Hypochloremic alkalosis
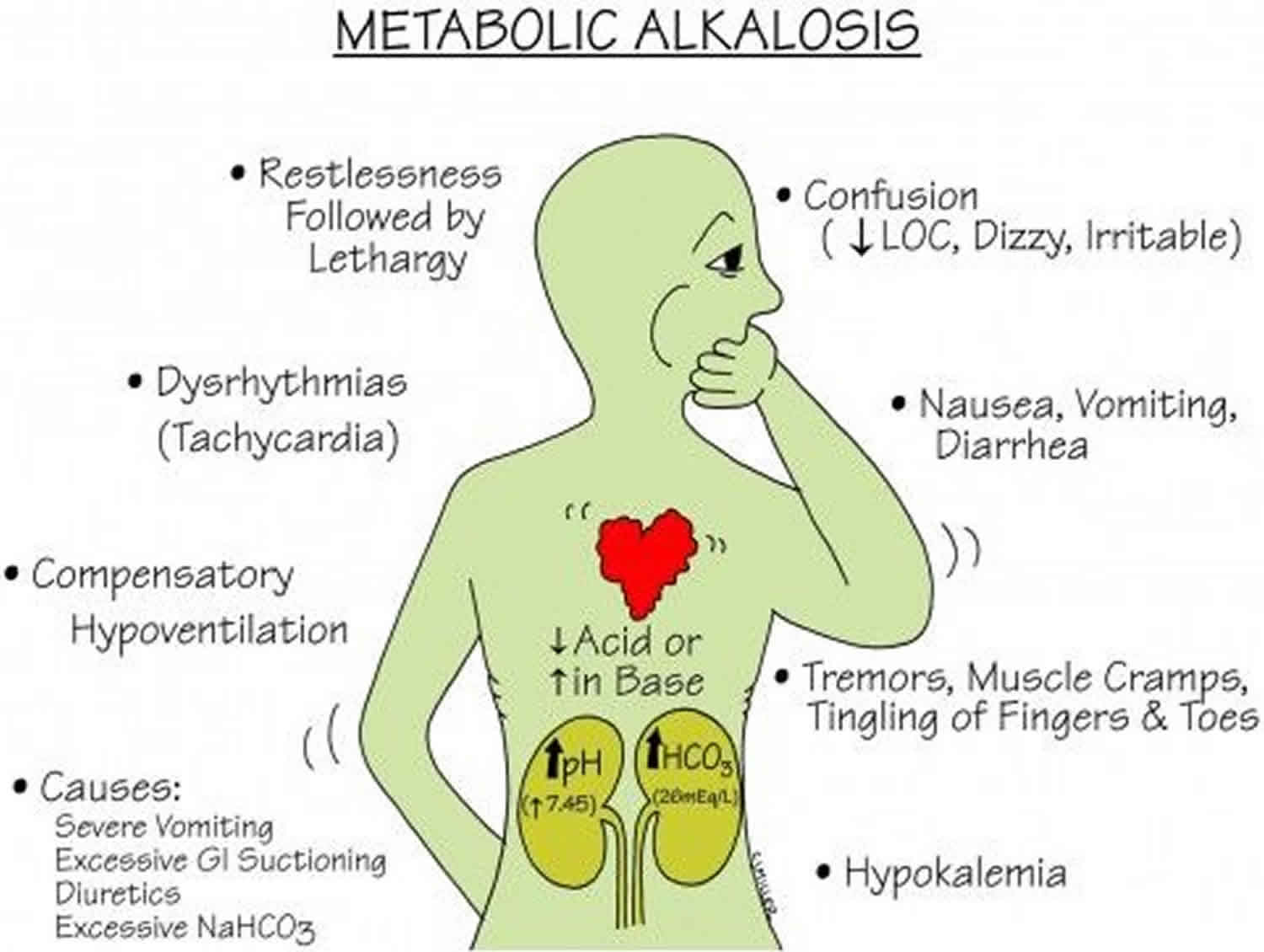
Hypochloremia is defined as a serum chloride level of less than 95 mEq/L. Hypochloremic alkalosis results from either low chloride intake or excessive chloride wasting. Whereas low chloride intake is very uncommon, excessive chloride wasting often occurs in hospitalized children, usually as a result of diuretic therapy or nasogastric tube suctioning 1. Hypochloremic alkalosis is also seen in hypertrophic pyloric stenosis where it occurs in only about half the patients 2. Diarrhea, when watery is also highly suggestive of chloride-losing diarrhea.
