Hepcidin
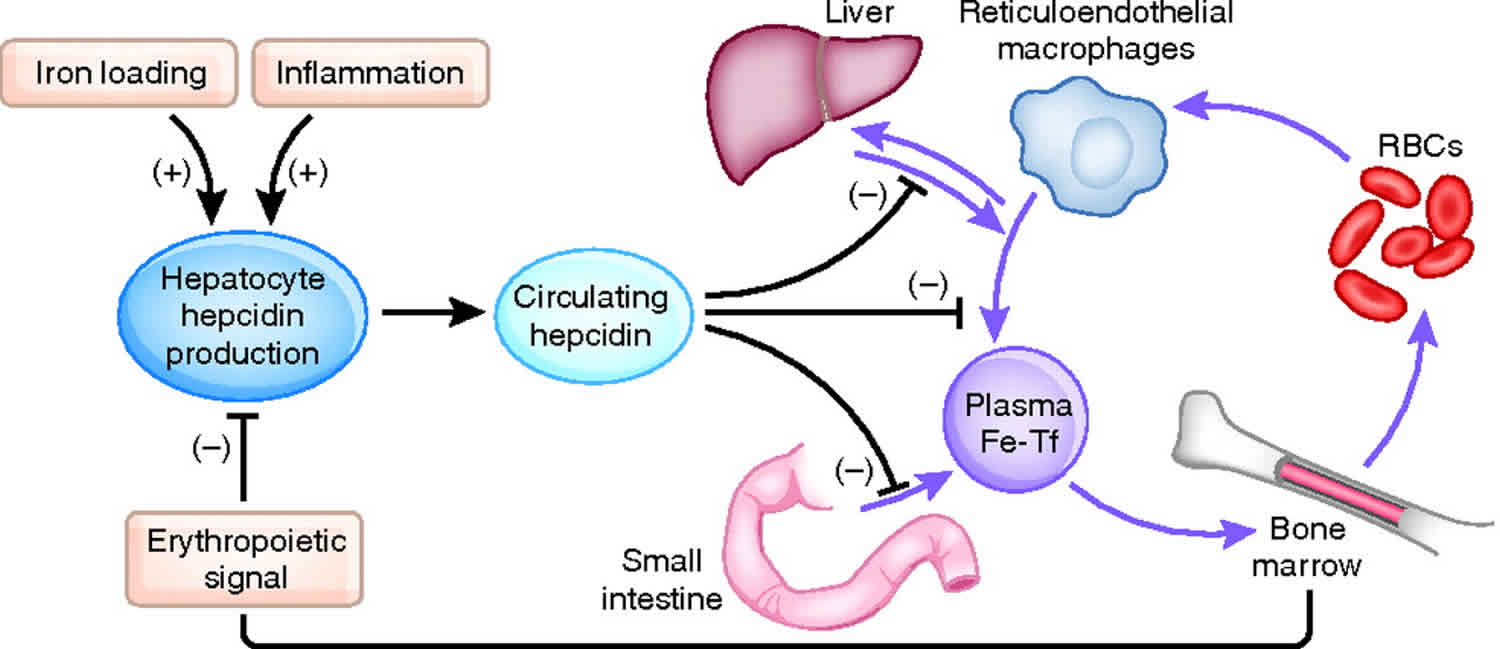
Hepcidin is a protein in humans that is encoded by the HAMP gene and hepcidin is a key hormonal regulator of systemic iron homeostasis and its expression is induced by iron or inflammatory stimuli 1. Serum iron levels are under the control of hepcidin, a liver-derived peptide hormone and master regulator of iron metabolism 2. Hepcidin operates by binding to the iron exporter ferroportin (FPN) in iron-releasing target cells, mainly tissue macrophages and duodenal enterocytes, but also other cell types (Figures 1 and 2). The binding of hepcidin occludes iron efflux 3 and triggers ubiquitination, internalization and lysosomal degradation of ferroportin 4. This leads to intracellular iron retention and eventually to hypoferremia.
The HAMP gene encodes pre-pro-hepcidin, an 84 amino acids long precursor, which is primarily expressed by hepatocytes in the liver, and at much lower levels by other cells in extrahepatic tissues. Pre-pro-hepcidin is processed to pro-hepcidin upon removal of its endoplasmic reticulum targeting sequence, consisting of 24 N-terminal amino acids. Further cleavage at the C-terminus yields matures, bioactive hepcidin, an evolutionary conserved, cysteine-rich peptide of 25-amino acids with antimicrobial properties. It folds to a distorted β-sheet with an unusual disulfide bridge between adjacent C13-C14 at the turn of a hairpin loop; according to this model, the structure is stabilized by further disulfide bonding between C7-C23, C10-C22 and C11-C19 5. An alternative structural model postulates disulfide bond connectivity between C7-C23, C10-C13, C11-C19 and C14-C22 6. Interestingly, the structural organization of hepcidin based on disulfide bonding is not essential for iron-regulatory function, since the substitution of cysteines or the deletion of cysteine-containing segments do not impair hormonal activity 7.
Hepcidin is an 84-amino-acid long prepropeptide mainly produced in hepatocytes and excreted in urine as a 25-amino-acid mature form 8. Hepcidin was originally thought to function solely as an antimicrobial peptide as it is upregulated under inflammatory conditions and as such is considered to be a type two acute-phase reactant due to its regulation via interleukin 6 (IL-6) 9. The 24-amino-acid N-terminal signal peptide of pre-prohepcidin is cleaved to produce prohepcidin, comprising 60 amino acids 10. The mature 25-amino-acid form of hepcidin 8 is generated via cleavage of the pro-region by furin-like prohormone convertases 10. The carboxy terminus of the mature hepcidin, comprising four highly conserved disulphide bonds, forms a β-hairpin; the N-terminus is unstructured and essential for ferroportin (FPN) interaction 11. This interaction results in reduced membrane concentrations of ferroportin, thus lowering levels iron availability to tissues 12.
