) protein chains. Hemoglobin F (Hb F, fetal hemoglobin) is the primary hemoglobin produced by a developing baby (fetus) during pregnancy. Its production usually falls to a low level within a year after after birth and reaches adult level within 1-2 years.
For hemoglobin (Hb), there are four genes in your DNA that code for the alpha (α) globin chains and two genes (each) for the beta (β), delta (δ), and gamma (γ) globin chains. Globin polypeptides are synthesized from separate α-like and β-like globin gene clusters located on human chromosomes 16 and 11, respectively. Since everyone inherits a set of chromosomes from each parent, each person inherits two alpha globulin genes and one beta globulin gene from each parent. A person may inherit mutations in either the alpha or beta globin genes.
The hemoglobin test measures the amount of hemoglobin in a person’s sample of blood. Serum free hemoglobin is a blood test that measures the level of free hemoglobin in the liquid part of the blood (the serum). Free hemoglobin is the hemoglobin outside of the red blood cells. Most of the hemoglobin is found inside the red blood cells, not in the serum. A hemoglobin level can be performed alone or with a hematocrit, a test that measures the proportion of blood that is made up of red blood cells, to quickly evaluate an individual’s red blood cells. Red blood cells, which make up about 40% (ranging 37-49%) of the blood’s volume, are produced in the bone marrow and are released into the bloodstream when they are, or nearly are, mature. The typical lifespan of an red blood cell is 120 days, and the bone marrow must continually produce new red blood cells to replace those that age and degrade or are lost through bleeding.
Several diseases and conditions can affect red blood cells and consequently the level of hemoglobin in the blood. In general, the hemoglobin level and hematocrit rise when the number of red blood cells increases. The hemoglobin level and hematocrit fall to less than normal when there is a drop in production of red blood cells by the bone marrow, an increase in the destruction of red blood cells, or if blood is lost due to bleeding. A drop in the red blood cell count, hemoglobin and hematocrit can result in anemia, a condition in which tissues and organs in the body do not get enough oxygen, causing fatigue and weakness. If too many red blood cells are produced, polycythemia results and the blood can become thickened, causing sluggish blood flow and related problems.