Hypochloremic alkalosis can be classified into two categories 3, 4, 5, 6, 7, 8, 9:
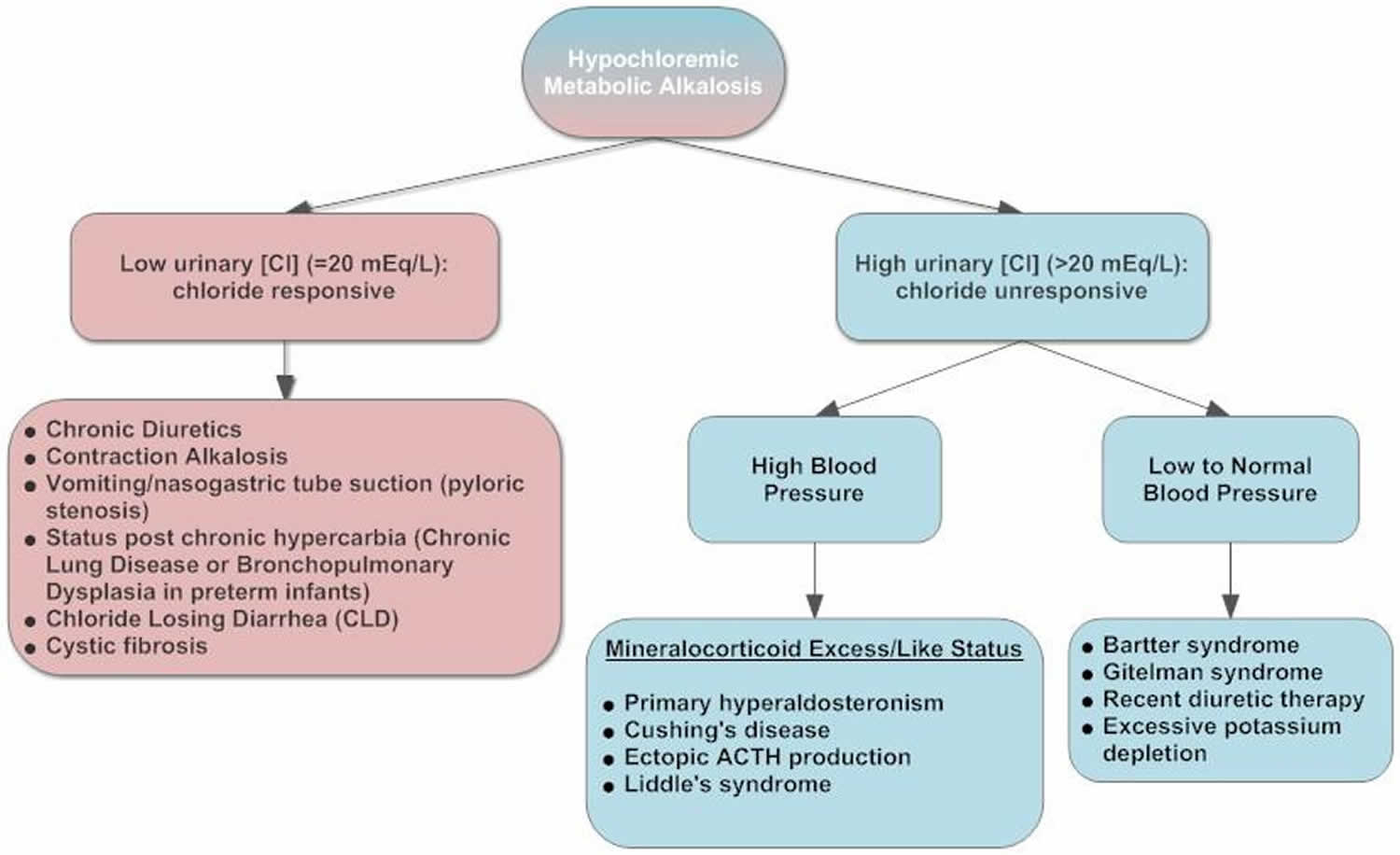
- Chloride-responsive alkalosis with urine chloride less than 20 mEq/L. Chloride-responsive alkalosis causes include loss of hydrogen via the gastrointestinal tract (vomiting, nasogastric suction), congenital chloride diarrhea syndrome (congenital chloridorrhea), loss of colonic secretions via villous adenoma, contraction alkalosis, diuretic therapy (thiazides and loop diuretics [after discontinuation]), post-hypercapnia syndrome, cystic fibrosis, exogenous alkalotic agent use and laxative abuse are also potential causes.
- Chloride-resistant alkalosis with urine chloride greater than 20 mEq/L.
- Causes of chloride-resistant alkalosis (urine chloride > 20 mEq/L) with hypertension include the following:
- Primary hyperaldosteronism: adrenal adenoma, bilateral adrenal hyperplasia, adrenal carcinoma, glucocorticoid-remediable hyperaldosteronism
- 11 beta-hydroxysteroid dehydrogenase type 2 (11 beta-HSD2): 11 β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) normally converts active cortisol to inactive cortisone and protects mineralocorticoid receptor (MR) occupied by cortisol. The Apparent Mineralocorticoid Excess syndrome is an ultrarare autosomal recessive disorder caused by 11β-hydroxysteroid dehydrogenase type 2 gene (HSD11B2) resulting in impairment of the enzyme 11β-HSD2 leading to excessive cortisol that is able to activate mineralocorticoid receptor (MR). The Apparent Mineralocorticoid Excess syndrome is typically characterized by hypertension with hypokalemia, metabolic alkalosis, low renin activity, and low aldosterone level. The cause is inherited in autosomal recessive form, mutations in 11β-HSD2 gene or acquired form by ingestion of competitive inhibitors of 11β-HSD2 such as liquorice, chewing tobacco or carbenoxolone. Uncontrolled hypertension and prolong hypokalemia are the leading causes of end organ damages and cardiovascular mortality 10.
- Congenital adrenal hyperplasia (CAH) 11-hydroxylase or 17-hydroxylase deficiency
- Current use of diuretics in hypertension
- Cushing syndrome
- Exogenous mineralocorticoids or glucocorticoids
- Liddle syndrome
- Renovascular hypertension
- Causes of chloride-resistant alkalosis (urine chloride >20 mEq/L) without hypertension include the following:
- Bartter syndrome
- Gitelman syndrome
- Severe potassium depletion (hypokalemia)
- Current use of thiazides and loop diuretics
- Hypomagnesemia
- Causes of chloride-resistant alkalosis (urine chloride > 20 mEq/L) with hypertension include the following:
Replacement of electrolytes with chloride salts is the most important mode of therapy for hypochloremic alkalosis. Nonsteroidal anti-inflammatory drugs (NSAIDs) are used in patients with Bartter syndrome. Hydrochloric acid (HCl) and carbonic anhydrase inhibitors may be used in some acute situations.
In patients with chloride-losing diarrhea, fluid intake should be encouraged so as to prevent renal damage resulting from recurrent dehydration. Patients or caregivers should be instructed to avoid long periods of exposure to hot climates, which may exacerbate dehydration episodes.
Constipation must be treated in patients with Bartter syndrome. Any intercurrent febrile illnesses, especially urinary tract infections, must be treated to prevent further renal damage.
