What is lymphomatoid papulosis
Lymphomatoid papulosis is a non-aggressive T-cell lymphoma characterized by recurrent, self-healing small bumps and spots on the skin that come and go and sometimes, necrotic lesions, often disseminated with histologic features suggestive of a CD30-positive lymphoma 1. Lymphomatoid papulosis can be persistent, with frequent, recurring eruptions, or it can disappear for an extended period of time before showing up again. Patients often report stress triggers breakouts.
Lymphomatoid papulosis accounts for about 12% of cutaneous lymphomas. It may occur at any age but, on average, earlier than CD30+ anaplastic large-cell lymphomas. It is less exceptional in children. In large series, the mean age of onset varies between 35 and 45 years. The male-to-female ratio is approximately 1 to 5.
Lymphomatoid papulosis belongs to a family of conditions called primary cutaneous CD30-positive lymphoproliferative disorders (pcCD30+LPD). The name “lymphoproliferative disorder” is used to define a broad range of diseases of the immune system that share a common biology (in this case CD30-positive T-cells). The family of primary cutaneous CD30-positive lymphoproliferative disorders includes diseases that are non-malignant and full-blown lymphomas. lymphomatoid papulosis is usually classified as non-malignant or as a cutaneous T-cell lymphoma precursor, though some experts consider it a very low-grade form of cutaneous T-cell lymphoma
The cause of lymphomatoid papulosis is not known. lymphomatoid papulosis is not contagious. There is no supportive research indicating that this is a genetic or hereditary disease. No single factor has been proven to cause lymphomatoid papulosis.
Lymphomatoid papulosis is classified, alongside primary cutaneous anaplastic large cell lymphoma, in the group of T-cell proliferations expressing CD30 2. Patients affected with lymphomatoid papulosis are at risk of developing another hematological disorder: mainly mycosis fungoides, erythrodermic T-cell lymphoma, Hodgkin disease or large-cell CD30+ lymphoma.
A diagnosis of lymphomatoid papulosis requires evaluating the symptoms and having a skin biopsy for various types of laboratory tests. While lymphomatoid papulosis usually is not classified as a cancer (although there has been some debate), it has characteristics of lymphoma under the microscope, and people with lymphomatoid papulosis have a life-long increased risk of developing lymphoma such as mycosis fungoides, primary cutaneous anaplastic large cell lymphoma or Hodgkin lymphoma 3. In 5 to 20 percent of people with lymphomatoid papulosis, the condition is either preceded by lymphoma, associated with lymphoma, or followed by lymphoma 4. When lymphomatoid papulosis is diagnosed it is important to rule out these cancers, and for this reason, various blood tests or imaging studies may also be recommended 5.
There is no cure for lymphomatoid papulosis and no standard treatment. Treatment is tailored to an individual’s symptoms and to addressing the skin issues to prevent scarring. Treatment for lymphomatoid papulosis may speed up the healing of existing lesions or prevent new lesions from forming, but it does not change the overall course or duration of the condition 5. People with only a few lesions or with no major symptoms or cosmetic concerns may opt to forego treatment. If treatment is desired, topical corticosteroids are an option. Options for people with extensive skin lesions or debilitating symptoms may include topical steroids, phototherapy, oral or topical retinoids, methotrexate, or other medications (alone or in combination) 6. Tetracycline is typically used if ulcerated lesions become infected 7. In most people with lymphomatoid papulosis-associated lymphomas, treatment of the lymphoma will also clear the lymphomatoid papulosis 6.
The long-term outlook (prognosis) in more than 90% of people with lymphomatoid papulosis is positive, as it usually does not affect overall health 6. People with lymphomatoid papulosis who do not develop cancer have a normal life expectancy. While there is a substantial risk to develop lymphoma, the reported mortality rates from associated lymphomas are low 5.