Since a hemoglobin level is often performed as part of a complete blood count (CBC), results from other components are taken into consideration. A rise or drop in the hemoglobin level must be interpreted in conjunction with other parameters, such as red blood cell count, hematocrit, reticulocyte count, and/or red blood cell indices. Age, sex, and race are other factors to be considered.
The hemoglobin test is a common test and is almost always done as part of a complete blood count (CBC). Reasons or conditions for ordering the hemoglobin test include:
- Symptoms such as fatigue, poor health, or unexplained weight loss
- Signs of bleeding
- Before and after major surgery
- During pregnancy
- Chronic kidney disease or many other chronic medical problems
- Monitoring of anemia and its cause
- Monitoring during treatment for cancer
- Monitoring medicines that may cause anemia or low blood counts
In general, hemoglobin mirrors the results of the red blood cell count and hematocrit.
- A recent blood transfusion can affect a person’s hemoglobin level.
- Hemoglobin decreases slightly during normal pregnancy.
Low hemoglobin with low red blood cell count and low hematocrit indicates anemia.
Figure 1. Red blood cell hemoglobin
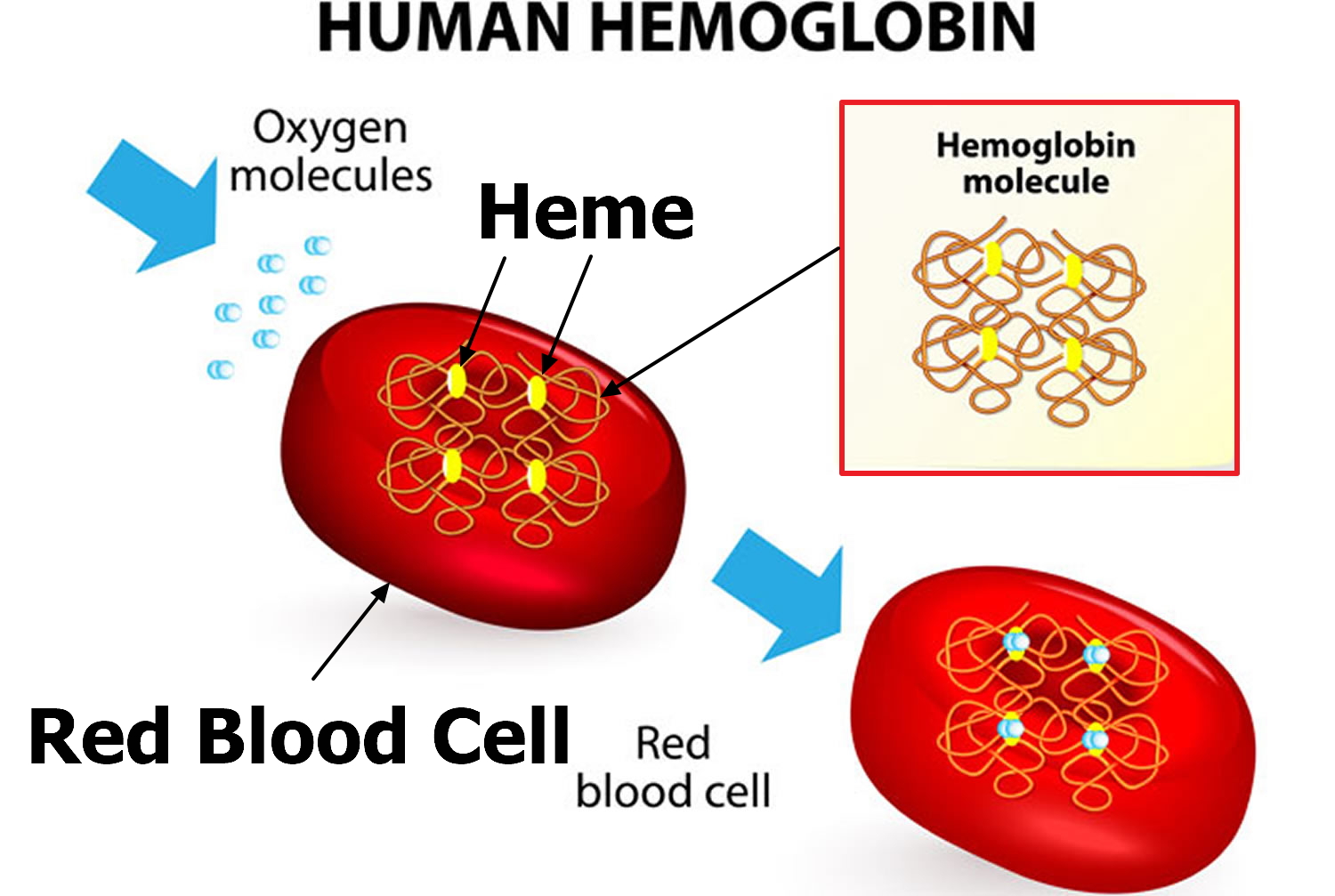
Figure 2. Structure of hemoglobin
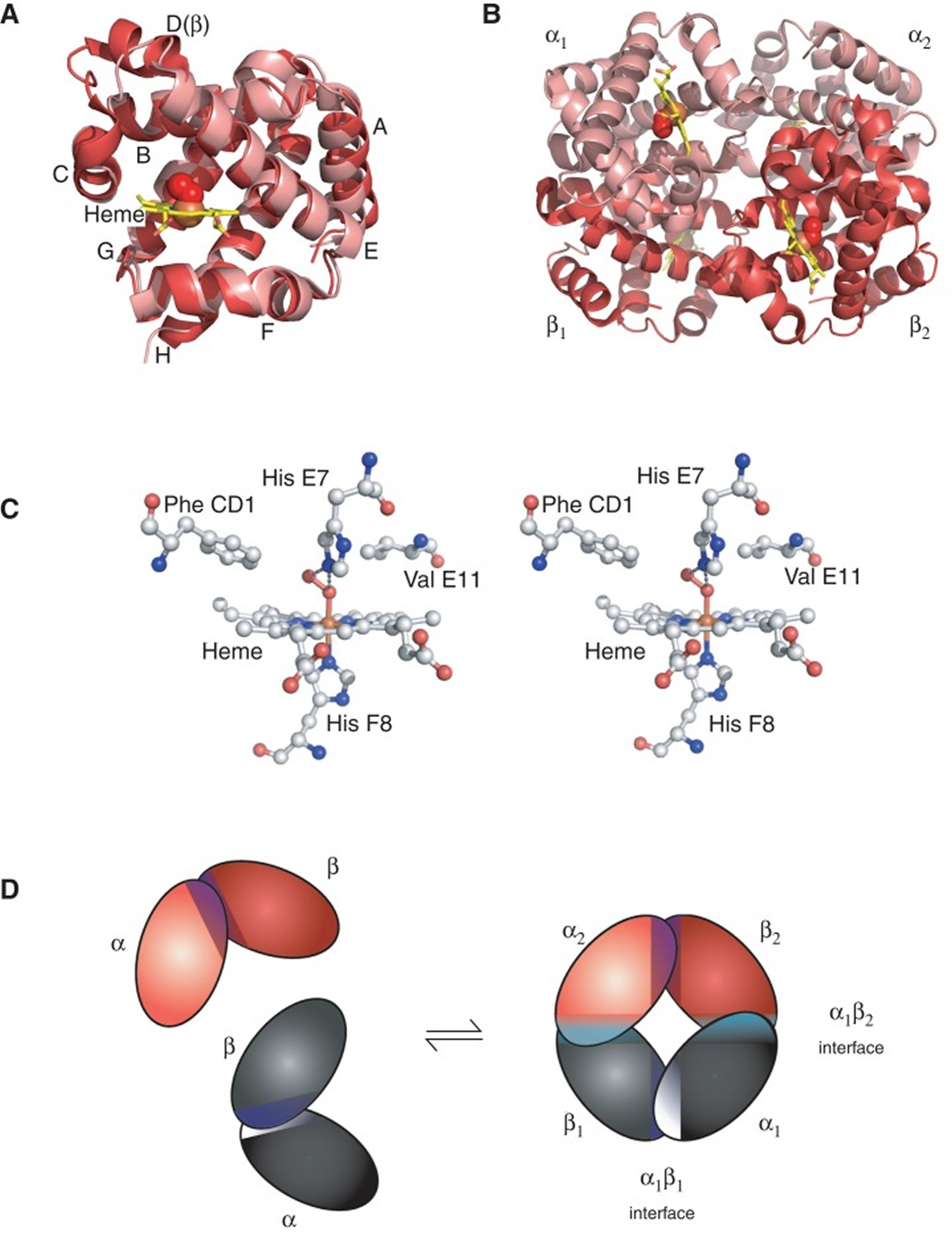
Footnotes: The structure of hemoglobin (Hb). (A) The α (pink) and β (red) hemoglobin (Hb) subunits have conserved α-helical folds. Helices are labeled A–H from the amino terminus. The α subunit lacks helix D. (B) The high O2 affinity R state quaternary structure of hemoglobin (Hb) with O2 (red spheres) bound at all four heme sites (protoporphyrin-IX as yellow sticks, with central iron atom as orange sphere). (C) Stereo (wall-eye) diagram of the heme pocket of β showing the proximal (F8) and distal (E7) histidines and selected residues in the distal heme pocket that influence ligand binding and autoxidation. (D) hemoglobin (Hb) tetramer is assembled from two identical αβ dimers (shown in red and gray for clarity). In the tetramer, each subunit makes contact with the unlike chain through a high affinity dimerization α1β1 interface and a lower affinity α1β2 dimer–tetramer interface (cyan).
