Hemophagocytic lymphohistiocytosis
HLH is medical abbreviation for hemophagocytic lymphohistiocytosis, covers a wide array of rare related life-threatening conditions in which the body makes too many activated immune cells (macrophages and lymphocytes) that commonly appears in infancy, although it has been seen in all age groups 1. HLH or hemophagocytic lymphohistiocytosis occurs as autosomal recessive familial HLH (familial hemophagocytic lymphohistiocytosis), familial erythrophagocytic lymphohistiocytosis, viral-associated hemophagocytic syndrome, and autoimmune-associated macrophage activation syndrome (MAS) 2. In hemophagocytic lymphohistiocytosis, the immune system responds to a stimulus or ‘trigger’, often an infection, but the response is ineffective and abnormal. This ineffective, abnormal response, causes a variety of signs and symptoms, which, if not treated, can potentially become life-threatening. These disorders feature severe cytopenias due to this uncontrolled hemophagocytosis. Other laboratory signs and clinical symptoms result from disordered immune regulation and cytokine storm. The term primary HLH refers to an underlying genetic abnormality causing the disorder, whereas secondary HLH indicates that the disorder is secondary to underlying conditions such as infection, autoimmune/rheumatologic, malignant, or metabolic conditions.
People with HLH usually develop symptoms within the first months or years of life. Symptoms may include fever, enlarged liver or spleen (hepatosplenomegaly), cytopenia (decreased number of blood cells), lymphadenopathy, rash and neurological abnormalities 3. Cutaneous involvement occurs in as many as 65% of patients 4. Varied skin manifestations of hemophagocytic lymphohistiocytosis are noted, including erythroderma, generalized purpuric macules and papules, and morbilliform eruptions. Detection of cutaneous involvement can assist in the initial diagnosis of hemophagocytic lymphohistiocytosis and potentially signify recurrences. HLH may be viewed as a marker for underlying cancer, which in adults is most often a lymphoma that may be rapidly progressive 5.
HLH may be inherited in an autosomal recessive manner or it can have non-genetic causes in which case it is called acquired HLH or secondary HLH. There are five subtypes of inherited HLH which are designated as familial HLH, types 1-5. Each subtype is caused by a change (mutation) in a different gene. The genetic cause of type 1 is currently unknown. Types 2-5 are caused by mutations in the PRF1 gene, the UNC13D gene, the STX11 gene and the STXBP2 gene, respectively 6. Treatment depends on a number of factors, including the severity of symptoms, the age of onset, and the underlying cause of the condition 6.
When HLH results from an inappropriate immune response to Epstein-Barr virus or another viral illness, it may be due to a separate genetic condition called X-linked lymphoproliferative disease (XLP). X-linked lymphoproliferative disease is caused by a mutation in the SH2D1A or XIAP gene and is inherited in an X-linked manner 7.
Primary hemophagocytic lymphohistiocytosis (familial HLH or familial erythrophagocytic lymphohistiocytosis [FEL]), an autosomal recessive inherited form of HLH, is a heterogeneous autosomal recessive disorder found to be more prevalent with parental consanguinity. Secondary hemophagocytic lymphohistiocytosis (ie, acquired hemophagocytic lymphohistiocytosis) occurs after strong immunologic activation, such as that which can occur with systemic infection, immunodeficiency, or underlying malignancy. Both forms are characterized by the overwhelming activation of normal T lymphocytes and macrophages, invariably leading to clinical and hematologic alterations and death in the absence of treatment 8. Drug reaction with eosinophilia and systemic symptoms (DRESS) is also a hypersensitivity reaction with overlapping syndromes with HLH, specifically dermatitis, lymphadenopathy, fever, eosinophilia, and visceral organ involvement 9. Drug reaction with eosinophilia and systemic symptoms (DRESS) is associated with reactivation of herpes viruses with activated CD8+ T lymphocytes directed against them. Thus, patients with DRESS should be evaluated for the development of HLH, including for reactivation of human herpes viruses such HHV-6, HHV-7 and EBV, and coagulation function evaluations.
It is difficult to assess the true epidemiology of HLH. The disease has been studied extensively in Sweden, where the incidence of familial HLH is estimated at 1 in 50,000 live births 10. Overall, an estimate of HLH in children <18 years old across ethnicities and races is approximately 1 in 100,000 11. Other large series have been described in Hong Kong and Taiwan; however, worldwide, the incidence of FHL is unknown, and even less epidemiologic data are available regarding acquired HLH cases 12. It is largely believed that the condition is underrecognized, as hemophagocytosis is often not pathologically evident until autopsy. The familial types are usually diagnosed in childhood, while secondary HLH can occur at any age.
The treatment of hemophagocytic lymphohistiocytosis is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, specialists in diagnosing and treating blood disorders (hematologists), specialists in diagnosing and treating cancer (oncologists), specialists in diagnosing and treating immune system diseases (immunologists), geneticists (for familial forms), social workers, and other healthcare professionals may need to systematically and comprehensively plan treatment. Psychosocial support for the entire family is essential as well. Genetic counseling may be of benefit for affected individuals and their families.
Specific therapeutic procedures and interventions may vary, depending upon numerous factors, such as the underlying cause; the presence or absence of certain symptoms; the overall severity of the symptoms and the disorder; an individual’s age and general health; and/or other elements. Decisions concerning the use of particular drug regimens and/or other treatments should be made by physicians and other members of the health care team in careful consultation with the patient based upon the specifics of his or her case; a thorough discussion of the potential benefits and risks, including possible side effects and long-term effects; patient preference; and other appropriate factors.
Affected individuals whose overall health is strong enough may undergo treatment for the underlying condition such as medications to treat an underlying infection, or appropriate treatment for autoimmune disorders or cancer. Treating the underlying condition may remove the “trigger” that has led to the abnormal immune system response.
Affected individuals who health is deteriorating require treated specific for hemophagocytic lymphohistiocytosis immediately. In 1994, the Histiocyte Society published treatment recommendations for this disorder (HLA-94). There were also published studies from 2004 (HLA-2004) that were slightly different.
These treatment regimens include chemotherapy and drugs that suppress the activity of the immune system (immunosuppressive drugs). They target and destroy the hyperactive immune system cells and reduce the life-threatening inflammation that characterizes hemophagocytic lymphohistiocytosis.