Hypochloremic alkalosis causes
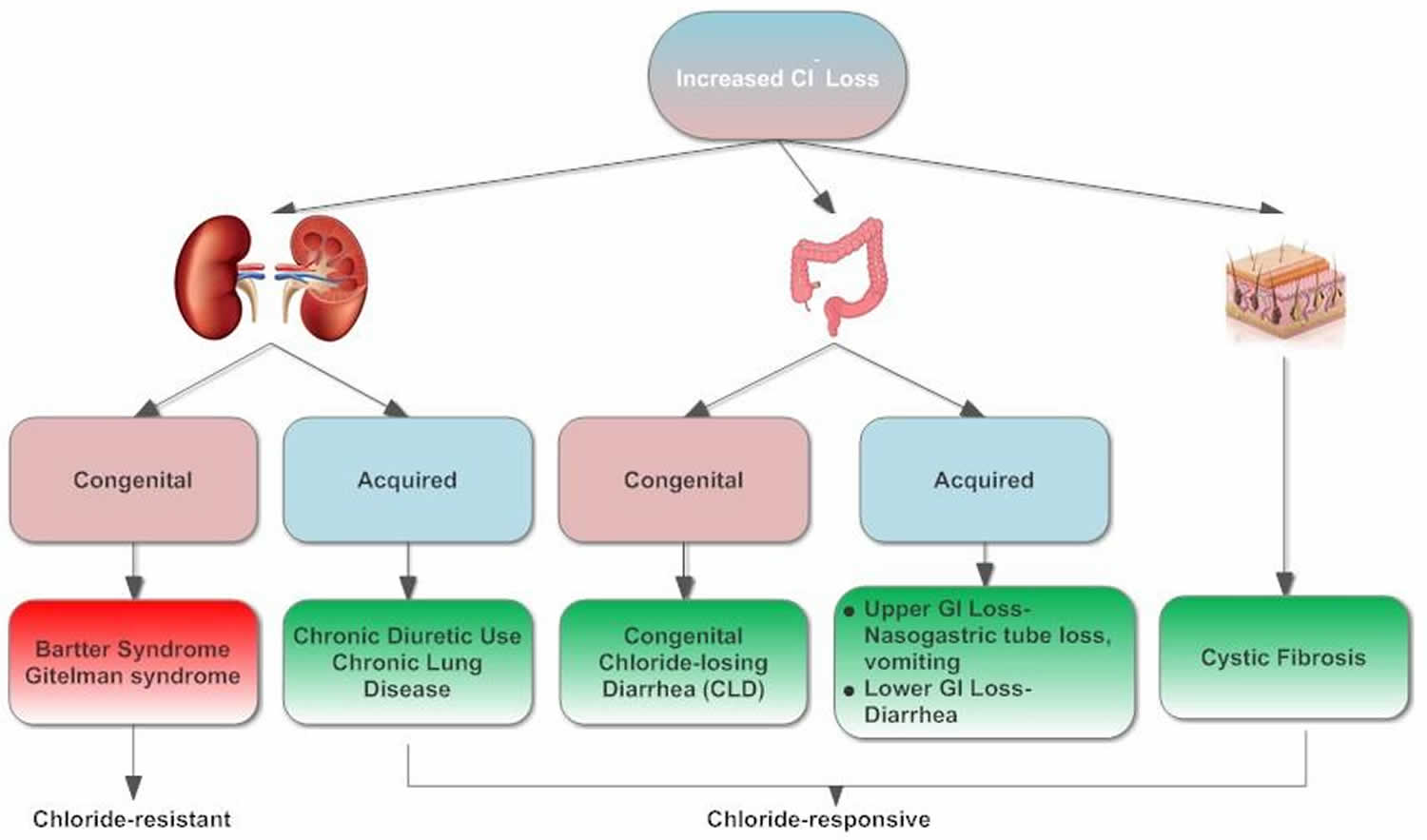
Hypochloremia results from either low chloride intake or excessive chloride wasting. Low chloride intake is very uncommon. Excessive chloride wasting often occurs in hospitalized children, usually as a consequence of diuretic therapy or nasogastric tube suctioning. Chloride-wasting syndromes, including Bartter syndrome, congenital chloride-losing diarrhea and cystic fibrosis, result from renal tubular loss, defective electrolyte transport across intestinal epithelia, and chloride loss via the skin, respectively 11. There have been reports of dietary deficiencies or formula lacking chloride causing metabolic alkalosis and severe neurological consequences 12. However, these causes are now rare since the introduction of standard nutritional guidelines for formulas and dietary requirements.
Chloride-losing diarrhea is caused by a defective anion exchange protein, an epithelial chloride/bicarbonate (Cl–/HCO3–) exchanger located in the brush border of the ileum and colon, resulting in defective intestinal chloride absorption and secretion of HCO3–, with a secondary defect in sodium/hydrogen (Na+/H+) transport, altogether leading to intestinal losses of both sodium and water, hypochloremia, hyponatremia, and metabolic alkalosis.
Many cases of Chloride-losing diarrhea have emerged from Eastern Europe and Middle Eastern Arab countries; indeed, the largest purported series is from Saudi Arabia 13. Fewer cases in the English language literature have been reported in the Far East and North America, perhaps because of cultural and academic barriers.
Chloride-losing diarrhea can manifest before birth as severe midtrimester polyhydramnios. Metabolic derangements may manifest as early as the first few days of life. Bartter syndrome may present at any age but primarily occurs in infants younger than 1 year. Hypochloremic alkalosis resulting from cystic fibrosis is infrequent in infancy but can become more severe in summer because of excessive chloride loss from sweating. Drug-related hypochloremic alkalosis is observed at all ages. Males and females are affected in equal numbers.
Figure 1. Causes of hypochloremic alkalosis in children
Hypochloremic metabolic alkalosis pathophysiology
The renal collecting duct plays an important role in acid-base balance by maintaining bicarbonate (HCO3–) reabsorption and secretion 14. Abundant evidence now supports pendrin as an important regulatory transporter in the cortical collecting ducts, which responds briskly in vivo 15 and in vitro 16 to alterations in chloride intake and to acid-base perturbations, including metabolic alkalosis and acidosis and respiratory acidosis. The figure below depicts a detailed explanation of ion exchange at collecting duct cells in a hypochloremic condition.
