
Hyperphenylalaninemia
Hyperphenylalaninemia is the term used to describe the mildest manifestation of phenylalanine hydroxylase deficiency and seldom by tetrahydrobiopterin (BH4) deficiency 1, with classic phenylketonuria (PKU) representing the more severe end of this spectrum 2. Hyperphenylalaninemia is broadly defined as the presence of blood phenylalanine levels that exceed the limits of the upper reference range (2 mg/dL or 120 µmol/L) without treatment but that are below the level found in patients with phenylketonuria (PKU) 3. Phenylalanine levels that exceed 20 mg/dL (1200 µmol/L) are considered typical of classic PKU (phenylketonuria). In 2014, the American College of Medical Genetics and Genomics released a practice statement that recommended that phenylketonuria (PKU) and hyperphenylalaninemia both be considered part of the spectrum of phenylalanine hydroxylase deficiency.
Phenylalanine hydroxylase deficiency results in intolerance to the dietary intake of the essential amino acid phenylalanine and produces a spectrum of disorders. The risk of adverse outcome varies based on the degree of phenylalanine hydroxylase deficiency. Without effective therapy, most individuals with severe phenylalanine hydroxylase deficiency (also known as classic phenylketonuria), develop profound and irreversible intellectual disability 4. Affected individuals on an unrestricted diet who have phenylalanine levels above normal but below 1,200 μmol/L (20 mg/dL) are at much lower risk for impaired cognitive development in the absence of treatment.
Hyperphenylalaninemia occurs in all races. Hyperphenylalaninemia affects approximately 15-75 cases per 1,000,000 births. Hyperphenylalaninemia is less prevalent than classic PKU and shows less variation in incidence among populations. Both sexes are equally affected because deficiency in phenylalanine hydroxylase activity is inherited as an autosomal-recessive trait.
Phenylalanine levels of 6 mg/dL (360 µmol/L) or less in patients consuming an unrestricted diet are generally considered to be a benign condition. No dietary phenylalanine restrictions are usually recommended in this instance. In contrast, dietary restriction is generally indicated among patients whose phenylalanine levels are more than 12 mg/dL (725 µmol/L); chronic phenylalanine levels in this range reportedly cause measurable intellectual impairment in children.
Pregnant women with phenylalanine levels that exceed 6 mg/dL risk having children with microcephaly, mental retardation, and birth defects (eg, maternal hyperphenylalaninemia) 5.
Treatment consists of dietary restriction of phenylalanine often with tyrosine supplementation. Other essential amino acids are supplemented using various medical foods, and vitamin, mineral, and other micronutrients are followed closely. Stringent phenylalanine-restricted diets have been reported to cause deficiencies of iron, zinc, selenium, and other nutrients and essential amino acids in patients with PKU. Therefore, the diet requires careful monitoring by a professional trained in PKU management and frequently requires supplementation of required nutrients.
Practices of dietary treatment vary in children with phenylalanine levels in the intermediate range of 7-11 mg/dL (425-660 µmol/L). Most centers in the United States recommend restricting dietary phenylalanine when levels exceed 10 mg/dL (600 µmol/L). Some also recommend treatment for levels that exceed 8-9 mg/dL (480-545 µmol/L). The British Medical Research Council Working Party on PKU recommends dietary phenylalanine restriction when levels consistently exceed 6.6-10 mg/dL (400-600 µmol/L).
Phenylalanine levels are followed at regular intervals, from 1-2 times weekly in neonates to perhaps once per month in older children and adults. Most US facilities recommend that phenylalanine levels be maintained in the range of 2-6 mg/dL (120-360 µmol/L). This requires expert care and close monitoring.
Benign hyperphenylalaninemia
Benign hyperphenylalaninemia is a mild form of phenylketonuria (PKU). Benign hyperphenylalaninemia is considered an amino acid condition because people with benign hyperphenylalaninemia have problems breaking down an amino acid, a building block of proteins, known as phenylalanine. Most people with this condition experience mild symptoms or no symptoms.