What is thalassemia?
Thalassemia is the name of a group of genetic blood disorders caused by decreased synthesis of alpha or beta chains of hemoglobin (Hb). The production can be diminished or can be absent for one or more of the globin chains. This imbalance of globin chain production impairs the production of normal hemoglobin. This impairment causes ineffective erythropoiesis with intramedullary hemolysis. Thalassemia is an inherited disease, meaning that at least one of the parents must be a carrier for the disease. It is caused by either a genetic mutation or a deletion of certain key gene fragments. Alpha thalassemia refers specifically to the abnormal or absent manufacturing of alpha-globin chains. These are associated with more than 15 different genetic mutations. The severity of the clinical condition is based on the mutation type. The severity of mutation is based on which of the two alpha-globin loci is affected. Alpha globin gene has 4 alleles and disease severity ranges from mild to severe depending on the number of deletions of the alleles. Mutations can also be deletion or non-deletion. In deletion mutation, there is an inheritance of a single alpha-globin gene. With the non-deletion type, a patient has inherited two alpha-globin genes, but one gene carries a non-deletion abnormality, for example, point mutation. In non-deletion, the severity of clinical expression is also affected depending on whether the mutation blocks the production of the remaining normal alpha chains partially or fully. Hemoglobin H disease occurs when only one normal alpha gene has been inherited. One of these most common non-deletion subtypes of Hemoglobin H is called Hemoglobin Constant Spring. Hemoglobin H disease tends to be more severe in patients with the non-deletion-type likely due to interference with the transcription of the normal alpha chain gene by the abnormal one. Four allele deletion is the most severe form in which no alpha globins are produced and the excess gamma chains (present during the fetal period) form tetramers. It is incompatible with life and results in hydrops fetalis.