The collecting duct plays the main role in acid-base balance by maintaining HCO3– reabsorption and secretion. In type-B intercalated cells of the cortical collecting duct, pendrin channel (Pn) activity is increased because of a low Cl– concentration in that segment (due to low chloride levels in ultrafiltrate from hypochloremia), but secretion of HCO3– is inhibited by insufficient Cl– for anion exchange. HCO3– reabsorption is continued by Na+/H+ exchange in the principal cell and in the type-A intercalated cells. In the medullary collecting duct, HCO3– reabsorption is continued because a decreased HCO3– concentration in that segment enhances H+ secretion in type-A cells 17. Rat studies have shown that, after Cl– delivery to the cortical collecting ducts increases, HCO3– secretion occurs and medullary HCO3– reabsorption diminishes, allowing correction of the hypochloremic alkalosis. In these studies, bicarbonaturia occurred within minutes of Cl– administration intravenously, and Cl– did not increase in the urine until correction of serum Cl– was nearly complete 16.
Figure 2. Hypochloremic metabolic alkalosis pathophysiology
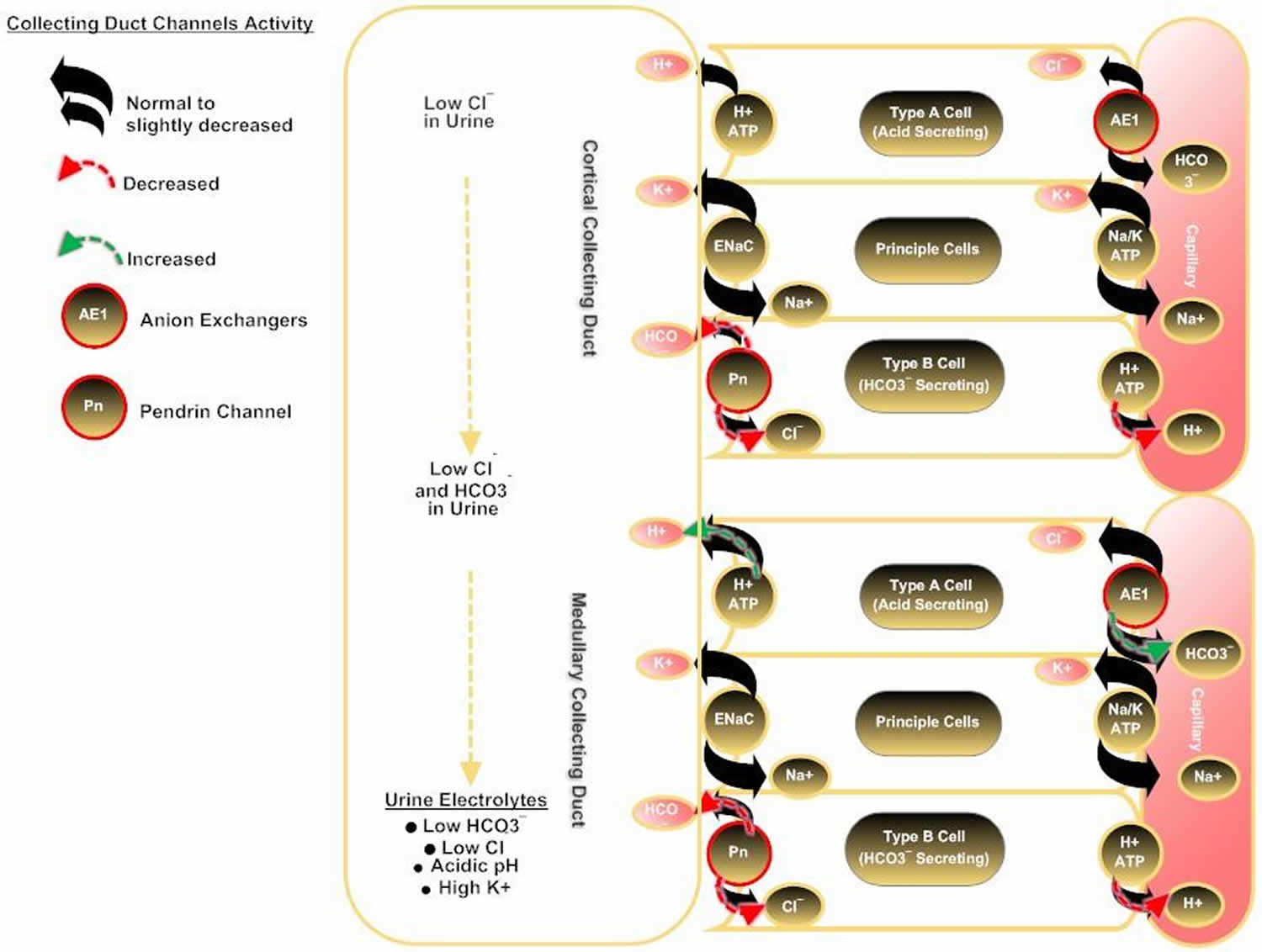
Footnote: Explanation of ion exchange at collecting duct cells in a hypochloremic condition.
Hypochloremic alkalosis differential diagnoses
- Bartter Syndrome
- Congenital chloride-losing diarrhea
- Glucocorticoid Therapy and Cushing Syndrome
- Hyperaldosteronism
- Hypercalciuria
- Hypomagnesemia
- Milk-Alkali Syndrome
- Pediatric Hypercalcemia
- Pediatric Hypokalemia
- Pediatric Hyponatremia
- Posthypercapnic Alkalosis
- Sinonasal Manifestations of Cystic Fibrosis
- Uric Acid Stones
In addition to the conditions listed in the differential diagnosis, other problems to be considered include the following:
- Enteric anendocrinosis or dysendocrinosis
- Gitelman syndrome
- Laxative abuse
- Loop or thiazide diuretic abuse
- Pyloric stenosis
Hypochloremic alkalosis signs and symptoms
Prenatal polyhydramnios is present in most patients with congenital forms of metabolic alkalosis, especially chloride-losing diarrhea. Premature birth resulting from polyhydramnios is common in patients with Bartter syndrome and chloride-losing diarrhea. Lack of meconium is highly suggestive of intrauterine diarrhea. Prolonged neonatal jaundice may be present. A history of hypotonia and lethargy without sepsis is significant in patients with early-onset hypochloremia and hypokalemia.