Babies with H-PHE make less PAH than babies without H-PHE. They can break down phenylalanine, but not as quickly as babies without H-PHE. If the body cannot break down phenylalanine quickly enough, phenylalanine can build up in the blood. Everyone has some phenylalanine in their blood, but high levels could be harmful. Babies with H-PHE have elevated levels of phenylalanine, but these levels are usually not dangerous.
Babies with benign hyperphenylalaninemia typically have no complications. They can have healthy growth and development.
However, some babies with benign hyperphenylalaninemia do have a small risk of brain damage without treatment. This is why newborn screening for benign hyperphenylalaninemia is important.
Hyperphenylalaninemia causes
The American College of Medical Genetics and Genomics recommended that hyperphenylalaninemia and phenylketonuria (PKU) both be considered part of the spectrum of phenylalanine hydroxylase (PAH) deficiency. The PAH gene provides instructions for making an enzyme called phenylalanine hydroxylase. Mutations in the PAH gene cause phenylketonuria. Phenylalanine hydroxylase (PAH) enzyme converts the amino acid phenylalanine to other important compounds in the body. If gene mutations reduce the activity of phenylalanine hydroxylase, phenylalanine from the diet is not processed effectively. As a result, this amino acid can build up to toxic levels in the blood and other tissues. Because nerve cells in the brain are particularly sensitive to phenylalanine levels, excessive amounts of this substance can cause brain damage.
More than 300 different changes (mutations) in the PKU gene have been identified 6. Because the different mutations result in varying degrees of PAH enzyme activity, and therefore varying degrees of phenylalanine elevation in blood, the diet of each child must be adjusted to the individual’s specific phenylalanine tolerance.
Classic PKU, the most severe form of the disorder, occurs when phenylalanine hydroxylase activity is severely reduced or absent. People with untreated classic PKU have levels of phenylalanine high enough to cause severe brain damage and other serious health problems. Mutations in the PAH gene that allow the enzyme to retain some activity result in milder versions of this condition, such as variant PKU or non-PKU hyperphenylalaninemia.
PKU is inherited in an autosomal recessive pattern. Recessive genetic disorders occur when an individual inherits an abnormal gene from each parent. If an individual receives one normal gene copy and one abnormal gene copy, they will be a carrier for the condition, but will not have symptoms. The risk for two carrier parents to both pass the abnormal gene and, therefore, have an affected child is 25% with each pregnancy. The risk is the same for males and females.
Changes in other genes may influence the severity of PKU, but little is known about these additional genetic factors.
In a few cases, defective synthesis or recycling of the biopterin cofactor is the cause. Tetrahydrobiopterin (BH4) deficiencies is a general term for a group of disorders characterized by abnormalities in the creation (biosynthesis) or regeneration of tetrahydrobiopterin, a naturally-occurring compound that acts as a cofactor 7. A cofactor is a non-protein substance in the body that enhances or is necessary for the proper function of certain enzymes. When tetrahydrobiopterin is deficient, the chemical balance within the body is upset. In most of these disorders, there are abnormally high levels of the amino acid phenylalanine (hyperphenylalaninemia).
Tetrahydrobiopterin deficiencies usually present within the first six months of life and can be detected upon newborn screening because of elevated levels phenylalanine. Infants usually appear normal at birth, although some newborns, particularly in 6-pyruvoyl tetrahydropterin synthase deficiency, may have a low birth weight. Failure to grow and gain weight (failure to thrive) may occur. Microcephaly, a condition defined as having a head circumference smaller than normally would be expected based on age and gender, is a common finding.