The interplay between the iron exporter ferroportin (FPN) and the hepatic peptide hormone, hepcidin, is central to iron homeostasis. Hepcidin binds to membrane-bound ferroportin and induces its internalisation and eventual degradation. The expression of these two proteins is highly regulated. The main regulatory pathway for liver hepcidin expression is the bone morphogenetic protein/Sma mothers against decapentaplegic (BMP/SMAD) pathway (Figure 1). The ligand BMP6, believed to be produced by liver sinusoidal endothelial cells 13, has been found to be the primary bone morphogenetic protein associated with hepcidin transcription 14. However, bone morphogenetic proteins 2, 4 and 9 have also been shown to increase hepcidin expression 15. Increased body iron stores induce BMP6 expression in the liver; BMP6 then binds to membrane-bound BMP receptors (BMPRs) with hemojuvelin acting as a co-receptor 16. Membrane-bound BMP receptors (BMPRs) type 1 (activin-like kinase (ALK) 2 and ALK3) and type 2 (BMPR2 and ActR2A) are critical for iron balance 17. BMP6 binding to BMPRs results in the phosphorylation of intracellular SMAD1/5/8 18. Phosphorylated SMAD1/5/8 then complexes with SMAD4, which translocates to the nucleus 19, where it binds to BMP-responsive elements (BMP-RE) on the hepcidin gene promoter inducing hepcidin expression 20.
In addition to the liver, hepcidin is also synthesised in a number of other organs including adipose tissue, brain kidney, heart, spleen, pancreas and stomach 21. Within the kidney, where hepcidin is produced in the cortical thick ascending limb, hepcidin was been shown to play a role in the absorption of non-haem iron through the down regulation of both divalent metal transport 1 (DMT1) and ferroportin (FPN) 21. The role of hepcidin within the spleen, stomach and the brain appear to be linked with its antimicrobial ability 22. Several macrophage cell types have been found to synthesise hepcidin when challenged with pathogens. Sow et al. 23 infected RAW264.7 macrophages, mouse bone marrow-derived macrophages and human THP-1 monocytic cell with Mycobacterium sp., which stimulated hepcidin mRNA and protein synthesis. Within the stomach, hepcidin expression is regulated by bacterial infection and involved with gastric acid production 24. Hepcidin in the brain has been found to be upregulated via inflammatory responses 25. Hepcidin synthesis in adipose tissue has also been reported; however, the exact role of this type of hepcidin remains largely unknown 21. Lastly, hepcidin production within the pancreas has been suggested to play a role in the regulation of insulin 21. These secondary roles of hepcidin will need to be considered when drug candidates undergo animal studies and clinical trials to ensure these critical roles of hepcidin are not altered.
Genetic defects in iron signaling to hepcidin lead to “hepcidinopathies” ranging from hereditary hemochromatosis to iron-refractory iron deficiency anemia, which are disorders caused by hepcidin deficiency or excess, respectively 1. Moreover, dysregulation of hepcidin is a pathogenic cofactor in iron-loading anemias with ineffective erythropoiesis and in anemia of inflammation. Experiments with preclinical animal models provided evidence that restoration of appropriate hepcidin levels can be used for the treatment of these conditions. This fueled the rapidly growing field of hepcidin therapeutics. Several hepcidin agonists and antagonists, as well as inducers and inhibitors of hepcidin expression have been identified to date. Some of them were further developed and are currently being evaluated in clinical trials.