In infants, a history of repeated vomiting may be suggestive of severe gastroesophageal reflux or pyloric stenosis. Failure to thrive is common. Constipation is very common in patients with Bartter syndrome. Diarrhea, when watery (see the image below), is highly suggestive of Chloride-losing diarrhea.
A salty taste upon being kissed may help identify patients with cystic fibrosis. Guidelines for newborn screening for cystic fibrosis have been established by the Centers of Disease Control and Prevention (CDC) 18. Central nervous system (CNS) dysfunctions (eg, lethargy, confusion, or seizure) are observed in patients with severe alkalosis. Neuromuscular symptoms include weakness and muscle cramps.
Central nervous system (CNS) manifestations range from mild to severe, depending on the severity of alkalosis, and may include the following:
- Confusion
- Apathy
- Disorientation
- Excessive sleeping
- Seizure
- Stupor
Other symptoms (eg, abdominal distention, dry skin, apathy, loss of interests, growth retardation 19 and frequent hospital admissions because of recurrent dehydration) are significant diagnostic clues during childhood.
Other history
A family history may be suggestive. Consanguinity, recurrent prematurity, neonatal demise, and psychomotor retardation are helpful clues to familial conditions.
A psychosocial history may reveal loss of interests and behavioral problems, which were reported in patients with chronic hypochloremic alkalosis. Difficulty in school performance may be a consequence of the disorder.
In hospitalized patients with hypochloremic metabolic alkalosis, the physician should always ask about nasogastric tube suctioning and oral secretions. Overzealous use of loop or thiazide diuretics, especially in the intensive care unit (ICU), is another important factor.
Physical examination
Patients with hypochloremic alkalosis commonly are small for their age, lethargic, or apathetic. Signs of chronic dehydration (eg, skin tenting and poor peripheral perfusion) may be evident upon presentation. One study reported that cystic fibrosis was diagnosed in an infant who presented with dehydration and metabolic alkalosis 20.
Weight and height usually fall below the reference range in patients with chronic disease but are not affected in patients with acute disease. In one series, both weight and height were in the lowest 3% in more than 60% of patients with chloride-losing diarrhea 13.
Depending on the cause of the hypochloremic alkalosis, the abdomen may be scaphoid (in Bartter syndrome) or distended (in chloride-losing diarrhea).
Additional abdominal findings that may be present are as follows:
- Peristaltic waves in children with chloride-losing diarrhea
- Exacerbated bowel sounds in patients with chloride-losing diarrhea
- Hard stools in patients with Bartter syndrome
- Hepatomegaly (suggesting cystic fibrosis)
Musculoskeletal findings include muscle wasting, atrophy, and hypotonia. Respiratory findings include shallow breathing and hypopnea in severely affected children.
Hypochloremic metabolic alkalosis complications
Disease-related complications of hypochloremic alkalosis include the following:
- Nephrocalcinosis and nephrolithiasis in patients with Bartter syndrome and in those with chloride-losing diarrhea
- Coexisting electrolyte abnormalities such as hypokalemia, hyponatremia, and hypercalcemia may be present
- Liver damage and recurrent chest infection leading to hepatic and pulmonary failure, respectively, in patients with cystic fibrosis
- End-stage renal disease (ESRD) in patients with poor compliance; ESRD can occur in all conditions mentioned, including Bartter syndrome and chloride-losing diarrhea
Hypochloremic alkalosis diagnosis
The flowchart in Figure 3 depicts a workup approach to hypochloremic alkalosis. Your healthcare provider will perform a physical examination and ask about your symptoms.
These tests can help diagnose hypochloremic alkalosis. Tests may include:
- Arterial blood gas (ABG). An arterial blood gas is a laboratory test used for the measurement of arterial pH, arterial partial pressure of oxygen (PaO2), arterial partial pressure of carbon dioxide (PaCO2), bicarbonate (HCO3–), base excess, total carbon dioxide (CO2) and oxygen (O2) saturation.
- A venous blood gas test is a laboratory test identical to an arterial blood gas test, except the blood is drawn from a venous site. This results in a slightly more acidic “normal” pH range.
- Basic metabolic panel, (a group of blood tests that measure your sodium, potassium, and chloride levels, kidney function, and other chemicals and functions)
- Urine and stool studies
- Urine chloride is a direct measurement of chloride being excreted into urine. This test is useful to help determine the etiology of metabolic alkalosis 21, 22, 23
- A complete blood count (CBC) to evaluate for an infectious cause with elevated white blood count and fluid body status with hemoglobin and hematocrit values is useful.
Other tests that may be needed to determine the cause of the acidosis include:
- Amniocentesis
- Genetic studies
- Pulmonary function test to measure breathing and how well the lungs are functioning
- Chest x-ray
- CT abdomen
- Ultrasonography
Figure 3. Hypochloremic metabolic alkalosis diagnostic algorithm
Laboratory studies
Amniocentesis
Amniotic fluid sodium and chloride concentrations may reflect fetal values; these are high in fetuses with chloride-losing diarrhea. Levels may also be elevated in patients with Bartter syndrome. Although testing for alpha1 -fetoprotein is not routine, levels may be elevated.
Blood workup
Serum electrolyte levels may be within the reference range, especially in neonates and treated patients. However, typical findings include low concentrations of serum chloride, sodium, and potassium. Attention must be paid in interpreting the serum potassium level in relation to the state of metabolic alkalosis. For example, the potassium shift from serum into the intracellular compartment increases as the serum pH rises; thus, the potassium level is less than normal by 0.6 mmol/L when measured at a serum pH of 7.5.