In the severe forms, common, but variable symptoms include neurological dysfunction including convulsions or seizures, swallowing difficulties, poor muscle tone of the trunk of the body (truncal hypotonia), and excess muscle tone of the arms and legs so that they may be stiff and difficult to move (limb hypertonia). Abnormal movements are common and can include abnormal slowness of movement (bradykinesia), rapid, involuntary, purposeless (chorea), slow, involuntary, writhing movements (athetosis), a type of spasm in which the head and feet bend the backward and the back arches (opisthotonus).
Affected children may also exhibit delays in reaching developmental milestones (developmental delays), delays in acquiring skills that require the coordination of mental and physical activities (psychomotor retardation), and, in some cases, intellectual disability.
Neurological dysfunction is progressive and, during the school years, affected individuals may appear uncoordinated or clumsy such as having an abnormal manner of walking (gait abnormalities). In some cases, this clumsiness is due, in part, to involuntary muscle contractions that force the body into abnormal, sometimes painful, movements and positions (dystonia).
Some affected individuals may develop abnormal movements of the eyes that can range from brief upward rolling of the eyes to oculogyric crises, in which the eyes roll upward for a sustained period of time. Sometimes, the eyes may roll downward or move toward each other (converge). Severe oculogyric crises can be associated with additional symptoms including the formation of tears (lacrimation), eye blinking, widening (dilation) of the pupils, drooling, backward flexion of the neck, restlessness, or a general feeling of poor health (malaise).
Additional symptoms that have been reported include excessive production of saliva, lethargy, and irritability. Recurrent episodes of elevated body temperature (hyperthermia) that is not associated with infection may also occur. Certain symptoms may become noticeably worse or more pronounced in the afternoon and evening than in the morning (marked diurnal fluctuation). Swallowing difficulties and poor sucking ability in infants can result poor feeding during infancy.
Hyperphenylalaninemia inheritance pattern
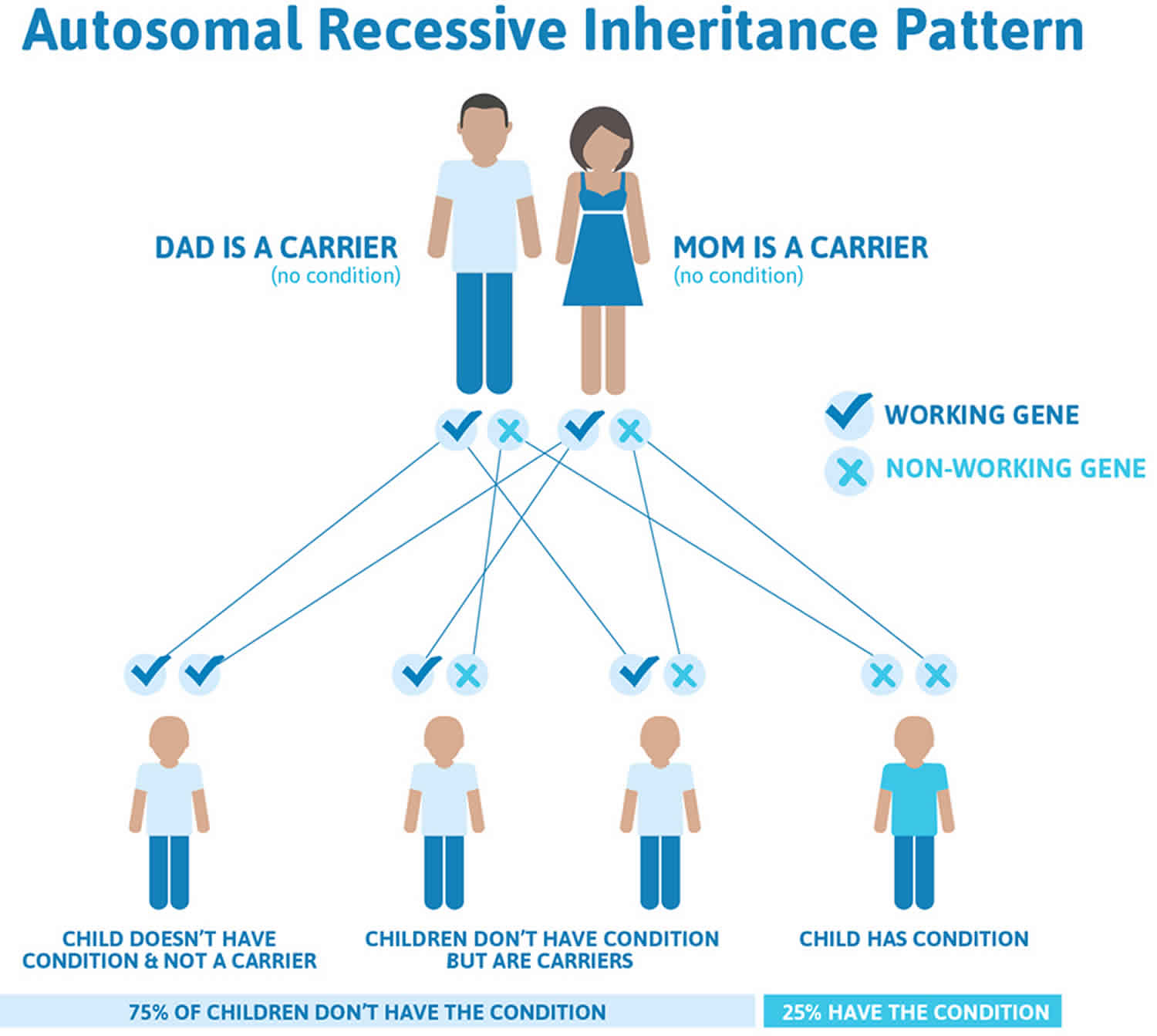