Lymphomatoid papulosis causes
The cause of lymphomatoid papulosis is unknown. A viral cause has been suggested. However, studies searching for an etiologic role for Epstein-Barr virus and other herpesviruses have been consistently negative 1. The mechanisms involved in the spontaneous regression of skin lesions have not yet been identified. Interactions between CD30 and its ligand (CD30L) may contribute to apoptosis of the neoplastic T cells and the subsequent regression of the skin lesions, but the exact mechanism is as yet unknown 8.
Lymphomatoid papulosis symptoms
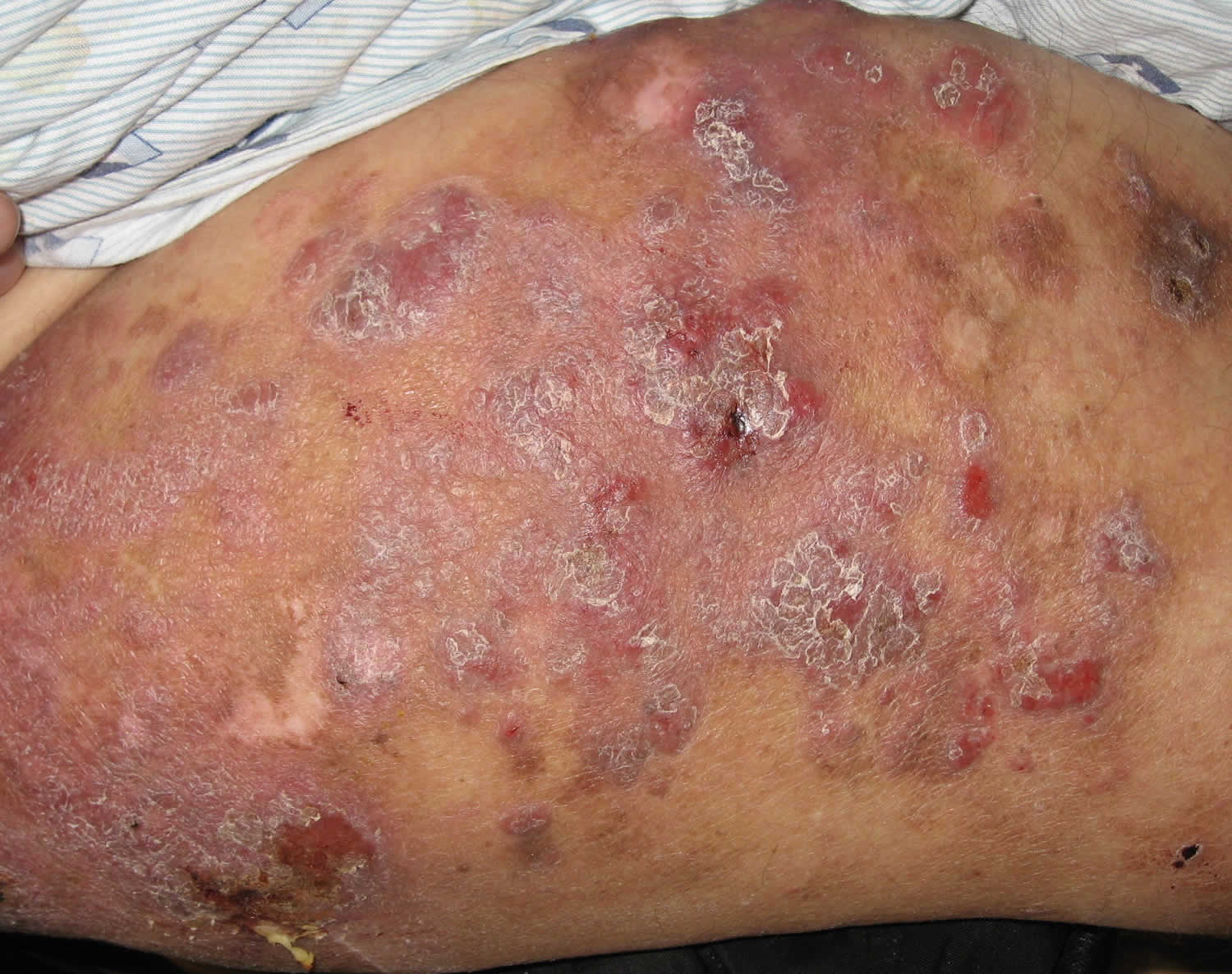
Lymphomatoid papulosis is a non-contagious, chronic skin condition characterized by the eruption of recurring, self-healing bumps (lesions) on the skin. The classic clinical presentation is characterized by a red-brown papulonodular eruption that may become hemorrhagic and necrotic. The lesions typically begin small and then become larger, and they may bleed or ulcerate before becoming scaly and crusty. These lesions go away on their own over a period of weeks to months (usually between 2 and 12 weeks), leaving hypopigmented, hyperpigmented or characteristic atrophic (varioliform) scars 9. Coexistence of different age elements is common. Recurrence is the rule with complete remissions of variable duration. Mucosal lesions, particularly oral, are possible but exceptional 10. The lesions vary in number from one individual to another and from one recurrence to another. Lesions may be localized, sometimes clustered within rather well-defined areas, or generalized. All areas of the skin may be affected with a predilection for the trunk and limbs. The eruption is generally asymptomatic.