During conditions in which the hepcidin level is abnormally high, such as inflammation, serum iron falls due to iron trapping within macrophages and liver cells and decreased gut iron absorption. This typically leads to anemia due to an inadequate amount of serum iron being available for developing erythrocytes. When the hepcidin level is abnormally low such as in hemochromatosis, iron overload occurs due to increased ferroportin mediated iron efflux from storage and increased gut iron absorption.
Figure 1. Major signalling pathways involved in Hepcidin regulation
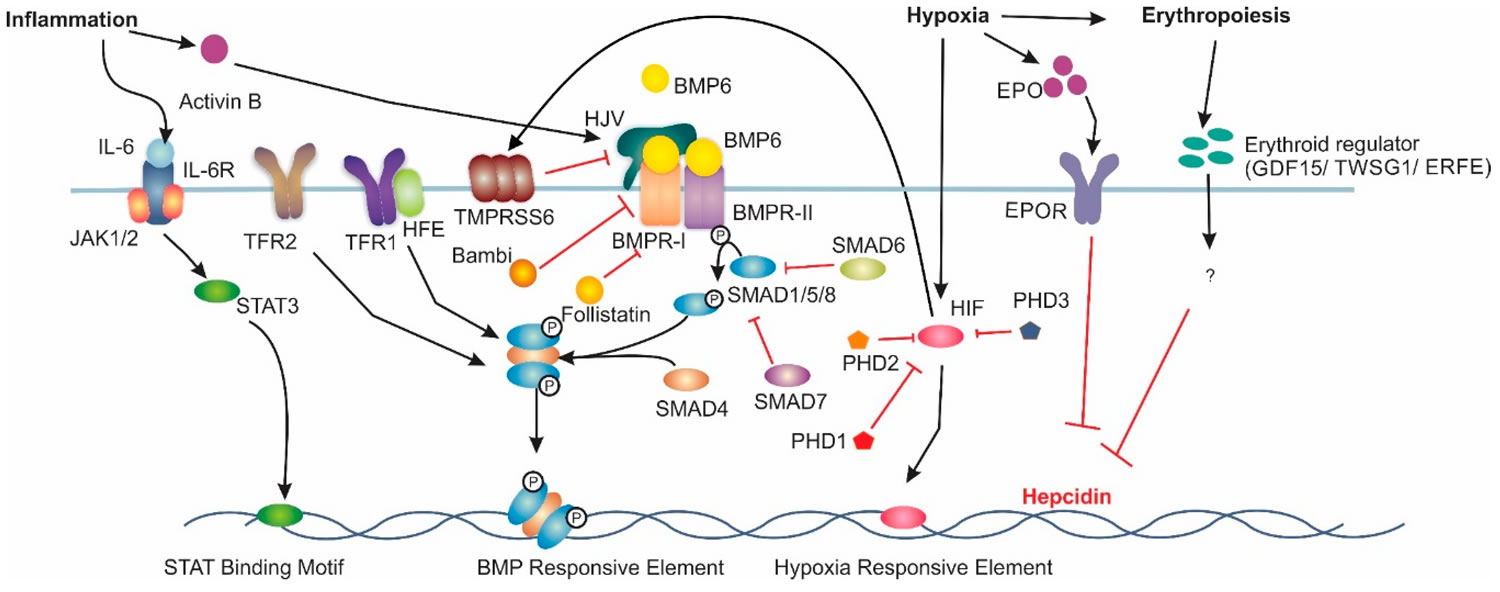
Footnote: Increased BMP6 levels induce hepcidin mRNA expression via the bone morphogenic protein (BMP)/sma and mothers against decapentaplegic homologue (SMAD) pathway. Under inflammatory conditions, interleukin 6 (IL-6) induces hepcidin mRNA via the Jak/signal transducer and activator of transcription (STAT) pathway. Hypoxia induces hepcidin downregulation through increases in erythropoietin (EPO) expression. Hypoxia Inducible Factor (HIF) can also directly downregulate hepcidin. Additionally, HIF upregulates matriptase-2 expression, which results in decreased hepcidin expression. Lastly, erythropoiesis has been theorised to reduce hepcidin expression acting through growth differentiation factor 15 (GDF-15), twisted gastrulation factor 1 (TWSG1) or erythroferrone (ERFE), however the exact mechanism is not understood. Interleukin-6 Receptor (IL6-R), Transferrin Receptor 1 (TFR1), Hemojuvelin (HJV), Bone Morphogenic Protein Receptor Type 1 (BMPR-I), Bone Morphogenic Protein Receptor Type 2 (BMPR-II), Erythropoietin Receptor (EPOR) and prolyl hydroxylase domain containing enzymes (PHD) are involved in hepcidin regulation.
Abbreviations: BMP = Bone Morphogenetic Protein; SMAD = Small Mothers Against Decapentaplegic; HFE = high iron (Fe); HJV = hemojuvelin; TfR2 = transferrin receptor 2; JAK = Janus kinase; STAT = Signal Transducer and Activator of Transcription.
[Source 22 ]Figure 2. Physiological and pharmacological regulation of hepcidin activity
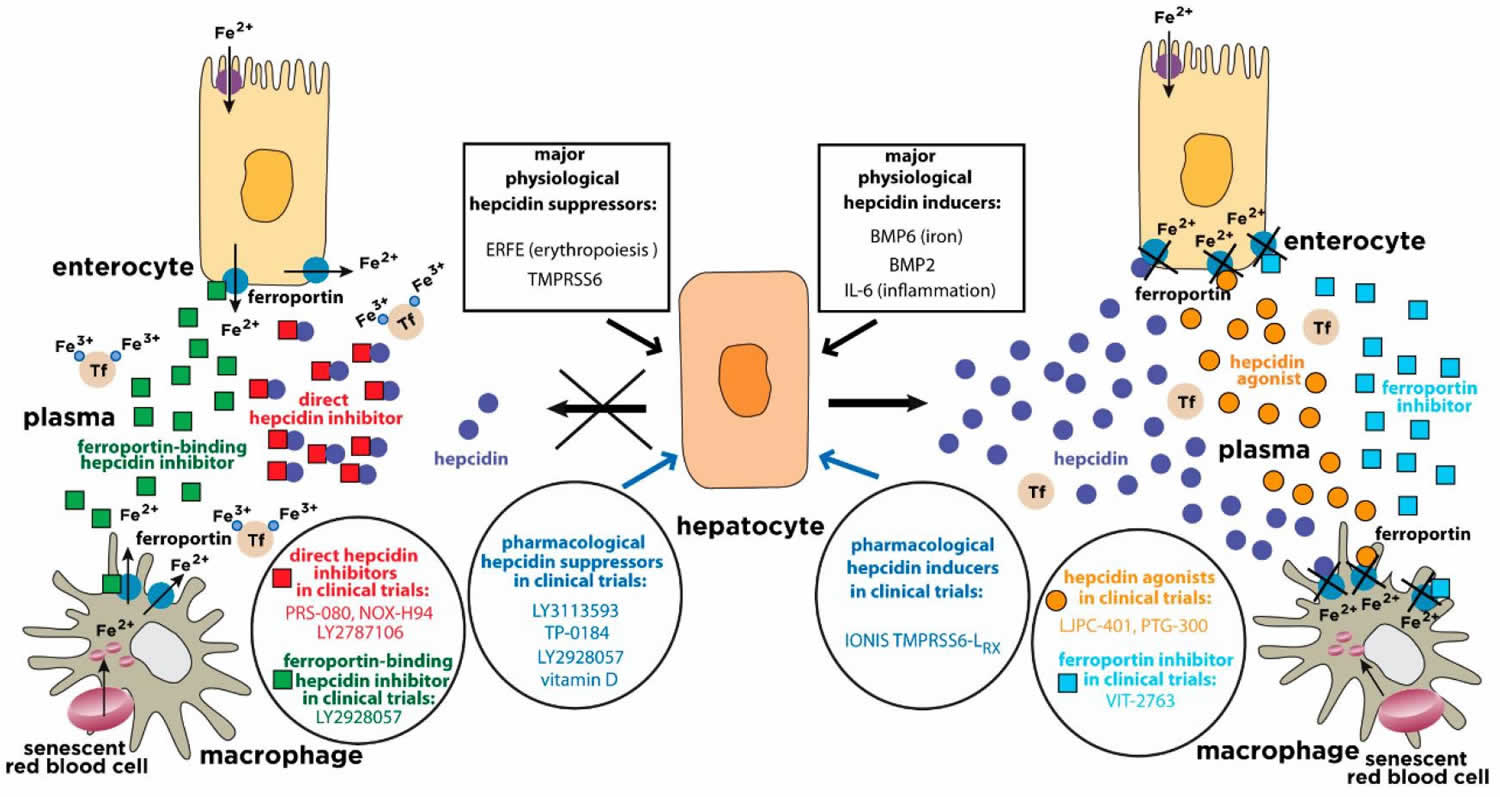
Footnote: Hepcidin is synthesized in hepatocytes in response to iron or inflammatory stimuli. It operates by targeting the iron exporter ferroportin in tissue macrophages and duodenal enterocytes. The binding of hepcidin promotes ferroportin degradation, iron retention in target cells and hypoferremia. Suppression of hepcidin by erythropoietic stimuli allows iron efflux from cells into plasma via ferroportin. The expression of hepcidin can be manipulated pharmacologically with drugs that target hepatocytes (blue arrows). Hepcidin responses can be mimicked by hepcidin agonists (orange circles) or ferroportin inhibitors (light blue squares). Conversely, hepcidin responses can be antagonized by direct hepcidin inhibitors (red squares) or ferroportin-binding hepcidin inhibitors (green squares). The insets highlight major physiological hepcidin inducers or suppressors, as well as clinically relevant drugs that modulate the hepcidin-ferroportin axis and are currently being evaluated in randomized controlled trials for the treatment of hepcidin-related disorders (“hepcidinopathies”). Tf = transferrin.