Beta thalassemia results from point mutations in the beta-globin gene. It is divided into three categories based on the zygosity of the beta-gene mutation. A heterozygous mutation (beta-plus thalassemia) results in beta-thalassemia minor in which beta chains are underproduced. It is mild and usually asymptomatic. Beta thalassemia major is caused by a homozygous mutation (beta-zero thalassemia) of the beta-globin gene, resulting in the total absence of beta chains. It manifests clinically as jaundice, growth retardation, hepatosplenomegaly, endocrine abnormalities, and severe anemia requiring life-long blood transfusions. The condition in between these two types is called beta-thalassemia intermedia with mild to moderate clinical symptoms.
- One mutated gene: Mild signs and symptoms. The condition is called thalassemia minor.
- Two mutated genes: Signs and symptoms will be moderate to severe. This condition is called thalassemia major, or Cooley anemia. Babies born with two mutated beta hemoglobin genes are usually healthy at birth but disease starts to manifest after 6 months of life when fetal hemoglobin (Hb-gamma) disappears and is replaced by adult hemoglobin.
The excess unpaired alpha-globin chains in beta-thalassemia aggregate and form precipitates that damage red cell membranes and result in intravascular hemolysis. This premature death of erythroid precursor cells leads to ineffective erythropoiesis and later results in extramedullary expansion of hematopoiesis.
Coinheritance of alpha thalassemia: Beta-thalassemia patients with coinheritance of alpha thalassemia have a milder clinical course due to a less severe alpha-beta chain imbalance.
Coexistence of sickle cell trait: The presence of sickle cell trait with beta-thalassemia is a major hemoglobinopathy and results in manifestations of sickle cell disease. Unlike sickle cell trait in which major hemoglobin is HbA, in the co-existence state the major hemoglobin is HbS which constitutes more than 60% of hemoglobin depending on the nature of the disease (beta-zero or beta-plus0.)
Hemoglobin-E (HbE) is also a common hemoglobin variant found in Southeast Asia population. It has a correlation with a beta-thalassemia phenotype, as people with thalassemia in this territory are commonly found to have HbE.
Two new terminologies being used more often in clinical settings are transfusion requiring and non-transfusion requiring thalassemias and all the basic classification falls into these two types depending on the requirement of frequent blood transfusions or not 6.
Hemoglobin H disease cause
Hemoglobin H disease forms when only one normal alpha gene has been inherited. This causes significantly impaired alpha globin production. In the neonatal period, this will cause an excess of gamma, and in adults, this leaves an excess of beta-globin chains. Free alpha chains are insoluble. Both gamma and beta chains are soluble and make homotetramers. Hemoglobin H is made of four beta chains, and hemoglobin Barts is made of four gamma chains. They are, however, unstable and some precipitate within the cell, leading to a variety of clinical manifestations. Hemoglobin H in adults can make up to 40% of circulating hemoglobin in affected individuals. This hemoglobin is more susceptible to oxidant injury and has poor oxygen-carrying capacity. Its affinity is ten times more than HbA. It has an abnormal oxyhemoglobin dissociation curve. This means that it can bind to oxygen, but does not deliver it to tissues normally.
HbH disease
- Most often caused by a large deletion on one allele in trans with a single α-globin-gene deletion (–/-α) or other non-deletion inactivating variant (–/αNDα or –/ααND)
- Individuals homozygous for the Constant Spring variant (αCSα/αCSα) or HBA2 poly (A) pathogenic variant may have HbH disease.
- Individuals who are homozygous or compound heterozygous for highly unstable α-globin gene variants may have HbH disease.
- Rarely, HbH disease is caused by a compound heterozygous or homozygous HS40 deletion 7.
Hemoglobin H can cause chronic hypochromic microcytic anemia and hemolytic anemia, which can worsen in periods of oxidant stress. This can be effectively broken down as ineffective erythropoiesis and increased hemolysis. The microcytic hypochromic anemia is due to impaired hemoglobin production due to decreased alpha chain synthesis and hyperhydration of the cell. The cause of the hyperhydration in alpha thalassemia is not clear. One theory argues that the K-Cl (potassium chloride) cotransporter stops early, thereby preventing the usual loss of K-Cl and water that is part of the red blood cell remodeling process.
Hemoglobin H has also shown shortened survival down to half of normal, 12 to 19 days versus normal of 28 to 37 days. This has accounted for two main factors: abnormal red blood cell membrane with increased rigidity and increased inclusion bodies. The inclusions were thought to be aggregates of beta chain tetramers, which precipitate in the red blood cell and cause damage, leading to their removal by the spleen. These inclusion bodies are also proposed to cause increased susceptibility to oxidant stress.