Lymphomatoid papulosis is not contagious.
Lymphomatoid papulosis diagnosis
A skin biopsy is critical in the diagnosis of lymphomatoid papulosis. The sample is processed and the cells examined under a microscope. The cutaneous biopsy reveals a dense dermal lymphocyte infiltrate of evocative triangular morphology with epidermal base seen at low magnification. The composition of the infiltrate is variable and correlated with the age of the lesion. The epidermis is frequently ulcerated. Several histologic types of lymphomatoid Papulosis have been described. According to the number of lymphocytes expressing CD30 and according to their size, some authors distinguish type A papulosis in which the CD30 cells are numerous, type B very close to the mycosis fungoides with no or few CD30+ cells and epidermotropism and finally, type C, with large CD30+ lymphocyte plaques. Other rare forms have been reported: forms expressing markers of cytotoxicity, CD8, TIA1, Granzyme B 11, forms with angiotropism 12 and strictly follicular forms mainly affecting the scalp 13. Different types, in particular types A and B, can be observed in the same patient, not only from one lesion to another but also within the same lesion. The vital prognosis and the risk of association with a second hemopathy do not seem to vary from one histological form to another.
Once the diagnosis of lymphomatoid papulosis is confirmed, no complementary assessments are needed. The blood count in search for circulating atypical cells is useless. The potentially associated hematological diseases being, almost exclusively, cutaneous lymphomas, only attentive clinical examination of the skin is necessary.
Lymphomatoid papulosis complications
Patients affected with lymphomatoid papulosis are at risk of developing another hematological disorder in 5% to 20% of the cases, mainly mycosis fungoides, erythrodermic T-cell lymphoma, Hodgkin’s lymphoma or large-cell CD30+ lymphoma 1. The hematological malignancy may precede, follow, or be concomitant with the appearance of lymphomatoid papulosis. This risk varies from 2% to 15% after 5 years of evolution, but it increases with the duration of the disease. Older age and especially the presence of a T-cell clone in the papulosis lesions are significant risk factors for the occurrence of a second hemopathy 14. The impact on the quality of life can be important, because of the chronic course of the disease and the possible localization in visible skin areas. Regardless of the risk of secondary disease, the prognosis of lymphomatous papulosis is excellent with a 10-year disease-specific survival of almost 100% 15.