[Source 1 ]Figure 3. Major mechanisms for hepcidin regulation
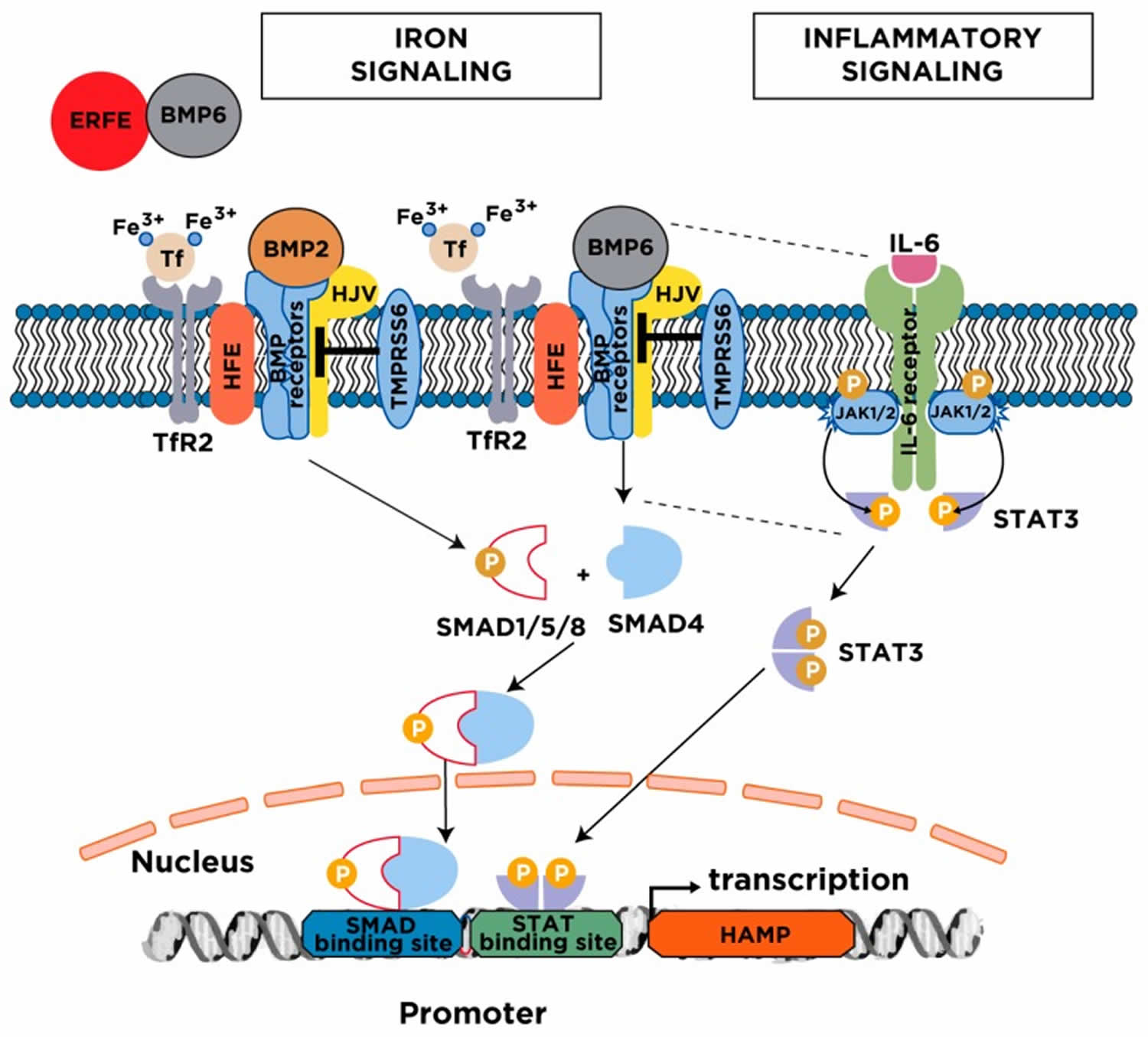
Footnote: Serum and tissue iron induce hepcidin transcription via the BMP/SMAD signaling pathway. The cascade is initiated following an increase in transferrin saturation and the secretion of BMP6 from liver sinusoidal endothelial cells; BMP2 is likewise secreted from liver sinusoidal endothelial cells but is less responsive to iron. Diferric transferrin binds to TfR2, while BMP6 and BMP2 bind to type I and II BMP receptors on hepatocytes. These events trigger phosphorylation of regulatory SMAD1/5/8, recruitment of SMAD4, and translocation of the SMAD complex to the nucleus for activating hepcidin transcription upon binding to BMP response elements in the HAMP promoter. Efficient iron signaling to hepcidin requires the BMP co-receptor HJV and the hemochromatosis protein HFE, and is negatively regulated by the transmembrane serine protease matriptase-2 (TMPRSS6). Under conditions of high iron demand for erythropoiesis, the erythropoietic regulator erythroferrone (ERFE) is released from bone marrow erythroblasts and suppresses hepcidin by sequestering BMP6. The inflammatory cytokine IL-6 induces hepcidin transcription via the JAK/STAT3 signaling pathway. The binding of IL-6 triggers dimerization of IL-6 receptors on hepatocytes, which leads to activation of associated JAK1/2 and subsequent phosphorylation of STAT3. Phospho-STAT3 dimerizes and translocates to the nucleus, where it activates hepcidin transcription upon binding to a STAT binding site in the HAMP promoter. Efficient hepcidin induction by the inflammatory pathway requires a threshold of BMP6/SMAD signaling (indicated by the dotted lines).