After initial treatment, which lasts about 8 weeks, affected individuals are gradually weaned off the drugs onto different medications. If affected individuals have not responded well to this treatment, an allogeneic stem cell transplant may be recommended. This treatment is also recommended for individuals with an abnormal variant in a known HLH gene, central nervous system involvement, and blood cancer (hematologic malignancy) that cannot be treated.
An allogeneic stem cell transplant is a procedure in which stem cells from an affected individual is replaced with the stem cells from a matched, healthy donor. Stem cells are special cells found in bone marrow that manufacture different types of blood cells (e.g. red blood cells, white blood cells, platelets).
Affected individuals undergo high-doses of chemotherapy or radiation to wipe out their stem cells. The stem cells are then replaced with those from a donor. Allogeneic stem cell transplant is a high-risk procedure with the potential for side effects.
Some affected individuals may need blood transfusion because they have low levels of circulating red blood cells or platelets. Some physicians may recommend antibiotics to prevent the development of an infection (prophylactic therapy).
In 2018, Gamifant (emapalumab) was approved for the treatment of pediatric and adult patients with primary HLH who have refractory, recurrent or progressive disease or cannot tolerate conventional HLH therapy.
HLH causes
Hemophagocytic lymphohistiocytosis is broadly broken down into primary and secondary (acquired) forms. The condition results from an ineffective, abnormal response of the immune system to a stimulus or ‘trigger’. The underlying mechanisms that cause signs and symptoms to develop are complex. There is overproduction and overactivity of immune system cells called histiocytes and T cells. These are types of white blood cells, which are the primary cell of the immune system and help the body to fight off infection.
Histiocytes (also called macrophages) are large phagocytic cells that normally play a role in responding to infection and injury. A phagocytic cell is any “scavenger” cell that engulfs and destroys invading microorganisms or cellular debris. Macrophages also secrete cytokines, which are proteins that stimulate or inhibit other immune system cells and promote inflammation in response to disease. Excessive cytokine production will eventually cause severe tissue damage. Macrophages may also mistakenly engulf and destroy healthy tissue including healthy blood cells, which is called hemophagocytosis. Cytotoxic lymphocytes, which include T cells and natural killer cells, do not function properly. These cells eliminate other cells that are damaged, stressed, or infected. In HLH, cytotoxic lymphocytes fail to eliminate activated macrophages allowing them to abnormally build up in the organs and tissues of the body, which further activates this ineffective immune response. These immune system abnormalities cause the excessive inflammation and tissue destruction that characterizes the condition.
Generally, the forms of HLH associated with infants and young children are caused by defects in immune regulation, such as mutations in genes controlling the function of cytotoxic T-lymphocytes and NK-cells. With older children and adults, HLH is more likely to be secondary to infection, malignancy, or autoimmune disease. While rare, some familial cases have been undetected until adulthood. Even children with a defined genetic cause of HLH often have a secondary assault such as infection that triggers HLH, in keeping with the two-hit hypothesis required for the development of many diseases.
Primary HLH
Primary HLH (familial hemophagocytic lymphohistiocytosis or FHL) is associated with abnormal variants in certain genes. Genes provide instructions for creating proteins that play a critical role in many functions of the body. When a mutation of a gene occurs, the protein product may be faulty, inefficient, absent, or overproduced. Depending upon the functions of the particular protein, this can affect many organ systems of the body.
At least four different genes have been identified that result in a genetic predisposition to developing familial hemophagocytic lymphohistiocytosis (FHL). A genetic predisposition means a person has a gene or genes for a particular disorder, but the disorder will not develop unless other factors help to trigger the disorder. The four genes are PRF1 (familial hemophagocytic lymphocytosis type 2), UNC13D (familial hemophagocytic lymphocytosis type 3), STX11 (familial hemophagocytic lymphocytosis type 4, and STXBP2 (familial hemophagocytic lymphocytosis type 5). The gene for familial hemophagocytic lymphocytosis type 1 has yet to be identified.
These genes produce proteins that have an essential role in the immune system. They play a role in turning off or destroying activated immune cells when they are no longer needed. Because of variations (mutations) in these genes, the genes do not produce enough or produce ineffective versions of these proteins. As a result, activated immune cells that should normally be turned off or destroyed persist and continue to work, eventually damaging healthy cells and tissue.
Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother. Disorders inherited in a recessive pattern occur when an individual inherits the same variant gene for the same trait from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
Some individuals may have different variants affecting each copy of the one of the disease genes (compound heterozygotes), while other individuals may have digenic inheritance. Digenic inheritance means they have an abnormal variant in two different genes known to be associated with hemophagocytic lymphohistiocytosis.
Some affected individuals have hemophagocytic lymphohistiocytosis as part of a broader genetic disorder. These disorders include Griscelli syndrome type 2, Chediak-Higashi syndrome, X-linked lymphoproliferative disorder, XMEN disease, interleukin-2-inductible T cell kinase deficiency, CD27 deficiency, Hermansky-Pudlak syndrome, lysinuric protein intolerance, and chronic granulomatous disease. In some individuals, hemophagocytic lymphohistiocytosis may be the only clinical problem that individuals with these disorders display.
Secondary HLH
Individuals with secondary (or acquired) HLH develop the disorder because of a heightened, abnormal immune system response that occurs for unknown reasons. There is no family history of the disorder and no known genetic factors can be identified. Conditions that can lead to secondary hemophagocytic lymphohistiocytosis include viral infections (29%) especially Epstein-Barr virus, other infections (20%) including bacterial and fungal infections, a weakened or depressed immune system (6%), autoimmune diseases, autoinflammatory diseases, rheumatological diseases such as juvenile idiopathic arthritis (7%), metabolic disorders, and cancer (27%) such as non-Hodgkin lymphoma 13.
The exact manner that these predisposing conditions cause the signs and symptoms, and specifically how they cause an ineffective, abnormal immune response, in hemophagocytic lymphohistiocytosis are not fully understood.
HLH symptoms
The signs and symptoms of HLH typically develop during the first months or years of life. However, in rare cases, affected people may not show symptoms until later in childhood or even into adulthood. The features of this condition may include 3:
- Fever
- Enlarged liver and/or spleen
- Skin rash
- Lymph node enlargement
- Breathing problems
- Easy bruising and/or abnormal bleeding
- Kidney abnormalities
- Heart problems
- Increased risk for certain cancers (leukemia, lymphoma)
Many people with HLA also develop neurologic abnormalities. The neurological symptoms vary but may include irritability, fatigue, abnormal muscle tone, seizures, neck stiffness, mental status changes, ataxia, blindness, paralysis, and/or coma 6.