Hyperphenylalaninemia is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. This means that a child must inherit two copies of the non-working gene for hyperphenylalaninemia, one from each parent, in order to have the condition. The parents of a child with an autosomal recessive condition each carry one copy of the non-working gene, but they typically do not show signs and symptoms of the condition. While having a child with hyperphenylalaninemia is rare, when both parents are carriers, they can have more than one child with the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 1 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 1. Hyperphenylalaninemia autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Hyperphenylalaninemia symptoms
An abnormal newborn screen is the most common history in patients with hyperphenylalaninemia. Infants are screened for elevated phenylalanine in every US state and in Puerto Rico. Several other countries also have established screening programs.
Affected individuals missed by screening may have mild-to-moderate neuropsychological effects, depending on the degree of phenylalanine elevation.
Measurable IQ deficits can be seen at phenylalanine levels in the hyperphenylalaninemia range, particularly as levels exceed 10 mg/dL (600 µmol/L). At phenylalanine levels near 20 mg/dL (1200 µmol/L), phenylketonuria (PKU)-like symptoms may emerge, including more pronounced developmental abnormalities, eczema, and vomiting. Preliminary evidence indicates milder attention and organizational problems may arise when levels exceed 6 mg/dL.
Most children have few abnormal findings on physical examination.
Some physical stigmata of PKU may be present in individuals who have phenylalanine levels near 20 mg/dL. PKU-like symptoms include eczema and fair hair and skin coloring.
Infants with PKU typically appear normal at birth. With early screening and dietary treatment, affected individuals may never show symptoms of PKU. However, untreated newborns not diagnosed in the first days of life may be weak and feed poorly. Other symptoms may include vomiting, irritability, and/or a red skin rash with small pimples. Developmental delay may be obvious at several months of age. The average IQ of untreated children is usually less than 50. Intellectual disability in PKU is a direct result of elevated levels of phenylalanine in the brain which causes the destruction of the fatty covering (myelin) of individual nerve fibers. It can also cause depression by reducing brain levels of dopamine and serotonin (neurotransmitters).
Untreated infants with PKU tend to have unusually light eye, skin, and hair color due to high phenylalanine levels interfering with production of melanin, a substance that causes pigmentation. They may also have a musty or “mousy” body odor caused by phenyl acetic acid in the urine or sweat.