Abbreviations: BMP = Bone Morphogenetic Protein; SMAD = Small Mothers Against Decapentaplegic; HFE = high iron (Fe); HJV = hemojuvelin; TfR2 = transferrin receptor 2; JAK = Janus kinase; STAT = Signal Transducer and Activator of Transcription.
[Source 1 ]Hepcidin function
Hepcidin plays a central role in iron transport and utilization and is, therefore, an important marker of iron bioavailability. Once released into circulation from hepatocytes, hepcidin regulates plasma iron levels through interactions with ferroportin, an iron export transmembrane protein. Specifically, hepcidin binds to ferroportin signaling a cascade of intracellular messengers that target the hepcidin-ferroportin complex for lysosomal degradation 26. The cell types most affected by this interaction are duodenal enterocytes and reticuloendothelial macrophages 26. Duodenal enterocytes absorb dietary iron, and reticuloendothelial macrophages release iron recovered from degraded erythrocytes in the bone marrow, liver, and spleen. The degradation of ferroportin blocks iron from entering circulation and instead remains in its intracellular storage form 27.
Hepcidin also plays a role in innate immunity through its interactions with IL-6 and other pro-inflammatory cytokines 26. The ability to sequester iron within cells to prevent its availability for pathogenic or neoplastic growth appears to be largely dependent on hepcidin stimulation by IL-6. This innate defense may help protect against many pathogens including streptococcal and malarial species 28
Several hepcidin agonists are currently in development and may become a viable treatment for hereditary hemochromatosis. Currently, phlebotomy is the mainstay of treatment for iron overload states, but a hepcidin agonist could help alleviate the symptoms from the deficient natural hepcidin 29.
Disorders with Hepcidin deficiency
Hereditary hemochromatosis
Hereditary hemochromatosis an endocrine disorder of systemic iron overload, is an autosomal recessive defect in the HFE gene, resulting in decreased hepcidin production 30. HFE mutations are more prevalent in individuals of European descent. Decreased hepcidin results in abnormal iron sensing. Continued iron absorption (up to 8–10 mg/day) despite adequate serum levels and uncontrolled release of iron to plasma, due to unrestricted expression of ferroportin in duodenal enterocytes and tissue macrophages can lead to iron overload when total body iron exceeds 20g. Symptoms of hemochromatosis are secondary to iron deposition in bodily tissue and typically present in the 4th and 5th decade of life for men and women respectively. The classic triad includes skin hyperpigmentation, liver cirrhosis, and diabetes mellitus. Additional findings include dilated cardiomyopathy, hepatocellular cancer, diabetes mellitus, hypogonadism, arthritis, osteoporosis, and hypothyroidism. Hemochromatosis patients also have increased infection risk with siderophilic bacteria but also various other pathogens now that serum iron levels cannot decrease during inflammatory states 31. Diagnosis is based on iron panel results showing an increased serum iron level with increased ferritin and transferrin saturation levels. Treatment involves the use of phlebotomy or an iron chelating agent like deferoxamine to remove iron from the circulation 32.
Hereditary hemochromatosis is genetically heterogenous and its severity depends on the degree of hepcidin suppression relative to body iron stores 33. The major form is linked to mutations in HFE (especially C282Y) and constitutes the most frequent genetic disorder in Caucasians. However, the clinical penetrance is variable and depends on further genetic and environmental factors. The disease phenotype is relatively mild, and symptoms typically develop after the fourth decade of life. Other forms of hereditary hemochromatosis are rare and typically associated with more severe phenotypes. Thus, inactivation of either hemojuvelin or hepcidin cause juvenile hemochromatosis with early-onset iron overload in the late teens or early twenties 1. Inactivation of TfR2 yields an intermediate clinical phenotype compared to HFE-related hereditary hemochromatosis and juvenile hemochromatosis 1.
Iron-loading anemias
Hereditary or acquired anemias, such as thalassemias, congenital dyserythropoietic anemias, sideroblastic anemias or myelodysplastic syndromes, are associated with bone marrow hyperplasia and ineffective erythropoiesis 34. Patients with severe forms of these diseases are often treated with blood transfusions, causing secondary iron overload. Ineffective erythropoiesis stimulates expression of erythropoietic regulator erythroferrone (ERFE) and other factors, such as GDF15, which suppress hepcidin expression. This in turn promotes a hemochromatosis-like phenotype of iron overload in non-transfused patients with milder forms of disease; in addition, it aggravates the already existing secondary iron overload in transfused patients. Importantly, repressive erythropoietic signals dominate over the stimulating iron signals creating a vicious cycle in iron homeostasis and hepcidin expression 35.
Chronic liver diseases
Patients with alcoholic liver disease and chronic hepatitis C often exhibit some degree of hepatocellular iron overload that aggravates liver disease progression to advanced stages (cirrhosis and hepatocellular carcinoma). This is linked to hepcidin suppression by oxidative stress and possibly additional mechanisms. Hepatic iron overload is also observed in patients with non-alcoholic fatty liver disease (NAFLD). The pattern of excessive iron distribution is variable with preference in either hepatocytes or macrophages, which may underlie differential hepcidin regulation by positive and negative stimuli 36. In advanced liver disease of any etiology, the expression of hepcidin decreases dramatically due to severe injury of hepatocytes, and hepcidin levels offer a potential biomarker for liver disease progression 37.