The onset and severity of hemophagocytic lymphohistiocytosis can vary greatly from one person to another. The specific symptoms that develop can also vary greatly, although the condition often causes multiorgan involvement. Generally, affected individuals develop fevers, a rash, an abnormally large liver (hepatomegaly), and an abnormally large spleen (splenomegaly). Fevers may be prolonged and persistent, often failing to respond to antibiotics. Sometimes, the lymph nodes are also abnormally large (lymphadenopathy). Lymph nodes are part of the lymphatic system, a circulatory network of vessels, ducts, and nodes that filter and distribute certain protein-rich (lymph) and blood cells throughout the body. Lymph nodes are small structures, found in groups throughout the body, that help to filter or drain out harmful substances from the body.
These initial sign and symptoms are described as nonspecific. This means that these signs and symptoms are common to many other different disorders or conditions, which can make getting a correct diagnosis difficult.
Affected individuals may also have low levels circulating red blood cells (anemia) and low levels of circulating platelets (thrombocytopenia). Red blood cells deliver oxygen to the body and platelets allow the body to form clots to stop bleeding. Individuals with anemia may experience tiredness, increased need for sleep, weakness, lightheadedness, dizziness, irritability, headaches, pale skin color, difficulty breathing (dyspnea), and cardiac symptoms. Individuals with thrombocytopenia are more susceptible to excessive bruising following minimal injury and to spontaneous bleeding from the mucous membranes, especially those of the gums and nose.
Some affected individuals may develop neurological symptoms including seizures, changes in mental status and irritability, paralysis (palsy) of certain cranial nerves, and problems coordinating voluntary movements (ataxia). Affected individuals are at risk of developing posterior reversible encephalopathy syndrome, which causes a rapid onset of headaches, altered consciousness, seizures, and disturbances in vision. Neurological problems are most common with familial hemophagocytic lymphohistiocytosis.
Additional symptoms can occur depending upon the specific organ system involved in an individual. These symptoms can include significant problems breathing (lung dysfunction), severe low blood pressure (hypotension), liver inflammation (hepatitis), kidney dysfunction, yellowing of the skin and whites of the eyes (jaundice), swelling due to fluid accumulation (edema), abdominal swelling due to fluid accumulation (ascites), and a variety of skin problems including widespread, reddening of the skin because of inflammation (erythroderma), rashes, blood spots (purpura), and tiny spots on the skin (petechiae).
HLH diagnosis
The diagnosis of autosomal recessive familial HLH or secondary HLH is based on a number of clinical signs and laboratory findings. Due to the relatively nonspecific nature of the clinical signs and symptoms, and significant overlap with other illnesses, diagnosis is often delayed. The official diagnosis of HLH, established by the Histiocyte Society, is based on fulfilling one or both of the following criteria 2.
HLH criteria
- A molecular diagnosis consistent with HLH
- Five out of the following nine diagnostic criteria for HLH: fever, splenomegaly, cytopenias (affecting two or more of three lineages in the peripheral blood), hypertriglyceridemia, hypofibrinogenemia, elevated ferritin, hemophagocytosis in bone marrow/spleen/lymph nodes, low or absent natural killer (NK)-cell activity, or elevated soluble CD25 (interleukin [IL]-2 receptor) 14.
All of the clinical and laboratory findings are readily linked to the pathophysiology of HLH. Fever is the result of high interleukin (IL) levels. Splenomegaly is the direct result of infiltration by lymphocytes and macrophages. Cytopenias can be explained by high concentrations of tumor necrosis factor (TNF)-α and interferon (IFN)-γ, as well as direct hemophagocytosis. High triglycerides are secondary to decreased lipoprotein lipase activity initiated by increased TNF-α levels. Elevated ferritin >10,000 μg/L has been demonstrated to be 90% sensitive and 96% specific for HLH 15. Ferritin is believed to accumulate during the anti-inflammatory process of macrophage scavenging of heme via the CD163 receptor. High concentrations of soluble IL-2 receptor are produced by activated lymphocytes 16. A summary of clinical and laboratory findings is provided in Table 1.
Coagulopathy is a prominent feature of HLH, as low fibrinogen is found in the majority of patients 2. Further coagulation studies have demonstrated normal factor V and VIII levels and an absence of fibrin split products. These findings provide evidence against disseminated intravascular coagulation, a diagnosis that may overlap with HLH due to the shared findings of thrombocytopenia and hypofibrinogenemia 17. It is believed that macrophages may secrete plasminogen activators which accelerate the conversion of plasminogen to plasmin, subsequently degrading fibrinogen. Fibrin split products may be phagocytized by macrophages in the reticuloendothelial system. Given the normal coagulation factors measured in HLH, liver failure, while often present, is not related to the coagulopathy. The hepatomegaly, elevated transaminases, and bilirubin are believed to be the direct result of organ infiltration by lymphocytes and histiocytes 18.