Neurological symptoms are present in some untreated patients with PKU, including seizures, abnormal muscle movements, tight muscles, increased reflexes, involuntary movements, or tremor.
Untreated females with PKU who become pregnant are at high risk for having a miscarriage or problems with fetal growth (intrauterine growth retardation). Children of women with untreated PKU may have an abnormally small head (microcephaly), congenital heart disease, developmental abnormalities, or facial abnormalities. There is a strong relationship between the severity of these symptoms and high levels of phenylalanine in the mother. As a result, all women with PKU who have stopped treatment should resume treatment before conception and continue on it throughout pregnancy, managed by a metabolic geneticist and dietician.
Hyperphenylalaninemia diagnosis
Phenylalanine hydroxylase (PAH) deficiency can be detected by newborn screening in virtually 100% of cases based on the presence of hyperphenylalaninemia using tandem mass spectrometry on a blood spot obtained from a heel prick and must be distinguished from classic PKU by confirmatory testing at an experienced center 8. The diagnosis of phenylalanine hydroxylase (PAH) deficiency is established in a proband with:
- A plasma phenylalanine concentration persistently above 120 µmol/L (2 mg/dL) and altered ratio of phenylalanine to tyrosine in the untreated state with normal BH4 cofactor metabolism;
and/or - The finding of biallelic pathogenic variants in phenylalanine hydroxylase (PAH) by molecular genetic testing.
Some cases in adult women have been detected using maternal screening programs or following birth of children with birth defects.
Low-grade phenylalanine elevations may require repeat screening. Phenylalanine levels can rise for several weeks after birth in children with hyperphenylalaninemia or phenylketonuria (PKU). A low-grade elevation 24-72 hours following birth might signal true PKU, not merely hyperphenylalaninemia.
Measure plasma phenylalanine and tyrosine levels as soon as possible after an abnormal screening result. An elevated phenylalanine level with low or normal tyrosine level is expected. Hepatic insufficiency and tyrosinemia can feature elevated phenylalanine levels, although typically in the context of elevated tyrosine.
Obtain blood and urine biopterins assays through a qualified laboratory to exclude a tetrahydrobiopterin defect.
Hyperphenylalaninemia treatment
In some children with mild enzyme deficits, excessive protein intake may elevate phenylalanine levels to a range requiring treatment. The problem may resolve when protein intake is reduced to more ordinary levels. For example, infants with nonphenylketonuric hyperphenylalaninemia who consume excessive infant formula (60-70 oz/d or 1800-2100 mL/d) may demonstrate phenylalanine levels exceeding 10-12 mg/dL. Levels may fall when formula intake is restricted to 32-40 oz/day.
In one study 9, 54% of patients with phenylalanine levels less than 600 µmol/L (10 mg/dL) demonstrated a decline of 30% or more in plasma phenylalanine levels when sapropterin (commercial tetrahydrobiopterin cofactor) was administered at a dose of 10 mg/kg/d. The percentage of patients who responded declined with increasing plasma phenylalanine levels. Response to sapropterin may improve at a dose of 20 mg/kg/d 10.
Preliminary studies are underway for injectable phenylalanine ammonium lyase, an enzyme substitute. This shows promise in animal studies as an alternative treatment to control phenylalanine levels 11.
If dietary treatment is necessary, refer the patient to a dietitian experienced with PKU.
Refer families of affected infants to a medical geneticist or genetic counselor to review the inheritance of hyperphenylalaninemia.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Diet
Determine the degree of dietary phenylalanine restriction for each patient based on untreated phenylalanine levels. The mainstay of the diet consists of phenylalanine restriction and supplementation of other essential amino acids, vitamins, minerals, and energy intake, using medical foods and low-protein foods 12.