Hemoglobin H disease symptoms
HbH disease is classified as moderate to severe in alpha thalassemia. Due to the variety of genetic mutations, as explained previously, there is marked variation in phenotypic expression. Patients can be asymptomatic, have episodic anemia requiring transfusions, and even fatal hydrops fetalis in utero.
Clinical presentation usually starts in the latter part of gestation with hemolytic anemia due to the formation of hemoglobin Barts. The most severe form will result in fatal hydrops fetalis. At birth, neonates present with jaundice and anemia.
As they age, patients will have fluctuating degrees of anemia. This anemia is due to a combination of ineffective erythropoiesis and hemolysis. Periods of increased oxidant stress, such as sepsis or certain medications, tend to increase the rate of hemolysis. Patients would then have the typical presentation of hemolytic anemia, indirect hyperbilirubinemia, elevated LDH, decreased haptoglobin, splenomegaly, hepatomegaly, jaundice, variable bony changes, premature biliary tract disease, leg ulcers, and eventually iron overload if multiple transfusions are required without adequate iron chelation.
In adults, the diagnosis is suspected in patients with microcytic anemia with normal iron stores and a normal hemoglobin A2 level on electrophoresis. Hemoglobin levels are usually 8 to 10 g/dL at baseline.
Hemoglobin H disease diagnosis
Several laboratory tests have been developed to screen and diagnose thalassemia:
- Complete blood count (CBC): CBC is often the first investigation in a suspected case of thalassemia. A CBC showing low hemoglobin (Hb Male: 10.9±1.0 g/dL; Female: 9.5±0.8 g/dL) and low MCV (low MCV [mean corpuscular volume] – Children: 56±5; Adults: 61±4 fl) is the first indication of thalassemia, after ruling out iron deficiency as the cause of anemia. Calculation of the Mentzer index (mean corpuscular volume divided by red cell count) is useful. A Mentzer lower than 13 suggests that the patient has thalassemia, and an index of more than 13 suggests that the patient has anemia due to iron deficiency 8.
- Peripheral blood smear: A blood smear also called peripheral smear and manual differential is next, to assess additional red cell properties. The peripheral blood film in hemoglobin H disease shows hypochromia (low MCH [mean corpuscular hemoglobin] 18.4±1.2 pg) and microcytosis (low MCV – Children: 56±5 fl; Adults: 61±4 fl) with inclusion bodies 9. Inclusion bodies are better visualized with a supravital dye such as methyl violet or brilliant cresyl blue.
- Bone marrow demonstrates erythroid hyperplasia with poorly hemoglobinized erythroblasts carrying inclusion bodies.
- Hemoglobin electrophoresis or chromatographic techniques can demonstrate HbH.
- DNA-based genotyping is required for precise diagnosis and is important in prenatal testing and genetic counseling. Since having relatives carrying mutations for thalassemia increases a person’s risk of carrying the same mutant gene, family studies may be necessary to assess carrier status and the types of mutations present in other family members.
- Genetic testing of amniotic fluid is useful in those rare instances where a fetus has an increased risk for thalassemia. This is particularly important if both parents likely carry a mutation because that increases the risk that their child may inherit a combination of abnormal genes, causing a more severe form of thalassemia. Prenatal diagnosis with chorionic villi sampling at 8 to 10 weeks or by amniocentesis at 14 to 20 weeks’ gestation can be carried out in high-risk families 10.
- Iron studies (serum iron, ferritin, unsaturated iron-binding capacity (UIBC), total iron-binding capacity (TIBC), and percent saturation of transferrin) are also done to rule out iron deficiency anemia as the underlying cause.
- Erythrocyte porphyrin levels may be checked to distinguish an unclear beta-thalassemia minor diagnosis from iron deficiency or lead poisoning. Individuals with beta-thalassemia will have normal porphyrin levels, but those with the latter conditions will have elevated porphyrin levels.
- Multisystem evaluation: Evaluation of all related systems should be done on a regular basis due to their frequent involvement in the disease progression. Biliary tract and gall bladder imaging, abdominal ultrasonography, cardiac MRI, serum hormone measurements are a few examples which can be done or repeated depending on the clinical suspicion and case description.
The most important differential diagnosis of all the thalassemia syndromes is iron deficiency, which should always be ruled out. Beta thalassemia and combinations of sickle cell disease and thalassemia should also be ruled out.