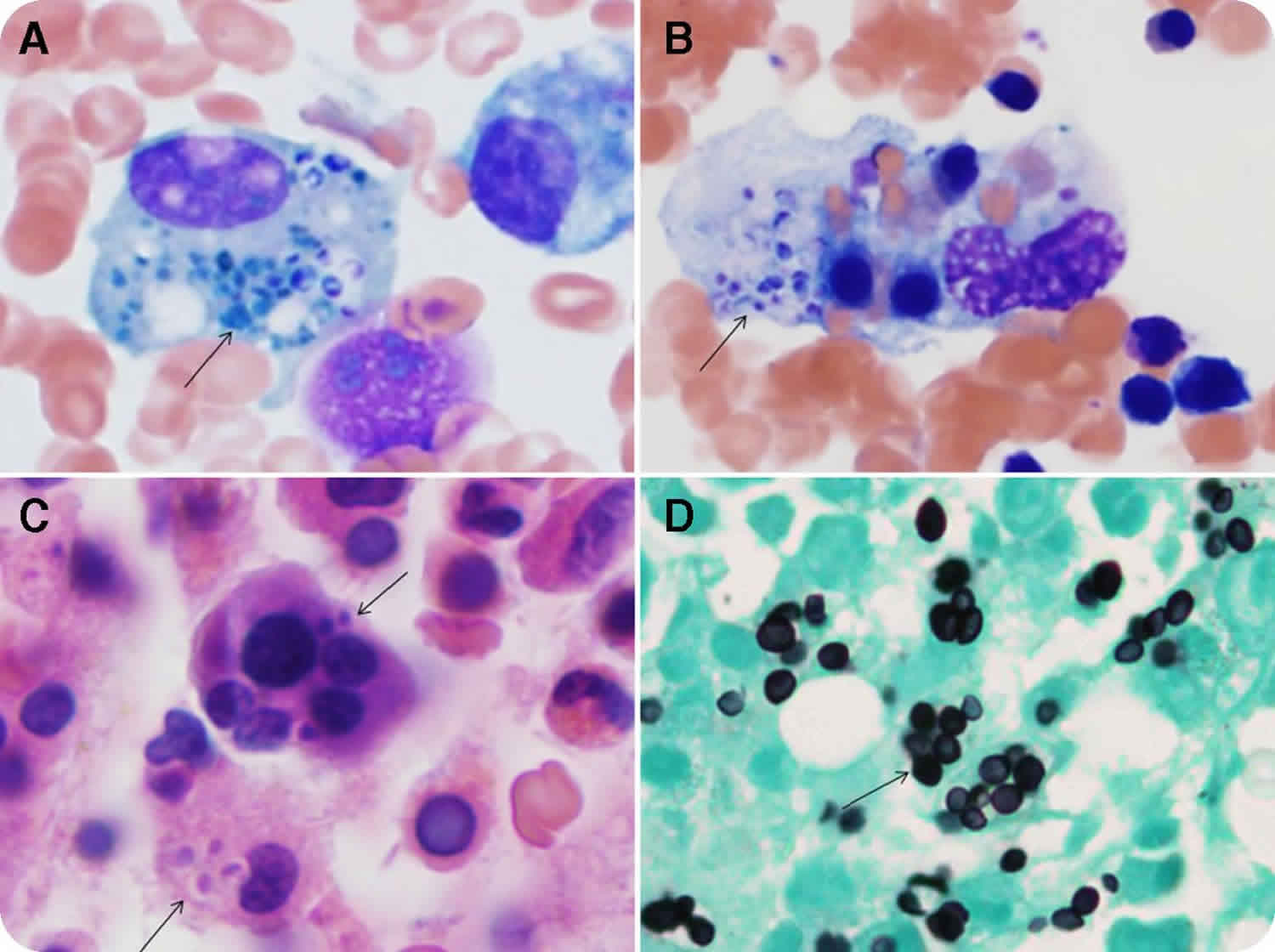
However, despite the fact that hemophagocytosis is prominently featured in the name of this disease, it is rarely found at presentation in secondary cases and may not be visible until late in disease progression. Bone marrow biopsies performed early in the course of secondary disease may be normal or demonstrate very nonspecific findings such as increased or decreased unilineage or multilineage hematopoiesis. Repeat studies may be necessary to show these findings. Familial HLH, on the other hand, may demonstrate prominent hemophagocytosis from the start. An immunohistochemical stain for CD163 may be useful, as upregulation of this receptor facilitates hemophagocytosis 19. In cases in which cerebrospinal fluid (CSF) is obtained, pleocytosis is sometimes noted with lymphocytes, histiocytes, and an increased protein level. Microscopic review of spun cerebrospinal fluid may demonstrate hemophagocytosis 20. While many of the laboratory tests are readily available, evaluation of IL-2 receptor and NK-cell activity may require sending specimens out to specialized reference laboratories and may not be a timely option for clinical diagnosis. When available, comparison of IFN-γ, IL-10, and IL-6 may be useful for distinguishing between bacterial sepsis, viral infections, and HLH. Using the criteria IFN-γ >75 pg/mL, and IL-10 60 pg/mL, sensitivity and specificity of diagnosing HLH is 98.9% and 93.0%, respectively 21. Additionally, measuring plasma levels of CD163, a receptor for hemoglobin-haptoglobin complexes, may also be helpful in distinguishing HLH from other purely infectious diseases 22.
Flow cytometry may be used to identify and quantify levels of several useful markers involved in the pathophysiology of HLH. A rapid flow cytometric analysis of intracellular X-linked inhibitor of apoptosis protein (XIAP) has been developed for detection for X-linked lymphoproliferative disease and carrier state, which has also proven useful following bone marrow transplant to monitor reconstitution 23. A prospective evaluation of degranulation assays was found to be useful in the differential diagnosis of familial HLH. CD107 may serve as a useful surrogate marker for reduced or absent NK-cell and cytotoxic T-cell activity. Using an assay for surface upregulation of CD107a on NK-cells and cytotoxic T-lymphocytes in a large cohort of patients under evaluation for HLH, the vast majority of patients with familial HLH subtypes 3–5 and Griscelli syndrome type 2 or Chediak–Higashi syndrome had abnormal resting NK-cell degranulation. NK-cell degranulation was found to be normal in the majority of patients with familial HLH type 2 and X-linked lymphoproliferative disease. Instead these patients were found to have diminished intracellular SAP (SLAM [signaling lymphocytic activation molecule]-associated protein), X-linked inhibitor of apoptosis protein (XIAP), and perforin expression. Most patients with secondary HLH did not have abnormalities of NK-cell degranulation. Thus, degranulation assays may help speed the diagnosis of HLH and allow treatment to begin more rapidly 24. Degranulation of CD107 may be associated with a specific type of familial HLH (FHL-5) in which missense mutations lead to decreased lymphocyte stability of syntaxin binding protein 2 (Munc18-2) and syntaxin 11. These proteins would normally be involved in regulating vesicle transport to the plasma membrane, which is key to the pathophysiology of HLH 25.
Flow cytometry for perforin staining in cytotoxic lymphocytes, including NK-cells, CD8+ T-cells, and CD56+ T-cells, seems to be useful as a quick and reliable marker for perforin gene mutations seen in HLH. In a study of eleven unrelated HLH patients and 19 family members, four of seven patients with FHL showed lack of intracellular perforin in all cytotoxic cell types, which corresponded to mutations in the perforin gene. The parents of these patients also had abnormal perforin staining, indicative of their carrier state for perforin mutations. Evaluation of cytotoxic T-cells from the other three patients with FHL demonstrated normal percentages of perforin staining cytotoxic T-cells. The four patients with Epstein–Barr virus (EBV)-associated secondary HLH had depressed numbers of NK-cells but increased CD8+ T-cells with perforin expression 26. Flow cytometry studies have proven to be a rapid and useful modality to reliably diagnose HLH and shorten the time to treatment 26.