Lymphomatoid papulosis treatment
As lymphomatoid papulosis is a rare and clinically polymorphic pathology, no controlled prospective therapeutic study could be conducted to validate a certain treatment 1. The treatments are only suspensive and poorly efficient. In patients with relatively few non-scarring lesions, therapeutic abstention can be considered. Disabling or cosmetically disturbing forms can be attenuated or controlled with weekly doses of 5 to 20 mg of methotrexate. Used orally, subcutaneously, or intramuscularly, methotrexate is the systemic treatment of choice for lymphomatoid papulosis, regardless of histological type 16. Starting with low doses (7.5 to 10 mg per week) and then increased in increments of 2.5 or 5 mg until remission of the disease is obtained. Doses higher than 25 mg per week are rarely needed. Once the remission is obtained, the dosage is gradually tapered until the lowest effective dose, or until discontinuation. In their recent publication on their experience with methotrexate in lymphomatoid papulosis, the Dutch Cutaneous Lymphoma Group reported 90% of good to very good results, but less than one-third of patients were able to discontinue treatment following complete and sustainable remission 17. Phototherapy can also be offered. PUVA therapy has been proposed for more than 30 years to treat lymphomatoid papulosis. The disappearance of the lesions is often obtained after about fifteen sessions, but recurrences are frequent. The local chemotherapies (solution or gel of mechlorethamine), bexarotene, interferon, are sometimes used as therapeutic alternatives. When larger skin tumors develop in the course of lymphomatoid papulosis, surgical excision or radiotherapy can be proposed if spontaneous resolution does not occur after a period of 4 to 12 weeks.
The treatment of the child’s papulosis poses difficulties because of the particular therapeutic risk of phototherapy at this age and the fear of using methotrexate in pediatrics. Most authors recommend therapeutic abstention or the use of very strong topical corticosteroids on papules at the initial inflammatory stages. The risk of scarring of the lesions may lead to general treatment, and UVB phototherapy will be preferred, although its results in children are inconsistent 18.
Because of the potential risk for developing a systemic lymphoma, long-term follow-up is required in all patients with lymphomatoid papulosis.
Lymphomatoid papulosis prognosis
In nearly all cases, lymphomatoid papulosis has an excellent prognosis and the condition remains benign or non-cancerous. A retrospective cohort analysis found that no patients with lymphomatoid papulosis died of the disease, and the overall survival rate was 92% at 5 and 10 years.
Physicians are guardedly optimistic about the prognosis because estimates indicate that as many as 4-25% of patients have a history of associated malignant lymphoma (anaplastic large cell lymphoma, Hodgkin’s lymphoma and mycosis fungoides) prior to, concurrent with, or subsequent to the diagnosis of lymphomatoid papulosis 19. Unfortunately, no clinical or histologic factors analyzed to date are predictive of worse outcomes in persons with lymphomatoid papulosis. A study suggested that fascin expression is increased in lymphomatoid papulosis cases associated with a malignant lymphoma 20. Alterations in transforming growth factor-beta signaling are hypothesized to play a role in the progression of lymphomatoid papulosis to malignant lymphoma. Additionally, data have shown an increased risk of associated lymphomas in lymphomatoid papulosis cases with CCR3+ atypical cells or the 30M362 allelic form of the CD30 promoter.
Primary cutaneous anaplastic large cell lymphoma is more likely than mycosis fungoides to manifest as an ulcerated tumor and palpable lymph nodes. Mycosis fungoides is the most common variant of cutaneous T-cell lymphoma and is characterized by the development of red patches or plaques in sun-protected areas. Mycosis fungoides is more likely to manifest as patches and plaques than tumors. Disease-specific survival at 5 and 10 years for primary cutaneous anaplastic large cell lymphoma was 85% in a 2003 study 21.
Associated lymphomas more rarely include immunoblastic lymphoma, lethal midline granuloma (currently considered as natural killer cell lymphoma in many patients), and systemic lymphocytic lymphoma. In most patients, the malignancy develops many years after the diagnosis of lymphomatoid papulosis.