Table 1. Hemoglobin Patterns in Alpha-Thalassemia (Age >12 Months)
| Hemoglobin Type | Normal | Affected | |
|---|---|---|---|
| Hb Bart hydrops fetalis syndrome 1 | HbH disease 2 | ||
| HbA | 96%-98% | 0 | 60%-90% |
| HbF | <1% | 0 | <1.0% |
| Hb Bart | 0 | 85%-90% | 2%-5% |
| HbH | 0 | 0 | 0.8%-40% |
| HbA2 | 2%-3% | 0 | <2.0% |
| Hb Portland | 0 | 10%-15% | 0 |
Footnote:
1. Deletion or inactivation of all four α-globin chains makes it impossible to assemble HbF and HbA. Fetal blood contains mainly Hb Bart (γ4) and 10%-15% of the embryonic hemoglobin Portland (ζ2γ2).
2. Deletion or inactivation of three α-globin chains
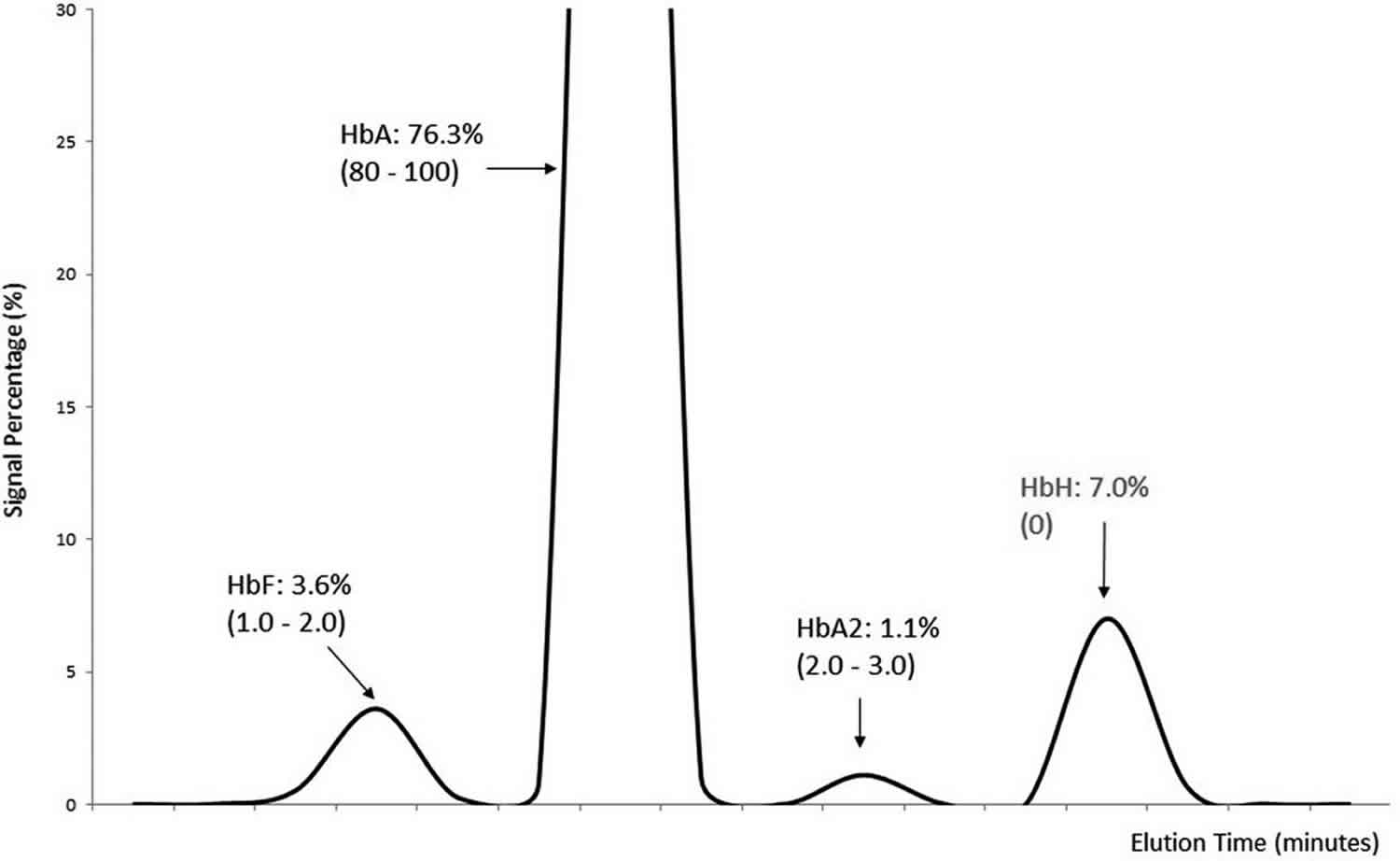
Figure 3. Hemoglobin H electrophoresis
Hemoglobin H (HbH) disease should be suspected in an infant or child with the following newborn screening results, clinical features, and/or laboratory features.