Serum pH and bicarbonate, calcium, uric acid, hemoglobin (if patient is not anemic), renin, and aldosterone levels may be elevated. The serum renin level is exponentially high, in line with secondary hyperaldosteronism due to chronic volume depletion, and this finding is supported by low or normal blood pressure measurements.
Urine and stool studies
In patients with Bartter syndrome, urine chloride, sodium, and potassium concentrations are usually measured. Urine calcium-to-creatinine and uric acid–to–creatinine ratios are usually high. Stool electrolytes cannot be measured because of well-formed or hard stool. Fractional excretion (Fex) studies are more reliable than absolute values. Usually, results are higher than reference range values, as follows:
- Fractional excretion (Fex) sodium concentration >1%
- Fractional excretion (Fex) potassium concentration >35%
- Fractional excretion (Fex) chloride concentration >2.5% (2.7% ± 1.1%)
In patients with chloride-losing diarrhea, urine chloride concentration is very low or undetectable (< 10 mmol/L). Stool is usually watery, and electrolyte studies are very helpful and diagnostic, as follows:
- Stool chloride concentration >100 mmol/L
- Stool sodium and potassium concentrations are elevated
- Stool chloride concentration is greater than stool sodium plus potassium concentrations, which is normally less than either; chloride concentrations are lowest in colonic secretions (usually < 35 mmol/L)
- The ratio of stool chloride to combined sodium and potassium concentrations is greater than 0.6
Patients with cystic fibrosis typically demonstrate high sweat chloride and sodium concentrations. Urine chloride concentration is usually very low, and stools are usually not watery, as they are in patients with chloride-losing diarrhea.
Kidney and liver function tests
- Renal function is usually normal. The glomerular filtration rate (GFR) may be low in patients with severe disease.
- Liver function test results are usually within the reference range in patients with chloride-losing diarrhea and Bartter syndrome but may be deranged in patients with cystic fibrosis.
Genetic studies
DNA diagnosis is available for most congenital disorders that cause hypochloremic metabolic alkalosis. For chloride-losing diarrhea, the chloride-losing diarrhea (SLC26A3) locus is on band 7q22-q31.1 24. Bartter syndrome is identified by NKCC2, ROMK, and CLCNKB 25; Bartter syndrome with deafness is identified by BSND; and Bartter syndrome with autosomal dominant hypocalcemia is identified by CASR. For cystic fibrosis, the CFTR locus is on band 7q31.2. For Gitelman syndrome, the NCCT locus is on 16q.
Ultrasonography
Prenatal ultrasonography may be useful in the detection of minimal polyhydramnios and assessment of intestinal fluid content, which is increased in patients with chloride-losing diarrhea.
Postnatal ultrasonography (see the images below) may be useful in the evaluation of a fluid-filled bowel, which is characteristically increased in patients with chloride-losing diarrhea. Ultrasonography may also assist in the evaluation of renal echogenicity, nephrocalcinosis, medullary or diffuse calcinosis, and renal growth.
Physiologic study of renal tubules
Physiologic study of renal tubules by performing maximal free water clearance during hypotonic saline diuresis is indicated.
Oral administration of water 20 mL/kg over 30 minutes is followed by administration of a one-half isotonic sodium chloride solution at a rate of 600 mL/m²/h for 2-3 hours. During this time, urine is collected in aliquots over 30-minute periods for 4-6 aliquots.
These samples are sent for evaluation of creatinine, sodium, potassium, and chloride levels, as well as for osmolality, pH, and volume. Usually, urine is diluted by oral administration of water. Halfway through each collection, a blood sample is obtained for evaluation of creatinine, sodium, potassium, and chloride levels, and for pH and osmolality. The clearance of each substance is calculated, and a ratio is derived by means of the following formula:
- Water clearance/(chloride clearance + water clearance)
Usually, the result of this formula reflects the percentage of distal tubule sodium and chloride reabsorption. Normal values are up to 85-90%, which means that the percentage of chloride and sodium excreted should be 10-15% (corrected to a GFR of 100 mL/min/1.73 m²). In patients with Bartter syndrome, the percentage of chloride and sodium excreted can reach 35% or more.
Other studies
Additional studies that may be considered include the following:
- Wrist radiography – This may be performed to determine bone age in infants with growth failure; it may also help assess bone density and the presence of rickets
- Upper gastrointestinal (GI) series – This helps detect gastroesophageal reflux and pyloric stenosis, which are case-dependent conditions 26
- Computed tomography (CT) of the brain – This is useful for evaluation of brain growth and calcifications
- Magnetic resonance imaging (MRI) of the brain – This is helpful in patients who present with seizures
- Electroencephalography (EEG) – This is also helpful in patients who present with seizures
- Renal nuclear scanning – This may facilitate assessment of renal function but is not useful in all patients
- Renal biopsy – This is not usually indicated, but if it is performed, it may reveal interstitial fibrosis and calcium/urate crystal deposition
Hypochloremic metabolic alkalosis treatment
Hydration status and electrolyte levels must be assessed. Replacement of electrolytes with chloride salts is the most important mode of therapy for hypochloremic alkalosis. A full nutritional assessment should be obtained, energy intake calculated, and adequate energy intake ensured through oral or nasogastric methods.
Nonsteroidal anti-inflammatory drugs (NSAIDs; eg, indomethacin) are used in patients with Bartter syndrome. Hydrochloric acid (HCl) and carbonic anhydrase inhibitors (eg, acetazolamide) may be used in some acute situations. Potential complications of pharmacotherapy include the following:
- Indomethacin-induced nephrotoxicity
- Acetazolamide treatment compromising respiratory function in children with lung disease
Discharge medication instructions should be clearly written, and a supply sufficiently large to last until the patient is seen in the outpatient clinic should be prescribed.