Interestingly, experimental data provided evidence for a protective role of hepcidin against liver fibrosis. Thus, adenoviral expression of hepcidin attenuated the development of liver fibrosis in mice subjected to CCl4 intoxication or to bile duct ligation 38. The underlying mechanism involves a hepcidin-mediated increase in the iron content of hepatic stellate cells, which prevents their differentiation from collagen-secreting myofibroblasts. Another study using Tmprss6-/- mice showed that genetic hepcidin overexpression protected these animals against high fat diet-induced obesity and liver steatosis by stimulating lipolytic pathways 39. These data highlight a critical role of hepcidin in liver disease pathogenesis.
Disorders with Hepcidin Resistance or Ferroportin Deficiency
Ferroportin hemochromatosis is a distinct clinical entity that develops as a result of “gain-of-function” ferroportin mutations that prevent the binding of hepcidin 40. Conversely, “loss-of-function” ferroportin mutations that inhibit intracellular trafficking of the protein are the hallmark of ferroportin disease, which is characterized by macrophage iron loading. Hepcidin levels are elevated rather than suppressed in these ferroportin-associated disorders of hepcidin resistance, or ferroportin deficiency, respectively. It should be noted that both ferroportin hemochromatosis and ferroportin disease are transmitted in an autosomal dominant fashion, contrary to all other forms of hereditary hemochromatosis, which are autosomal recessive.
Disorders with systemic Hepcidin Overexpression
Systemic overexpression of hepcidin occurs in anemias with iron-restricted erythropoiesis, such as iron-refractory iron deficiency anemia, anemia of inflammation (also known as anemia of chronic disease) or Castleman disease. Local overexpression of hepcidin has been reported in tumors.
Iron-refractory iron deficiency anemia
Iron-refractory iron deficiency anemia is an autosomal recessive disease caused by loss-of-function mutations in the hepcidin suppressor matriptase-2 (TMPRSS6), which lead to hepcidin overexpression 41. Iron-refractory iron deficiency anemia is characterized by hypochromic microcytic anemia, hyperhepcidinemia, hypoferremia, low transferrin saturation and unresponsiveness to oral iron therapy.
Anemia of chronic disease
Anemia of chronic disease also known as anemia of inflammation, is the second most common cause of anemia after iron deficiency anemia. Anemia of chronic disease or anemia of inflammation is a heterogenous disorder caused by chronic inflammation due to infections, inflammatory bowel disease, inflammatory rheumatic disease, autoimmune conditions like systemic lupus erythematosus, chronic kidney disease or obesity 42. Anemia of chronic disease is also observed in cancer patients and in frail, elderly persons. Anemia of chronic disease or anemia of inflammation is the most common type of anemia among hospitalized patients in the developed world. Anemia of chronic disease or anemia of inflammation is normochromic and normocytic, but this phenotype can be affected by chronic blood loss or dietary iron malabsorption, which result in smaller and hypochromic red blood cells 43.
Hepcidin gets upregulated by IL-6 and other proinflammatory cytokines and leads to degradation of ferroportin. Serum iron levels decline in an attempt to deprive rapidly dividing cells and invading microbes from nutrients. Other contributing factors are reduced proliferation of erythroblasts, reduced expression of erythropoietin, impaired erythropoietin signaling and increased erythrophagocytosis. In chronic kidney disease, hyperhepcidinemia is aggravated by ineffective renal clearance 44.
Anemia of chronic disease typically begins as a mild to moderate normocytic normochromic anemia denoted by a hemoglobin concentration of 8 to 9.5 g/dL. The anemia can progress to microcytic and hypochromic if the inflammatory conditions remain. Presenting symptoms are often nonspecific signs of anemia including fever, pallor, and fatigue. An iron panel would show a decrease in serum iron level despite an increase in ferritin because of intracellular iron sequestration. Treatment with iron supplementation is often not beneficial as the issue lies with iron availability and not an iron deficiency. It is crucial to treat the underlying condition to prevent further inflammation 45.
Castleman disease
Castleman disease is a rare lymphoproliferative disorder characterized by generalized lymphadenopathy and multiple organ involvement that is linked to chronic overproduction of IL-6 46. Castleman disease patients present with iron-refractory hypochromic microcytic anemia, which develops in response to IL-6-mediated upregulation of hepcidin 47.
Disorders with local Hepcidin Overexpression
Hepcidin is produced at lower levels in several extrahepatic tissues, including the heart or the brain, where it exerts local cell autonomous functions 48. Interestingly, many tumor cells produce and utilize hepcidin for their own advantage. Thus, hepcidin targets ferroportin in tumor cells in an autocrine manner and thereby promotes retention of iron, which is essential for cell proliferation and tumor growth 49. This mechanism has been documented in breast 50, prostate 51 and thyroid 52 cancers. It should also be noted that in advanced disease stages, the immune response against primary or metastatic tumor cells may trigger hepcidin induction in hepatocytes and thereby lead to systemic hepcidin overproduction. This may result in anemia of chronic disease due to hepcidin-mediated iron sequestration in tissues and inhibition in iron absorption.
References- Katsarou A, Pantopoulos K. Hepcidin Therapeutics. Pharmaceuticals (Basel). 2018;11(4):127. Published 2018 Nov 21. doi:10.3390/ph11040127 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6316648
- Ganz T. Systemic iron homeostasis. Physiol. Rev. 2013;93:1721–1741. doi: 10.1152/physrev.00008.2013
- Aschemeyer S., Qiao B., Stefanova D., Valore E.V., Sek A.C., Ruwe T.A., Vieth K.R., Jung G., Casu C., Rivella S., et al. Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin. Blood. 2018;131:899–910. doi: 10.1182/blood-2017-05-786590.