Table 1. Incidences of clinical and laboratory findings in familial HLH and secondary HLH
| Finding | Percentage of FHL cases with finding at diagnosis | Percentage of secondary HLH cases with finding at diagnosis | Diagnostic criteria |
|---|---|---|---|
| Fever | ∼100% | ∼100% | >37°C |
| Hepatosplenomegaly | ∼100% | ∼80%–90% | Radiographic or physical exam evidence |
| Cytopenias | ∼100% | ∼80% | Hemoglobin <9 g/dL Platelets <100 × 109/L Neutrophils <1.0 × 109/L |
| Hypertriglyceridemia (fasting) | 70% | 40% | >3 mmol/L |
| Hypofibrinogenemia | 60%–65% | 40% | <1.5 g/L |
| Elevated ferritin | 70% | 95% | >500 μg/L |
| Hemophagocytosis | 85% | Variable and not necessary to make initial diagnosis if other features present | Bone marrow or other tissue biopsy |
| Decreased NK-cell activity | 100% | 30% | <10% activity by flow cytometric assays |
| Elevated sCD25 | 90% | Percentage not found in secondary HLH literature | >2,400 U/mL |
| LDH | 40%–45% | 100% | ≥500 U/L |
| ALT | 30%–35% | Percentage not found in secondary HLH literature | ≥100 U/L |
| AST | 30%–35% | Percentage not found in secondary HLH literature | ≥100 U/L |
| Bilirubin | ∼30% | Percentage not found in secondary HLH literature | ≥34 μmol/L |
| CSF cells | 35%–40% | Percentage not found in secondary HLH literature | ≥5/μL |
| CSF protein | 45% | Percentage not found in secondary HLH literature | ≥0.5 g/L |
Abbreviations: ALT = alanine transaminase; AST = asparate aminotransferase; CSF = cerebrospinal fluid; FHL = familial HLH; HLH = hemophagocytic lymphohistiocytosis; LDH = lactate dehydrogenase; NK = natural killer; sCD25 = soluble CD25.
[Source 2 ]HLH treatment
Prior to the use of modern treatment regimens, survival with HLH was close to 0% 27. Broadly, treatment of HLH involves immune-suppressive and modulatory agents, biological response modifiers, treatment of the inciting illness if secondary, and subsequent stem-cell transplantation. Therapy is aimed at suppressing the hyperinflammatory state and immune dysregulation that leads to life-threatening organ damage and susceptibility to deadly infections. It is also important to kill infected antigen-presenting cells to remove the stimulus for ongoing immune activation. Treatment of HLH may vary according to cause. Discussion of treatment is subdivided into familial HLH, infection-related, malignancy-associated, and autoimmune diseases. In 2018, Gamifant (emapalumab) was approved for the treatment of pediatric and adult patients with primary HLH (familial HLH) who have refractory, recurrent or progressive disease or cannot tolerate conventional HLH therapy.
Familial HLH
Early attempts at treating HLH included vinblastine (a vinca alkaloid) and corticosteroids. The epipodophyllotoxins, etoposide (VP-16), and teniposide (VM-26), in combination with steroids showed some promise in achieving prolonged remissions. In 1994, the Histiocyte Society proposed the first protocol for the treatment of HLH (HLH-94). The protocol began in 1994, 5 years before genetic markers for familial HLH were found. At the time, familial HLH was defined by having an affected sibling. HLH-94 offered an established chemotherapy regimen (epidodophyllotoxin and corticosteroids) in conjunction with immunotherapy with cyclosporin A (CsA). The original eligibility requirements for the trial included only patients under the age of 16 years. HLH-94 included 8 weeks of initial chemotherapy and immunotherapy, attempting to achieve complete remission, followed by continuation therapy until an acceptable bone marrow donor could be found. The first 8 weeks consisted of dexamethasone at a starting dosage of 10 mg/m2 body surface area, then tapering down by half in 2-week increments for 6 weeks, then 1 week of 1.25 mg/m², and a subsequent week of taper. Concurrently with the dexamethasone taper, VP-16 therapy was initiated twice weekly during the first 2 weeks, then weekly. After week 8, cyclosporin A (CsA) therapy began and extended for the duration of therapy. Intrathecal methotrexate was included in selected patients with evidence of central nervous system (CNS) involvement. After 8 weeks, pulses of dexamethasone were given at regular intervals. HSCT was recommended for all children with a suitable allogeneic donor. Supportive care may have included an intensive care unit stay, broad-spectrum antibiotics until appropriate culture results were available, microbiological surveillance, HLA testing of patient and family in anticipation of hematopoietic stem cell transplant (HSCT), and prophylactic antifungals 28. Conditioning regimens prior to hematopoietic stem cell transplant (HSCT) included busulfan, cyclophosphamide, etoposide, and if the donor was unrelated, antithymocyte globulin (ATG) 29.
Retrospective reviews of the HLH-94 treatment protocol morbidity and mortality for 249 patients with long-term follow-up revealed that overall survival and response to therapy did not differ in patients with versus without family history. Overall survival rates were 54%. In this study, 114 (46%) died, and 72 did not receive transplant. Of patient deaths, 64 (89%) occurred in the first year. Of the eight deaths occurring after 1 year of therapy, two had progressive disease without an available hematopoietic stem cell transplant donor, one could not be transplanted due to severe neurological disease, and five relapsed. Overall, HLH-94 achieved complete remission or allowed survival to hematopoietic stem cell transplant in 71% of patients 29.
Given the success of the HLH-94 protocol, in 2004, a revised protocol called HLH-2004 was proposed, which aimed to 1) evaluate a revised initial and continuation therapy with an end goal of hematopoietic stem cell transplant, 2) evaluate and improve results of hematopoietic stem cell transplant with various types of donors, 3) evaluate the prognostic importance of state of remission at the time of hematopoietic stem cell transplant, 4) evaluate long-term neurologic sequelae, and 5) improve understanding of the pathophysiology of HLH by including genotype–phenotype studies and evaluate the prognostic value of NK-cell activity subtyping. HLH-2004 essentially took HLH-94 and moved cyclosporine from later in the regimen to an initial therapy, concurrent with dexamethasone and etoposide (VP-16). Dexamethasone and VP-16 dosing was the same as in HLH-94. Intrathecal methotrexate was still included for select patients with CNS involvement. In general, HLH-2004 approaches all patients with an initial 8 weeks of chemotherapy, and supports the patient until hematopoietic stem cell transplant can be performed in the case of patients with genetically determined disease or persistent non-genetic disease. Cases which resolve or are non-genetic can cease therapy unless relapse occurs, in which case hematopoietic stem cell transplant would be undertaken. Eligibility requirements include children <18 years old. Separate concurrent studies have been undertaken on patients >18 years old, and those with Chediak–Higashi syndrome, Griscelli syndrome, or X-linked lymphoproliferative syndrome 28. The HLH-2004 protocol officially ceased enrolment at the end of 2011. Long-term outcomes are still under evaluation.