Acute emergency management (6 hours or less)
Initial management includes assessment of dehydration status and severity of hypochloremia, hypokalemia, hyponatremia, and metabolic alkalosis. If the patient is in shock, treatment should be directed toward aggressive resuscitation with isotonic fluid, preferably normal saline. Blood and urine samples for testing of electrolytes should always be obtained before any form of therapy is initiated; this is of great help in differentiating etiologic factors in new cases.
Chronic acid-base disturbances must not be treated too rapidly; more serious complications may be prevented by meticulous and slow correction. For example, consider the case of a child whose initial blood work shows the following results:
- Sodium 120 mmol/L
- Potassium 2 mmol/L
- Chloride 80 mmol/L
- Bicarbonate 40 mmol/L
- pH 7.5
In this child, assessment of cardiac function is indicated. If there is no dysrhythmia, rapid correction of this severe hypokalemia is unnecessary. Administration of 5% dextrose in 0.9 isotonic sodium chloride solution plus potassium chloride 20 mEq/L at a maintenance rate can be a safe measure.
Maintenance management (7-72 hours)
Maintenance therapy depends on how much improvement occurred after 6 hours of initial fluid and electrolyte administration. The aim is to increase the serum potassium concentration very slowly as the serum bicarbonate level drops. This helps prevent a sharp increase in serum potassium concentration and its subsequent detrimental effects on cardiac conductivity.
Long-term management (after 72 hours)
For long-term management, intravenous (IV) fluids can be discontinued. The physician should calculate the average daily amounts of chloride, sodium, and potassium that were required to correct the serum electrolyte levels. The total amounts can then be administered orally in 3-4 divided doses per day. In most patients, the average chloride dose required is 4-10 mEq/kg/day in the form of sodium and potassium salts.
Other management procedures depend on the primary cause of hypochloremic alkalosis.
Surgical or endoscopic intervention
Surgical intervention is usually unnecessary. If ileus is suspected in a child with severe hypokalemia, the appropriate treatment is administration of potassium chloride, not surgical intervention. However, if the cause of hypochloremic alkalosis is an upper gastrointestinal (GI) tract abnormality, such as gastroesophageal reflux or pyloric stenosis, surgical or endoscopic intervention is indicated.
Diet
Kilojoule intake should be appropriate for the patient’s catabolic status, usually 100-150% of the recommended daily allowance (RDA). Additional protein should be ingested to prevent malnutrition. Fat requirements depend on the individual patient. For example, patients with cystic fibrosis have special dietary needs that should be met.
Multivitamins and hematinic agents should be provided as required. Supplemental trace elements (eg, zinc) should be provided to patients with a trace-element deficiency, such as some patients with chloride-losing diarrhea (chloride-losing diarrhea). High sodium and potassium diets are required for all children with chronic metabolic alkalosis secondary to Bartter syndrome or chloride-losing diarrhea.
Activity
Normal activity should be recommended for children with hypochloremic alkalosis unless central nervous system (CNS) damage is severe, in which case special restrictions are required.
Children with refractory severe hypokalemia should avoid extended exposure to heat, especially in hot climates. Exposure to heat may cause dehydration and may exacerbate the condition.
Genetic counseling
Genetic counseling should be considered when prenatal diagnosis is offered to mothers with familial diseases, such as cystic fibrosis, Bartter syndrome, or chloride-losing diarrhea.
Long-term monitoring
Patients should receive regular follow-up examinations by a physician and nurse clinician. Such examinations should take place at least once every month in infants but may be less frequent in older children and children who are more stable.
The preclinic laboratory workup includes a biochemical profile and monitoring of urine electrolytes. The pharmacotherapeutic regimen should be reviewed at each visit. Medications should be refilled and dosages adjusted in accordance with the patient’s clinical status and laboratory results.
Diagnostic imaging studies should be repeated as necessary. For example, kidney ultrasonography may be needed to assess the degree of nephrocalcinosis in children with Bartter syndrome.
Growth parameters should be assessed, and the question of whether growth hormone therapy is needed should be evaluated in consultation with a pediatric endocrinologist. Renal function should be assessed, and every effort should be made to minimize the use of nephrotoxic agents if possible.
Patients with chronic diseases, such as Bartter syndrome, chloride-losing diarrhea and cystic fibrosis, should have lifelong follow-up care.
Future pregnancies in women with a child with hypochloremic alkalosis should be monitored in a tertiary care center so that early diagnosis and intervention are available at delivery.
Hypochloremic metabolic alkalosis prognosis
Hypochloremic alkalosis prognosis is usually good for patients with Bartter syndrome, provided the patient complies well with treatment. Children who receive effective treatment have minimal risk of severe renal damage.
In patients with chloride-losing diarrhea, renal failure and end-stage renal disease (ESRD) may complicate the picture if diagnosis and treatment are delayed.
In patients with cystic fibrosis, prognosis depends on the severity of lung and liver involvement.
Anorexia and polyuria eventually lead to malnutrition and growth failure. Chronic dehydration frequently causes constipation. A small muscle mass and muscle wasting are frequently seen in patients following a late diagnosis or in untreated patients.
Central nervous system (CNS) effects include cerebral dysfunction and defective cognitive function resulting from chronic hypoperfusion in moderate-to-severe metabolic alkalosis due to hypokalemic and hypochloremic states. Hypopnea is due to depression of respiratory drive. CNS calcification occurs in some patients for unclear reasons. Seizure disorder, brain atrophy, and mental retardation are other known complications.
Depending on the renal disorder, complications may include nephrocalcinosis, interstitial nephropathy, hypercalcemia, hyperuricemia, hypertension during the late stages of renal damage, and renal failure. Children with end-stage renal disease require renal replacement therapy in the form of hemodialysis or peritoneal dialysis. Kidney transplantation with the consequences of graft loss due to metabolic derangements adds more morbidity in these patients.