Aspartame must also be avoided. Phenylalanine is one of the primary components of aspartame. Aspartame is found in many artificially sweetened foods and soft drinks, as well as some vitamins and medicines. A 12-oz can of aspartame-sweetened diet drink contains approximately 105 mg of phenylalanine (ie, 25-50% of the usual daily intake). A pharmacist can help determine if a medication has a significant amount of aspartame. The amount of aspartame in a children’s vitamin or in a teaspoon of antibiotic may be significant for a child who can tolerate only 200 mg/d of phenylalanine, yet such a dose may be insignificant for a child who can tolerate more than 1000 mg/d.
Breastfeeding is usually possible and should not be stopped unless instructed by a local health official or treatment center.
Stringent phenylalanine-restricted diets have been reported to cause deficiencies of iron, zinc, selenium, and other nutrients and essential amino acids in patients with PKU. The diet requires careful monitoring by a professional trained in PKU management.
Most newborns with PKU require 40-60 mg/kg/d of dietary phenylalanine to maintain normal growth and development. As growth slows, the phenylalanine requirement falls, and most older children and adults tolerate 200-400 mg/d.
Providing some natural phenylalanine is essential in order to prevent deficiency of this essential amino acid. The diet requires virtual elimination of all high-protein foods, such as meat, dairy, nuts, and legumes. Starches, including bread, potatoes, corn, and beans, also must be restricted (a slice of bread or small order of fries contains approximately 120-150 mg of phenylalanine).
Essential amino acids, vitamins, and minerals must be supplemented by using medical foods. Currently, most are consumed as a powder dissolved in liquid (ie, formula). Newer supplements, including capsules, amino acid bars, and amino acids cooked into foods, are becoming available.
Energy and variety are provided by low-protein foods, including fruits and nonstarchy vegetables, as well as specially ordered low-protein foods. Low-protein foods include pastas, breads, imitation cheese, baking mixes, and other foods especially designated for low-protein diets. These foods are covered by medical benefits in some states.
As patients with PKU transition into adolescence, their caregivers have a less direct influence on their diet. It is common to see these patients “cheat” by failing to limit foods such as potatoes, pasta, and bread.
Pharmacologic therapy
Some patients with PKU experience significant lowering of plasma phenylalanine levels after administration of sapropterin, a commercially available, FDA-approved form of the tetrahydrobiopterin (BH4) cofactor 9. One study 9 found that, although 54% of those with plasma phenylalanine levels lower than 600 µmol/L (10 mg/dL) had a reduction in plasma phenylalanine levels of 30% or more after 10 mg/kg/day of sapropterin, only 10% of those with phenylalanine levels higher than 1200 µmol/L had such a response.
In May 2018, the FDA approved the first enzyme substitute, pegvaliase (Palynziq), to reduce phenylalanine levels in adults with PKU who have uncontrolled phenylalanine levels of more than 600 µmol/L. It is administered as a pegylated subcutaneous injection of phenylalanine ammonium lyase, an enzyme capable of substituting for phenylalanine hydroxylase (PAH).
Approval of pegvaliase was based on two phase 3 studies, PRISM-1 and PRISM-2, which evaluated the efficacy and safety of pegvaliase treatment using an induction, titration, and maintenance dosing regimen in adults with PKU. Of 261 participants who received pegvaliase, 72% and 32.6% reached ≥12 months and ≥24 months of study treatment, respectively, and 65% are still actively receiving treatment. Mean (SD) blood phenylalanine levels were 1232.7 (386.4) μmol/L at baseline, 564.5 (531.2) μmol/L at 12 months, and 311.4 (427) μmol/L at 24 months, a decrease from baseline of 51.1% and 68.7%, respectively 13.
Research into gene therapy for the treatment of PKU has been ongoing over the last 2 decades. The focus has been on replacement of the human mutant PAH gene in somatic cells of PKU patients 14.