An alternative regimen for familial HLH was first described by Stephan et al 30 in 1993 and in an expanded trial described by Mahlaoui et al 31 in 2007. The development of these regimens was based on the recognition that T-lymphocytes play a role in the pathology of HLH as well as macrophages. This discovery was based on the detection of major histocompatibility complex class II positive T-cells, high levels of soluble serum IL-2, CD8, and IFN-γ in the serum of familial HLH patients. This regimen featured a combination of ATG with corticosteroids, cyclosporin A (CsA), and intrathecal methotrexate. The Stephan study 30 evaluated six patients and was successful in achieving remission quickly, although two patients died from CNS disease. In the expanded study, the regimen was used to treat 38 consecutive patients with familial HLH over the course of 14 years. The patients received 45 courses of antithymocyte globulin (ATG), with a total dosage of 50 mg/kg or 25 mg/kg varying by severity of disease over the course of 5 days. Methylprednisolone at 4 mg/kg/day was administered with the antithymocyte globulin for 5 days then tapered. Intrathecal methotrexate and corticosteroids were given at various dosages determined by patient age and at intervals varying according to the severity of CNS involvement. Patients also received supportive care with fibrinogen infusions, irradiated packed red blood cells, and platelets. Broad spectrum antibiotics and intravenous immunoglobulins were also given. Cyclosporin A (CsA) was added to reach a plasma concentration of 150 ng/mL prior to hematopoietic stem cell transplant. Immediate adverse effects of antithymocyte globulin were relatively minor and presented as fever and chills during infusion that rapidly resolved and did not preclude further treatment. After approximately 2 weeks of antithymocyte globulin treatment, bacterial, viral, or fungal infections were encountered. Infections were more common in patients receiving antithymocyte globulin as a secondary treatment for relapse than in first-line therapy. Once a complete response was achieved as evidenced by normalization of clinical and biological parameters, hematopoietic stem cell transplant was performed for patients with an available HLA identical donor. Patients without a complete response or without a viable donor were given maintenance therapy. Overall, the efficacy of antithymocyte globulin therapy in achieving complete remission was 73%. Using antithymocyte globulin as a first-line treatment had a higher success rate of 82% of patients achieving complete remission versus only about 50% achieving complete remission with second-line antithymocyte globulin treatment. The best outcomes were seen with patients who received hematopoietic stem cell transplant shortly after starting antithymocyte globulin therapy 31.
Infection-associated HLH
While treating the inciting infectious agent is important in the treatment of infection-related HLH, treating the identified organism alone is not enough. Most cases of infection-related HLH should be treated aggressively with standard HLH protocols. The exception to this rule is in Leishmania-related HLH, which has been treated successfully with liposomal amphotericin alone 32. In particular, the prognosis for EBV-associated cases has improved dramatically with chemotherapy and immune modifying agents. In a multivariate analysis of patients on regimens consisting of corticosteroids alone, intravenous immunoglobulins alone, CSA alone, or a combination of treatments without etoposide versus another group of patients receiving etoposide, early introduction of etoposide was the only significant variable for improved survival 33. Etoposide appears to interfere with EBV-induced lymphocyte transformation and suppresses formation of EBV nuclear antigen. Despite its potential risks, the benefits of etoposide justify its early use in light of the fact that even seemingly mild cases may deteriorate quickly.
Macrophage activation syndrome
Macrophage activation syndrome is rather unique in that these HLH cases may respond quite well to high dose corticosteroids alone. One of the first reports of treating macrophage activation syndrome associated HLH with corticosteroids, was a case series of systemic Juvenile Idiopathic Arthritis (SJIA) in France which described seven children with hemorrhagic, hepatic, and neurologic features, later realized to be macrophage activation syndrome. The children were treated with high-dose steroids, and five out of seven survived 34. Several other case series had similar outcomes 35. Cyclosporin A (CsA) therapy has also become a prominent therapy in addition to corticosteroids in macrophage activation syndrome associated with systemic Juvenile Idiopathic Arthritis (SJIA), as cyclosporin A may preferentially inhibit lymphocytes by targeting transcription factors that activate various cytokine genes 36. Cyclosporin A (CsA) likely inhibits the cytokine storm of macrophage activation syndrome. Cyclophosphamide has also been used to target lymphocytes in macrophage activation syndrome 37. Etoposide-based regimens such as HLH-94 and HLH-2004 can be used in macrophage activation syndrome, but the risks must be weighed carefully.
Hematopoietic stem cell transplant
Virtually all genetic cases of HLH and many secondary cases should be treated with hematopoietic stem cell transplant. The first report of successful hematopoietic stem cell transplant was reported in 1986 38. Several studies have demonstrated that hematopoietic stem cell transplant is the only true hope for permanent control of the disease or essentially a cure 39. A study of 86 children treated with HLH-94 followed by hematopoietic stem cell transplant demonstrated similar long-term disease-free survival (70% at 3 years) with matched unrelated donor transplants as with matched sibling transplants. Survival with family haploidentical donor transplants or mismatched unrelated transplants showed much less favorable results with long-term disease-free survival of only 50% 40. Cord blood transplant has been successful in some patients. However, overall transplant morbidity and mortality remains high. The same pediatric study showed a mortality rate of 26 out of 86 patients, with deaths resulting from pulmonary and liver complications 40. Patients responding well to pre-transplant induction therapy appear to respond best to hematopoietic stem cell transplant. Pre-transplant conditioning regimens generally include busulfan, etoposide, and cyclophosphamide. Busulfan levels must be carefully monitored, and clonazepam or phenytoin may be useful as anticonvulsive prophylaxis. Dexamethasone may be used to prevent etoposide-induced anaphylactic-like symptoms. Mesna can be used for protection against cyclophosphamide-induced bladder injury. Trimethoprim/sulfamethoxazole may be used for pneumocystis prophylaxis, and acyclovir prophylaxis is recommended 28.