References- Hypochloremic Alkalosis. https://emedicine.medscape.com/article/945263-overview
- Breaux CW Jr, Hood JS, Georgeson KE. The significance of alkalosis and hypochloremia in hypertrophic pyloric stenosis. J Pediatr Surg. 1989;24(12):1250-1252. doi:10.1016/s0022-3468(89)80561-5
- Siegler JC, Marshall P. The effect of metabolic alkalosis on central and peripheral mechanisms associated with exercise-induced muscle fatigue in humans. Exp Physiol. 2015 Apr 20;100(5):519-30. doi: 10.1113/EP085054
- Khanna A, Kurtzman NA. Metabolic alkalosis. J Nephrol. 2006 Mar-Apr;19 Suppl 9:S86-96.
- Seldin DW, Rector FC Jr. Symposium on acid-base homeostasis. The generation and maintenance of metabolic alkalosis. Kidney Int. 1972 May;1(5):306-21. doi: 10.1038/ki.1972.43
- Oppersma E, Doorduin J, van der Hoeven JG, Veltink PH, van Hees HWH, Heunks LMA. The effect of metabolic alkalosis on the ventilatory response in healthy subjects. Respir Physiol Neurobiol. 2018 Feb;249:47-53. doi: 10.1016/j.resp.2018.01.002
- Wang MX, Cuevas CA, Su XT, Wu P, Gao ZX, Lin DH, McCormick JA, Yang CL, Wang WH, Ellison DH. Potassium intake modulates the thiazide-sensitive sodium-chloride cotransporter (NCC) activity via the Kir4.1 potassium channel. Kidney Int. 2018 Apr;93(4):893-902. doi: 10.1016/j.kint.2017.10.023
- Galla JH, Gifford JD, Luke RG, Rome L. Adaptations to chloride-depletion alkalosis. Am J Physiol. 1991 Oct;261(4 Pt 2):R771-81. doi: 10.1152/ajpregu.1991.261.4.R771
- Metabolic Alkalosis. https://emedicine.medscape.com/article/243160-overview#a6
- Yau M, Haider S, Khattab A, Ling C, Mathew M, Zaidi S, Bloch M, Patel M, Ewert S, Abdullah W, Toygar A, Mudryi V, Al Badi M, Alzubdi M, Wilson RC, Al Azkawi HS, Ozdemir HN, Abu-Amer W, Hertecant J, Razzaghy-Azar M, Funder JW, Al Senani A, Sun L, Kim SM, Yuen T, Zaidi M, New MI. Clinical, genetic, and structural basis of apparent mineralocorticoid excess due to 11β-hydroxysteroid dehydrogenase type 2 deficiency. Proc Natl Acad Sci U S A. 2017 Dec 26;114(52):E11248-E11256. doi: 10.1073/pnas.1716621115
- Akil I, Ozen S, Kandiloglu AR, Ersoy B. A patient with Bartter syndrome accompanying severe growth hormone deficiency and focal segmental glomerulosclerosis. Clin Exp Nephrol. 2010 Jun. 14(3):278-82.
- Miyahara J, Aramaki S, Yokochi K. Dietary chloride deficiency due to new liquid nutritional products. Pediatr Int. 2009 Apr. 51 (2):197-200.
- Al-Abbad A, Nazer H, Sanjad SA, Al-Sabban E. Congenital chloride diarrhea: A single center experience with ten patients. Ann Saudi Med. 1995 Sep. 15(5):466-9.
- Luke RG, Galla JH. It is chloride depletion alkalosis, not contraction alkalosis. J Am Soc Nephrol. 2012 Feb. 23 (2):204-7.
- Purkerson JM, Tsuruoka S, Suter DZ, Nakamori A, Schwartz GJ. Adaptation to metabolic acidosis and its recovery are associated with changes in anion exchanger distribution and expression in the cortical collecting duct. Kidney Int. 2010 Nov. 78 (10):993-1005.
- Gifford JD, Ware MW, Luke RG, Galla JH. HCO3- transport in rat CCD: rapid adaptation by in vivo but not in vitro alkalosis. Am J Physiol. 1993 Mar. 264 (3 Pt 2):F435-40.
- Galla JH, Rome L, Luke RG. Bicarbonate transport in collecting duct segments during chloride-depletion alkalosis. Kidney Int. 1995 Jul. 48 (1):52-5.
- [Guideline] Grosse SD, Boyle CA, Botkin JR, et al. Newborn screening for cystic fibrosis: evaluation of benefits and risks and recommendations for state newborn screening programs. MMWR Recomm Rep. 2004 Oct 15. 53:1-36.
- Querfeld U, Lechner S, Janecke AR. Hypochloremic metabolic alkalosis and failure to thrive: answer. Pediatr Nephrol. 2011 Jun. 26 (6):895-6.
- Aranzamendi RJ, Breitman F, Asciutto C, Delgado N, Castanos C. [Dehydration and metabolic alkalosis: an unusual presentation of cystic fibrosis in an infant]. Arch Argent Pediatr. 2008 Oct. 106(5):443-6.
- Stimson L, Reynolds T. Differential diagnosis for chronic hypokalaemia. BMJ Case Rep. 2018 Jun 5;2018:bcr2017223680. doi: 10.1136/bcr-2017-223680
- Galla JH. Metabolic alkalosis. J Am Soc Nephrol. 2000 Feb;11(2):369-375. doi: 10.1681/ASN.V112369
- Hopkins E, Sanvictores T, Sharma S. Physiology, Acid Base Balance. [Updated 2022 Sep 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK507807
- Makela S, Kere J, Holmberg C. SLC26A3 mutations in congenital chloride diarrhea. Hum Mutat. 2002 Dec. 20(6):425-38.
- Simon DB, Bindra RS, Mansfield TA, et al. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat Genet. 1997 Oct. 17(2):171-8.
- Hulka F, Campbell TJ, Campbell JR, Harrison MW. Evolution in the recognition of infantile hypertrophic pyloric stenosis. Pediatrics. 1997 Aug. 100(2):E9.





{kind=link}