- Nemeth E., Tuttle M.S., Powelson J., Vaughn M.B., Donovan A., Ward D.M., Ganz T., Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742
- Hunter H.N., Fulton D.B., Ganz T., Vogel H.J. The solution structure of human hepcidin, a peptide hormone with antimicrobial activity that is involved in iron uptake and hereditary hemochromatosis. J. Biol. Chem. 2002;277:37597–37603. doi: 10.1074/jbc.M205305200
- Jordan J.B., Poppe L., Haniu M., Arvedson T., Syed R., Li V., Kohno H., Kim H., Schnier P.D., Harvey T.S., et al. Hepcidin revisited, disulfide connectivity, dynamics, and structure. J. Biol. Chem. 2009;284:24155–24167. doi: 10.1074/jbc.M109.017764
- Pandur E., Fekete Z., Tamasi K., Grama L., Varga E., Sipos K. The C19S Substitution Enhances the Stability of Hepcidin While Conserving Its Biological Activity. Protein J. 2018;37:113–121. doi: 10.1007/s10930-018-9759-9
- Park, C.H.; Valore, E.V.; Waring, A.J.; Ganz, T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem. 2001, 276, 7806–7810.
- Krause, A.; Sillard, R.; Kleemeier, B.; Kluver, E.; Maronde, E.; Conejo-Garcia, J.R.; Forssmann, W.G.; Schulz-Knappe, P.; Nehls, M.C.; Wattler, F.; et al. Isolation and biochemical characterization of LEAP-2, a novel blood peptide expressed in the liver. Protein Sci. 2003, 12, 143–152.
- Valore, E.V.; Ganz, T. Posttranslational processing of hepcidin in human hepatocytes is mediated by the prohormone convertase furin. Blood Cells Mol. Dis. 2008, 40, 132–138.
- Nemeth, E.; Preza, G.C.; Jung, C.L.; Kaplan, J.; Waring, A.J.; Ganz, T. The N-terminus of hepcidin is essential for its interaction with ferroportin: Structure-function study. Blood 2006, 107, 328–333.
- Ganz, T. Systemic iron homeostasis. Physiol. Rev. 2013, 93, 1721–1741
- Canali, S.; Zumbrennen-Bullough, K.B.; Core, A.B.; Wang, C.Y.; Nairz, M.; Bouley, R.; Swirski, F.K.; Babitt, J.L. Endothelial cells produce bone morphogenetic protein 6 required for iron homeostasis in mice. Blood 2017, 129, 405–414.
- Andriopoulos, B.; Corradini, E.; Xia, Y.; Faasse, S.A.; Chen, S.Z.; Grgurevic, L.; Knutson, M.D.; Pietrangelo, A.; Vukicevic, S.; Lin, H.Y.; et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat. Genet. 2009, 41, 482–487
- Canali, S.; Wang, C.-Y.; Zumbrennen-Bullough, K.B.; Bayer, A.; Babitt, J.L. Bone morphogenetic protein 2 controls iron homeostasis in mice independent of Bmp6. Am. J. Hematol. 2017, 92, 1204–1213.
- Babitt, J.L.; Huang, F.W.; Wrighting, D.M.; Xia, Y.; Sidis, Y.; Samad, T.A.; Campagna, J.A.; Chung, R.T.; Schneyer, A.L.; Woolf, C.J.; et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat. Genet. 2006, 38, 531–539.
- Mayeur, C.; Leyton, P.A.; Kolodziej, S.A.; Yu, B.L.; Bloch, K.D. BMP type II receptors have redundant roles in the regulation of hepatic hepcidin gene expression and iron metabolism. Blood 2014, 124, 2116–2123.
- Miyazono, K.; Kamiya, Y.; Morikawa, M. Bone morphogenetic protein receptors and signal transduction. J. Biochem. 2010, 147, 35–51.
- Wang, R.H.; Li, C.; Xu, X.; Zheng, Y.; Xiao, C.; Zerfas, P.; Cooperman, S.; Eckhaus, M.; Rouault, T.; Mishra, L.; et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005, 2, 399–409.
- Truksa, J.; Lee, P.; Beutler, E. Two BMP responsive elements, STAT, and bZIP/HNF4/COUP motifs of the hepcidin promoter are critical for BMP, SMAD1, and HJV responsiveness. Blood 2009, 113, 688–695.
- Daher, R.; Lefebvre, T.; Puy, H.; Karim, Z. Extrahepatic hepcidin production: The intriguing outcomes of recent years. World J. Clin. Cases 2019, 7, 1926–1936.
- Therapeutic Advances in Regulating the Hepcidin/Ferroportin Axis. Pharmaceuticals 2019, 12(4), 170; https://doi.org/10.3390/ph12040170
- Sow, F.B.; Florence, W.C.; Satoskar, A.R.; Schlesinger, L.S.; Zwilling, B.S.; Lafuse, W.P. Expression and localization of hepcidin in macrophages: A role in host defense against tuberculosis. J. Leukoc. Biol. 2007, 82, 934–945.
- Schwarz, P.; Kübler, J.A.M.; Strnad, P.; Müller, K.; Barth, T.F.E.; Gerloff, A.; Feick, P.; Peyssonnaux, C.; Vaulont, S.; Adler, G.; et al. Hepcidin is localised in gastric parietal cells, regulates acid secretion and is induced by Helicobacter pylori infection. Gut 2012, 61, 193–201.
- Qian, Z.M.; He, X.; Liang, T.; Wu, K.C.; Yan, Y.C.; Lu, L.N.; Yang, G.; Luo, Q.Q.; Yung, W.H.; Ke, Y. Lipopolysaccharides upregulate hepcidin in neuron via microglia and the IL-6/STAT3 signaling pathway. Mol. Neurobiol. 2014, 50, 811–820.
- Chambers K, Sharma S. Physiology, Hepcidin. [Updated 2019 Mar 16]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK538257
- Nemeth E, Ganz T. The role of hepcidin in iron metabolism. Acta Haematol. 2009;122(2-3):78-86.