Newborn screening. Hb Bart >15% at birth
- Newborn screening for sickle cell disease offered by several states/countries may detect Hb Bart in the newborn with α-thalassemia.
- Reference ranges may vary among laboratories performing newborn screening.
- Low concentrations of Hb Bart (1%-8%) are indicative of the carrier states and usually do not need further evaluation.
Clinical features
- Hepatosplenomegaly
- Mild thalassemia-like bone changes (e.g., hypertrophy of the maxilla, bossing of the skull, and prominence of the malar eminences)
- Mild jaundice
Laboratory features
- Red blood cell indices: mild-to-moderate (rarely severe) microcytic hypochromic hemolytic anemia
- Moderate reticulocytosis: 3%-6%
- Peripheral blood smear with anisopoikilocytosis, and very rare nucleated red blood cells (i.e., erythroblasts)
- Red blood cell supravital stain showing HbH inclusions (β4 tetramers) in 5% to 80% of erythrocytes following incubation of fresh blood smears with 1% brilliant cresyl blue for one to three hours
The diagnosis of hemoglobin H disease is established in a proband with the above clinical and laboratory features. Identification of pathogenic variants in HBA1 and HBA2 on molecular genetic testing that results in deletion or inactivation of three α-globin genes (e.g., a 2 α-globin-deletion allele in trans with a single α-globin-deletion allele; –/-α3.7) confirms the diagnosis and allows for family studies 2.
Hemoglobin H disease treatment
Supplementation with folic acid should be given because there is hemolytic anemia. HbH patients are at risk of clinical manifestations with oxidative damage. Blood counts should be monitored every six to 12 months to determine the steady state levels of hemoglobin, and occasional red blood cell transfusions may be required during periods of oxidant stress, such as infection or oxidant drug use.
In children, assessment of growth and development every six to 12 months
Chronic red blood cell transfusions should be considered in selected individuals only. Clear indications for red blood cell transfusions are severe anemia affecting cardiac function and massive erythroid expansion, resulting in severe bone changes and extramedullary erythropoiesis. These events are quite rare in HbH disease.
Patients must be monitored carefully for complications of chronic transfusion and given iron chelation therapy as needed in individuals with iron loading caused by regular blood transfusion, inappropriate iron therapy, or abnormal iron absorption, especially during the second and third decades of life. Since serum ferritin may underestimate the degree of iron overload, a periodic quantitative measurement of liver iron concentration is also recommended 12. In individuals with HbH disease who are not red blood cell transfusion dependent, the only iron chelator specifically approved is deferasirox, which has been shown to be superior to placebo in reducing liver iron concentration in those older than age ten years who have beta-thalassemia intermedia, HbE/beta-thalassemia, and HbH disease 13.
Other complications, such as gallstones and leg ulcers, require appropriate medical or surgical treatment.
Patients require careful genetic counseling 14. Test the siblings of a proband as soon as possible after birth for HbH disease so that monitoring can be instituted.
Genetic counseling
Alpha-thalassemia is usually inherited in an autosomal recessive manner. At conception, each sib of an individual with Hb Bart syndrome has a 25% chance of having Hb Bart syndrome, a 50% chance of having α-thalassemia trait with a two-gene deletion or inactivation in cis (–/αα), and a 25% chance of being unaffected and not a carrier.
At conception, if one parent has α-thalassemia trait with a two-gene deletion in cis (–/αα) and the other parent is an α-thalassemia silent carrier (1-gene deletion; -α/αα), each sib of an individual with HbH disease has a 25% chance of having HbH disease, a 25% chance of having α-thalassemia trait, a 25% chance of being an α-thalassemia silent carrier, and a 25% chance of being unaffected and not a carrier. Each child of an individual with HbH disease inherits either the two-gene deletion in cis and has α-thalassemia trait or is an α-thalassemia silent carrier and is thus an obligate heterozygote; risk to the child of having the disease depends on the allele inherited from the other parent.
Family members, members of ethnic groups at risk, and gamete donors should be considered for carrier testing. Couples who are members of populations at risk for α-thalassemia trait with a two-gene deletion in cis (–/αα) can be identified prior to pregnancy as being at risk of conceiving a fetus with Hb Bart syndrome. Prenatal testing may be carried out for couples who are at high risk of having a fetus with Hb Bart syndrome or for a pregnancy in which one parent is a known α-thalassemia carrier with a two-gene deletion in cis (–/αα) when the other parent is either unknown or unavailable for testing.