Acute graft versus host disease (GVHD) appears to be the most common complication post-transplant, with rates as high as 32% and chronic graft versus host disease rates at about 9% 41. Additionally, some patients may develop mixed chimerism necessitating regular donor lymphocyte infusions 10. With reduced intensity conditioning at an experienced transplant center, patients surviving to hematopoietic stem cell transplant have an approximate survival rate of 92% 27. The unifying thread of all treatments is that the best success rates occur when complete remission is achieved rapidly and hematopoietic stem cell transplant closely follows.
Salvage therapy
Despite advances in treatment regimens, up to 25% of children with HLH cannot undergo hematopoietic stem cell transplant due to advancing disease. Due to the rarity of HLH and short survival, salvage therapies have been described in various case reports but few large studies, leaving clinicians with few evidence-based options for refractory HLH. Removal of cytokines via plasmapheresis has been described to support patients until other therapies have reached therapeutic effect 42. Recombinant human thrombopoietin has been used as supportive therapy for thrombocytopenia in HLH 43. The use of monoclonal antibodies such as alemtuzumab, infliximab, and daclizumab has been described in various case reports. Alemtuzumab targets the CD-52 antigen, which is expressed on most lymphocytes, monocytes, macrophages, and dendritic cells 44. Infliximab targets TNF, and daclizumab targets CD-25. Both have been used with reported success 45. Additionally, etanercept, a TNF inhibitor, was used with success in a patient with acute lupus hemophagocytic syndrome 46. Various case reports have elaborated on treatment of refractory cases with splenectomy 47 and even liver transplant for the damage caused by the unbridled macrophage activity 48. However, given the scarcity of literature on the subject, it is unclear what role these measures will play in the treatment of HLH.
HLH prognosis
Although the prognosis varies between studies and with different approaches to treatment, the disease is invariably fatal if not treated. The median survival rate has been reported to be 2-6 months without treatment, but survival time has dramatically improved with the advent of the HLH-94 protocol.
A study of 122 patients from the International Registry for hemophagocytic lymphohistiocytosis found that the overall estimated 5-year survival rate was 21%, with 66% of patients who received bone marrow transplantation (hematopoietic stem cell transplant) surviving 5 years versus only 10.1% of patients treated with chemotherapy alone 49. More recent studies have shown that the HLH-94 protocol resulted in an overall survival rate of 55%. Success or failure of an allogeneic bone marrow transplantation is the most important long-term prognostic factor. Unfortunately, many cases are diagnosed late in the course of the disease, after irreversible damage has occurred.
Although patients with hemophagocytic lymphohistiocytosis are at high risk for death early in their disease course, steroids, intravenous immunoglobulin (IVIG), or both may be sufficient as first-line therapy for selected patients 50.
The ratio of C16 –ceramide to sphingosine was elevated in those who died despite appropriate treatment, but remained low in survivors, implying that this ratio may be of prognostic significance 51. The balance of ceramide and sphingosine may be pivotal in the clinical outcome for these patients.
References- Lymphohistiocytosis (Hemophagocytic Lymphohistiocytosis). https://emedicine.medscape.com/article/986458-overview
- George MR. Hemophagocytic lymphohistiocytosis: review of etiologies and management. J Blood Med. 2014;5:69–86. Published 2014 Jun 12. doi:10.2147/JBM.S46255 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4062561
- George MR.. Hemophagocytic lymphohistiocytosis: review of etiologies and management.. J Blood Med. June 2014; 5:69-86. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4062561
- Morrell DS, Pepping MA, Scott JP, et al. Cutaneous manifestations of hemophagocytic lymphohistiocytosis. Arch Dermatol. 2002 Sep. 138(9):1208-12.
- Pasvolsky O, Zoref-Lorenz A, Abadi U, Geiger KR, Hayman L, Vaxman I, et al. Hemophagocytic lymphohistiocytosis as a harbinger of aggressive lymphoma: a case series. Int J Hematol. 2019 Mar 8.
- Kejian Zhang, MD, MBA, Alexandra H Filipovich, MD, Judith Johnson, MS, Rebecca A Marsh, MD, and Joyce Villanueva, MT, MBA.. Hemophagocytic Lymphohistiocytosis, Familial. GeneReviews. January 17, 2013; http://www.ncbi.nlm.nih.gov/books/NBK1444
- Zhang K, Wakefield E, Marsh R. Lymphoproliferative Disease, X-Linked. 2004 Feb 27 [Updated 2016 Jun 30]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1406
- Feldmann J, Le Deist F, Ouachee-Chardin M, et al. Functional consequences of perforin gene mutations in 22 patients with familial haemophagocytic lymphohistiocytosis. Br J Haematol. 2002 Jun. 117(4):965-72.
- Liang J, Qu H, Wang X, Wang A, Liu L, Tu P, et al. Drug Reaction with Eosinophilia and Systemic Symptoms Associated with Reactivation of Epstein-Barr Virus and/or Cytomegalovirus Leading to Hemophagocytic Syndrome in One of Two Patients. Ann Dermatol. 2018 Feb. 30 (1):71-74.
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med. 2012;63:233–246.
- Niece JA, Rogers ZR, Ahmad N, Langevin AM, McClain KL. Hemophagocytic lymphohistiocytosis in Texas: observations on ethnicity and race. Pediatr Blood Cancer. 2010;54(3):424–428.
- Janka GE. Hemophagocytic lymphohistiocytosis. Hematology. 2005;10(Suppl 1):104–107.
- Dhote R, Simon J, Papo T, et al. Reactive hemophagocytic syndrome in adult systemic disease: report of twenty-six cases and literature review. Arthritis Rheum. 2003;49(5):633–639.
- HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski J, Janka G. Pediatr Blood Cancer. 2007 Feb; 48(2):124-31.
- Lin TF, Ferlic-Stark LL, Allen CE, Kozinetz CA, McClain KL. Rate of decline of ferritin in patients with hemophagocytic lymphohistiocytosis as a prognostic variable for mortality. Pediatr Blood Cancer. 2011;56(1):154–155.
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Eur J Pediatr. 2007;166(2):95–109.
- McClure PD, Strachan P, Saunders EF. Hypofibrinogenemia and thrombocytopenia in familial hemophagocytic reticulosis. J Pediatr. 1974;85(1):67–70.
- Janka G, zur Stadt U. Familial and acquired hemophagocytic lymphohistiocytosis. Hematology Am Soc Hematol Educ Program. 2005:82–88.