- Rodriguez R, Jung CL, Gabayan V, Deng JC, Ganz T, Nemeth E, Bulut Y. Hepcidin induction by pathogens and pathogen-derived molecules is strongly dependent on interleukin-6. Infect. Immun. 2014 Feb;82(2):745-52.
- Blanchette NL, Manz DH, Torti FM, Torti SV. Modulation of hepcidin to treat iron deregulation: potential clinical applications. Expert Rev Hematol. 2016;9(2):169-86.
- Brissot P., Pietrangelo A., Adams P.C., de Graaff B., McLaren C.E., Loreal O. Haemochromatosis. Nat. Rev. Dis. Primers. 2018;4:18016. doi: 10.1038/nrdp.2018.16
- Frank K.M., Schneewind O., Shieh W.J. Investigation of a researcher’s death due to septicemic plague. N. Engl. J. Med. 2011;364:2563–2564. doi: 10.1056/NEJMc1010939
- Means RT. Hepcidin and iron regulation in health and disease. Am. J. Med. Sci. 2013 Jan;345(1):57-60.
- Pantopoulos K. Inherited Disorders of Iron Overload. Front. Nutr. 2018;5:103. doi: 10.3389/fnut.2018.0010
- Gupta R., Musallam K.M., Taher A.T., Rivella S. Ineffective Erythropoiesis: Anemia and Iron Overload. Hematol. Oncol. Clin. N. Am. 2018;32:213–221. doi: 10.1016/j.hoc.2017.11.009
- Papanikolaou G., Pantopoulos K. Systemic iron homeostasis and erythropoiesis. IUBMB Life. 2017;69:399–413. doi: 10.1002/iub.1629
- Kowdley K.V. Iron Overload in Patients with Chronic Liver Disease. Gastroenterol. Hepatol. (N. Y.) 2016;12:695–698
- Vela D. Low hepcidin in liver fibrosis and cirrhosis; a tale of progressive disorder and a case for a new biochemical marker. Mol. Med. 2018;24:5. doi: 10.1186/s10020-018-0008-7
- Han C.Y., Koo J.H., Kim S.H., Gardenghi S., Rivella S., Strnad P., Hwang S.J., Kim S.G. Hepcidin inhibits Smad3 phosphorylation in hepatic stellate cells by impeding ferroportin-mediated regulation of Akt. Nat. Commun. 2016;7:13817. doi: 10.1038/ncomms13817
- Folgueras A.R., Freitas-Rodriguez S., Ramsay A.J., Garabaya C., Rodriguez F., Velasco G., Lopez-Otin C. Matriptase-2 deficiency protects from obesity by modulating iron homeostasis. Nat. Commun. 2018;9:1350. doi: 10.1038/s41467-018-03853-1
- Pietrangelo A. Ferroportin disease: Pathogenesis, diagnosis and treatment. Haematologica. 2017;102:1972–1984. doi: 10.3324/haematol.2017.170720
- Heeney M.M., Finberg K.E. Iron-Refractory Iron Deficiency Anemia (IRIDA) Hematol. Oncol. Clin. N. Am. 2014;28:637–652. doi: 10.1016/j.hoc.2014.04.009
- Fraenkel P.G. Anemia of Inflammation: A Review. Med. Clin. N. Am. 2017;101:285–296. doi: 10.1016/j.mcna.2016.09.005
- Nairz M., Theurl I., Wolf D., Weiss G. Iron deficiency or anemia of inflammation?: Differential diagnosis and mechanisms of anemia of inflammation. Wien. Med. Wochenschr. 2016;166:411–423. doi: 10.1007/s10354-016-0505-7
- Ueda N., Takasawa K. Role of Hepcidin-25 in Chronic Kidney Disease: Anemia and Beyond. Curr. Med. Chem. 2017;24:1417–1452. doi: 10.2174/0929867324666170316120538
- Madu AJ, Ughasoro MD. Anaemia of Chronic Disease: An In-Depth Review. Med Princ Pract. 2017;26(1):1-9.
- Yoshizaki K., Matsuda T., Nishimoto N., Kuritani T., Taeho L., Aozasa K., Nakahata T., Kawai H., Tagoh H., Komori T., et al. Pathogenic significance of interleukin-6 (IL-6/BSF-2) in Castleman’s disease. Blood. 1989;74:1360–1367
- Arlet J.B., Hermine O., Darnige L., Ostland V., Westerman M., Badoual C., Pouchot J., Capron L. Iron-deficiency anemia in Castleman disease: Implication of the interleukin 6/hepcidin pathway. Pediatrics. 2010;126:e1608–e1612. doi: 10.1542/peds.2010-1123
- You L.H., Yan C.Z., Zheng B.J., Ci Y.Z., Chang S.Y., Yu P., Gao G.F., Li H.Y., Dong T.Y., Chang Y.Z. Astrocyte hepcidin is a key factor in LPS-induced neuronal apoptosis. Cell Death Dis. 2017;8:e2676. doi: 10.1038/cddis.2017.93
- Torti S.V., Manz D.H., Paul B.T., Blanchette-Farra N., Torti F.M. Iron and Cancer. Annu. Rev. Nutr. 2018;38:97–125. doi: 10.1146/annurev-nutr-082117-051732
- Pinnix Z.K., Miller L.D., Wang W., D’Agostino R., Jr., Kute T., Willingham M.C., Hatcher H., Tesfay L., Sui G., Di X., et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci. Transl. Med. 2010;2:43ra56. doi: 10.1126/scitranslmed.3001127
- Tesfay L., Clausen K.A., Kim J.W., Hegde P., Wang X., Miller L.D., Deng Z., Blanchette N., Arvedson T., Miranti C.K., et al. Hepcidin regulation in prostate and its disruption in prostate cancer. Cancer Res. 2015;75:2254–2263. doi: 10.1158/0008-5472.CAN-14-2465
- Zhou Q., Chen J., Feng J., Wang J. E4BP4 promotes thyroid cancer proliferation by modulating iron homeostasis through repression of hepcidin. Cell Death Dis. 2018;9:987. doi: 10.1038/s41419-018-1001-3





{kind=link}