Surveillance
For HbH disease, hematologic evaluation every six to 12 months; assessment of growth and development in children every six to 12 months; monitoring of iron load with serum ferritin concentration and periodic quantitative measurement of liver iron concentration.
Agents and circumstances to avoid
In HbH disease, inappropriate iron therapy, oxidant drugs such as sulphonamides, and some antimalarials because of the risk of hemolytic crisis.
Pregnancy management
Complications reported in pregnant women with HbH disease include worsening anemia, preeclampsia, congestive heart failure, and threatened miscarriage; monitoring for these issues during pregnancy is recommended.
References- Harewood J, Azevedo AM. Alpha Thalassemia (Hemoglobin H Disease) [Updated 2019 Nov 14]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK441826
- Origa R, Moi P. Alpha-Thalassemia. 2005 Nov 1 [Updated 2016 Dec 29]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1435
- Byrd KA, Williams TN, Lin A, Pickering AJ, Arnold BF, Arnold CD, Kiprotich M, Dentz HN, Njenga SM, Rao G, Colford JM, Null C, Stewart CP. Sickle Cell and α+-Thalassemia Traits Influence the Association between Ferritin and Hepcidin in Rural Kenyan Children Aged 14-26 Months. J. Nutr. 2018 Dec 01;148(12):1903-1910.
- Thom CS, Dickson CF, Gell DA, Weiss MJ. Hemoglobin Variants: Biochemical Properties and Clinical Correlates. Cold Spring Harbor Perspectives in Medicine. 2013;3(3):a011858. doi:10.1101/cshperspect.a011858. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3579210/
- Chen X, Hu J, Zhu J, Xu W, Yao H, Wu A, Xiao M, Lu Z, Yin L, Fu S. Severe hemolytic anemia due to combined α thalassemia and de novo Hemoglobin Sabine. Ann. Hematol. 2019 Mar;98(3):783-785.
- He LN, Chen W, Yang Y, Xie YJ, Xiong ZY, Chen DY, Lu D, Liu NQ, Yang YH, Sun XF. Elevated Prevalence of Abnormal Glucose Metabolism and Other Endocrine Disorders in Patients with β-Thalassemia Major: A Meta-Analysis. Biomed Res Int. 2019;2019:6573497.
- Coelho A, Picanço I, Seuanes F, Seixas MT, Faustino P. Novel large deletions in the human alpha-globin gene cluster: Clarifying the HS-40 long-range regulatory role in the native chromosome environment. Blood Cells Mol Dis. 2010;45:147–53.
- Singha K, Taweenan W, Fucharoen G, Fucharoen S. Erythrocyte indices in a large cohort of β-thalassemia carrier: Implication for population screening in an area with high prevalence and heterogeneity of thalassemia. Int J Lab Hematol. 2019 Aug;41(4):513-518.
- Galanello R, Aru B, Dessì C, Addis M, Paglietti E, Melis MA, Cocco S, Massa P, Giagu N, Barella S, Turco MP, Maccioni L, Cao A. HbH disease in Sardinia: molecular, hematological and clinical aspects. Acta Haematol. 1992;88:1–6.
- Ansari S, Rashid N, Hanifa A, Siddiqui S, Kaleem B, Naz A, Perveen K, Hussain Z, Ansari I, Jabbar Q, Khan T, Nadeem M, Shamsi T. Laboratory diagnosis for thalassemia intermedia: Are we there yet? J. Clin. Lab. Anal. 2019 Jan;33(1):e22647
- Chanchlani N, Rack D, Leigh A. G18(P) Haemoglobin H disease detected during presentation of Idiopathic Thrombocytopenic Purpura in a 14-month old boy. Archives of Disease in Childhood 2016;101:A13-A14. https://adc.bmj.com/content/101/Suppl_1/A13.2
- Musallam KM, Cappellini MD, Wood JC, Taher AT. Iron overload in non-transfusion-dependent thalassemia: a clinical perspective. Blood Rev. 2012;26 Suppl 1:S16–9.
- Taher AT, Porter J, Viprakasit V, Kattamis A, Chuncharunee S, Sutcharitchan P, Siritanaratkul N, Galanello R, Karakas Z, Lawniczek T, Ros J, Zhang Y, Habr D, Cappellini MD. Deferasirox reduces iron overload significantly in nontransfusion-dependent thalassemia: 1-year results from a prospective, randomized, double-blind, placebo-controlled study. Blood. 2012;120:970–7.
- Lin TF, Huang JN, Cash HL. Investigation of Pediatric Anemia in the Commonwealth of the Northern Mariana Islands. Matern Child Health J. 2019 Mar;23(3):416-421.





{kind=link}