Pregnancy management
To minimize or prevent teratogenic effects of phenylalanine, women with phenylalanine hydroxylase (PAH) deficiency should follow a phenylalanine-restricted diet for at least several months prior to conception in order to maintain plasma phenylalanine concentrations between 120 and 360 µmol/L (2-6 mg/dL); after conception, continuous nutritional guidance and weekly or biweekly measurement of plasma phenylalanine concentration to assure that target levels are met in addition to adequate energy intake with the proper proportion of protein, fat, and carbohydrates. Evaluation for fetal anomalies using high-resolution ultrasound and fetal echocardiogram.
Hyperphenylalaninemia prognosis
Prognosis is excellent for normal development when treated as indicated. Most individuals with hyperphenylalaninemia have normal life expectancy. Several studies have identified a linear relationship between the phenylalanine level and intelligence testing and performance. Intelligence quotients seem less affected by benign hyperphenylalaninemia than by PKU, even at the same levels of serum phenylalanine. This effect may be due to smaller fluctuations of serum phenylalanine concentration.
References- Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. Lancet. 2010;376(9750):1417‐1427.
- Procházková D, Jarkovský J, Haňková Z, Konečná P, Benáková H, Vinohradská H, et al. Long-term treatment for hyperphenylalaninemia and phenylketonuria: a risk for nutritional vitamin B12 deficiency?. J Pediatr Endocrinol Metab. 2015 Nov 1. 28 (11-12):1327-32.
- Hyperphenylalaninemia. https://emedicine.medscape.com/article/945180-overview
- Regier DS, Greene CL. Phenylalanine Hydroxylase Deficiency. 2000 Jan 10 [Updated 2017 Jan 5]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1504
- Prick BW, Hop WC, Duvekot JJ. Maternal phenylketonuria and hyperphenylalaninemia in pregnancy: pregnancy complications and neonatal sequelae in untreated and treated pregnancies. Am J Clin Nutr. 2012 Feb. 95(2):374-82.
- Phenylketonuria. https://rarediseases.org/rare-diseases/phenylketonuria
- Tetrahydrobiopterin Deficiency. https://rarediseases.org/rare-diseases/tetrahydrobiopterin-deficiency
- Mak CM, Ko CH, Lam CW, et al. Phenylketonuria in Hong Kong Chinese: a call for hyperphenylalaninemia newborn screening in the Special Administrative Region, China. Chin Med J (Engl). 2011 Aug. 124(16):2556-8.
- Burton BK, Grange DK, Milanowski A, et al. The response of patients with phenylketonuria and elevated serum phenylalanine to treatment with oral sapropterin dihydrochloride (6R-tetrahydrobiopterin): a phase II, multicentre, open-label, screening study. J Inherit Metab Dis. 2007 Oct. 30(5):700-7.
- Matalon R, Michals-Matalon K, Koch R, Grady J, Tyring S, Stevens RC. Response of patients with phenylketonuria in the US to tetrahydrobiopterin. Mol Genet Metab. 2005 Dec. 86 Suppl 1:S17-21.
- Sarkissian CN, Gamez A, Wang L, et al. Preclinical evaluation of multiple species of PEGylated recombinant phenylalanine ammonia lyase for the treatment of phenylketonuria. Proc Natl Acad Sci U S A. 2008 Dec 30. 105(52):20894-9.
- Yannicelli S, Ryan A. Improvements in behaviour and physical manifestations in previously untreated adults with phenylketonuria using a phenylalanine-restricted diet: a national survey. J Inherit Metab Dis. 1995. 18(2):131-4.
- Thomas J, Levy H, Amato S, Vockley J, Zori R, Dimmock D, et al. Pegvaliase for the treatment of phenylketonuria: Results of a long-term phase 3 clinical trial program (PRISM). Mol Genet Metab. 2018 May. 124 (1):27-38.
- Pietz J, Kreis R, Rupp A, et al. Large neutral amino acids block phenylalanine transport into brain tissue in patients with phenylketonuria. J Clin Invest. 1999 Apr. 103(8):1169-78.





{kind=link}