- Filipovich AH. Hemophagocytic lymphohistiocytosis and related disorders. Curr Opin Allergy Clin Immunol. 2006;6(6):410–415.
- Janka GE. Familial hemophagocytic lymphohistiocytosis. Eur J Pediatr. 1983;140(3):221–230.
- Xu XJ, Tang YM, Song H, et al. Diagnostic accuracy of a specific cytokine pattern in hemophagocytic lymphohistiocytosis in children. J Pediatr. 2012;160(6):984–990. e1.
- Filipovich AH. Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology Am Soc Hematol Educ Program. 2009:127–131.
- Marsh RA, Bleesing JJ, Filipovich AH. Using flow cytometry to screen patients for X-linked lymphoproliferative disease due to SAP (SLAM [signaling lymphocytic activation molecule]-associated protein) deficiency and X-linked inhibitor of apoptosis protein (XIAP) deficiency. J Immunol Methods. 2010;362(1–2):1–9.
- Bryceson YT, Pende D, Maul-Pavicic A, et al. A prospective evaluation of degranulation assays in the rapid diagnosis of familial hemophagocytic syndromes. Blood. 2012;119(12):2754–2763.
- zur Stadt U, Rohr J, Seifert W, et al. Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18-2 and impaired binding to syntaxin 11. Am J Hum Genet. 2009;85(4):482–492.
- Kogawa K, Lee SM, Villanueva J, Marmer D, Sumegi J, Filipovich AH. Perforin expression in cytotoxic lymphocytes from patients with hemophagocytic lymphohistiocytosis and their family members. Blood. 2002;99(1):61–66.
- Weitzman S. Approach to hemophagocytic syndromes. Hematology Am Soc Hematol Educ Program. 2011;2011:178–183.
- Henter JI, Horne A, Arico M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–131.
- Trottestam H, Horne A, Arico M, et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. 2011;118(17):4577–4584.
- Stephan JL, Donadieu J, Ledeist F, Blanche S, Griscelli C, Fischer A. Treatment of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins, steroids, and cyclosporin A. Blood. 1993;82(8):2319–2323.
- Mahlaoui N, Ouachee-Chardin M, de Saint Basile G, et al. Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: a single-center retrospective report of 38 patients. Pediatrics. 2007;120(3):e622–e628.
- Cancado GG, Freitas GG, Faria FH, de Macedo AV, Nobre V. Hemophagocytic lymphohistiocytosis associated with visceral leishmaniasis in late adulthood. Am J Trop Med Hyg. 2013;88(3):575–577.
- Imashuku S, Kuriyama K, Teramura T, et al. Requirement for etoposide in the treatment of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. J Clin Oncol. 2001;19(10):2665–2673.
- Hadchouel M, Prieur AM, Griscelli C. Acute hemorrhagic, hepatic, and neurologic manifestations in juvenile rheumatoid arthritis: possible relationship to drugs or infection. J Pediatr. 1985;106(4):561–566.
- Ravelli A, Grom AA, Behrens EM, Cron RQ. Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: diagnosis, genetics, pathophysiology and treatment. Genes Immun. 2012;13(4):289–298.
- Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Ann Rev Immunol. 1997;15:707–747.
- Wallace CA, Sherry DD. Trial of intravenous pulse cyclophosphamide and methylprednisolone in the treatment of severe systemic-onset juvenile rheumatoid arthritis. Arthritis Rheum. 1997;40(10):1852–1855.
- Fischer A, Cerf-Bensussan N, Blanche S, et al. Allogeneic bone marrow transplantation for erythrophagocytic lymphohistiocytosis. J Pediatr. 1986;108(2):267–270.
- Baker KS, Filipovich AH, Gross TG, et al. Unrelated donor hematopoi-etic cell transplantation for hemophagocytic lymphohistiocytosis. Bone Marrow Transplant. 2008;42(3):175–180.
- Horne A, Janka G, Maarten Egeler R, et al. Haematopoietic stem cell transplantation in haemophagocytic lymphohistiocytosis. Br J Haematol. 2005;129(5):622–630.
- Horne A, Zheng C, Lorenz I, et al. Subtyping of natural killer cell cytotoxicity deficiencies in haemophagocytic lymphohistocytosis provides therapeutic guidance. Br J Haematol. 2005;129(5):658–666.
- Raschke RA, Garcia-Orr R. Hemophagocytic lymphohistiocytosis: a potentially underrecognized association with systemic inflammatory response syndrome, severe sepsis, and septic shock in adults. Chest. 2011;140(4):933–938.
- Wang Y, Wang Z, Wu L, Zhang J, Wang J, Yan L. Recombinant human thrombopoietin is an effective treatment for thrombocytopenia in hemophagocytic lymphohistiocytosis. Ann Hematology. 2013;92(12):1695–1699.
- Marsh RA, Allen CE, McClain KL, et al. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer. 2013;60(1):101–109.
- Oda Y, Urushidani Y, Ooi S, et al. Hemophagocytic lymphohistiocytosis in a rheumatoid arthritis patient treated with infliximab. Intern Med. 2012;51(6):655–657.
- Takahashi N, Naniwa T, Banno S. Successful use of etanercept in the treatment of acute lupus hemophagocytic syndrome. Mod Rheumatol. 2008;18(1):72–75.
- Machaczka M. Splenectomy as a therapeutic approach in refractory hemophagocytic lymphohistiocytosis. Biomed Pharmacother. 2012;66(2):159–160.
- Wright G, Wilmore S, Makanyanga J, et al. Liver transplant for adult hemophagocytic lymphohistiocytosis: case report and literature review. Exp Clin Transplant. 2012;10(5):508–512.
- Arico M, Janka G, Fischer A, et al. Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia. 1996 Feb. 10(2):197-203.
- Gupta AA, Tyrrell P, Valani R, Benseler S, Abdelhaleem M, Weitzman S. Experience with hemophagocytic lymphohistiocytosis/macrophage activation syndrome at a single institution. J Pediatr Hematol Oncol. 2009 Feb. 31(2):81-4.
- Jenkins RW, Clarke CJ, Lucas JT Jr, Shabbir M, Wu BX, Simbari F, et al. Evaluation of the role of secretory sphingomyelinase and bioactive sphingolipids as biomarkers in hemophagocytic lymphohistiocytosis. Am J Hematol. 2013 Jul 5.





{kind=link}