References- Toumi A, Litaiem N. Lymphomatoid Papulosis. [Updated 2019 Jan 25]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK532295
- Willemze R, Jaffe ES, Burg G, Cerroni L, Berti E, Swerdlow SH, Ralfkiaer E, Chimenti S, Diaz-Perez JL, Duncan LM, Grange F, Harris NL, Kempf W, Kerl H, Kurrer M, Knobler R, Pimpinelli N, Sander C, Santucci M, Sterry W, Vermeer MH, Wechsler J, Whittaker S, Meijer CJ. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005 May 15;105(10):3768-85
- Lymphomatoid Papulosis. https://www.clfoundation.org/lymphomatoid-papulosis
- Lymphomatoid papulosis. https://emedicine.medscape.com/article/1098954-overview
- Lymphomatoid papulosis. https://www.uptodate.com/contents/lymphomatoid-papulosis
- Lymphomatoid papulosis. https://www.clfoundation.org/lymphomatoid-papulosis
- Lymphomatoid papulosis. https://www.dermcoll.edu.au/atoz/lymphomatoid-papulosis
- Mori M, Manuelli C, Pimpinelli N, Mavilia C, Maggi E, Santucci M, Bianchi B, Cappugi P, Giannotti B, Kadin ME. CD30-CD30 ligand interaction in primary cutaneous CD30(+) T-cell lymphomas: A clue to the pathophysiology of clinical regression. Blood. 1999 Nov 01;94(9):3077-83.
- Willemze R, Jaffe ES, Burg G, Cerroni L, Berti E, Swerdlow SH, Ralfkiaer E, Chimenti S, Diaz-Perez JL, Duncan LM, Grange F, Harris NL, Kempf W, Kerl H, Kurrer M, Knobler R, Pimpinelli N, Sander C, Santucci M, Sterry W, Vermeer MH, Wechsler J, Whittaker S, Meijer CJ. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005 May 15;105(10):3768-85.
- Allabert C, Estève E, Joly P, Troussard X, Comoz F, Courville P, Morice A, Verneuil L, Leroy D, Dompmartin A. [Mucosal involvement in lymphomatoid papulosis: four cases]. Ann Dermatol Venereol. 2008 Apr;135(4):273-8.
- Bertolotti A, Pham-Ledard AL, Vergier B, Parrens M, Bedane C, Beylot-Barry M. Lymphomatoid papulosis type D: an aggressive histology for an indolent disease. Br. J. Dermatol. 2013 Nov;169(5):1157-9.
- Kempf W, Kazakov DV, Schärer L, Rütten A, Mentzel T, Paredes BE, Palmedo G, Panizzon RG, Kutzner H. Angioinvasive lymphomatoid papulosis: a new variant simulating aggressive lymphomas. Am. J. Surg. Pathol. 2013 Jan;37(1):1-13.
- Kempf W, Kazakov DV, Baumgartner HP, Kutzner H. Follicular lymphomatoid papulosis revisited: a study of 11 cases, with new histopathological findings. J. Am. Acad. Dermatol. 2013 May;68(5):809-16.
- Cordel N, Tressières B, D’Incan M, Machet L, Grange F, Estève É, Dalac S, Ingen-Housz-Oro S, Bagot M, Beylot-Barry M, Joly P., French Study Group on Cutaneous Lymphoma. Frequency and Risk Factors for Associated Lymphomas in Patients With Lymphomatoid Papulosis. Oncologist. 2016 Jan;21(1):76-83.
- Bekkenk MW, Geelen FA, van Voorst Vader PC, Heule F, Geerts ML, van Vloten WA, Meijer CJ, Willemze R. Primary and secondary cutaneous CD30(+) lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000 Jun 15;95(12):3653-61.
- Bruijn MS, Horváth B, van Voorst Vader PC, Willemze R, Vermeer MH. Recommendations for treatment of lymphomatoid papulosis with methotrexate: a report from the Dutch Cutaneous Lymphoma Group. Br. J. Dermatol. 2015 Nov;173(5):1319-22.
- Wieser I, Oh CW, Talpur R, Duvic M. Lymphomatoid papulosis: Treatment response and associated lymphomas in a study of 180 patients. J. Am. Acad. Dermatol. 2016 Jan;74(1):59-67.
- Miquel J, Fraitag S, Hamel-Teillac D, Molina T, Brousse N, de Prost Y, Bodemer C. Lymphomatoid papulosis in children: a series of 25 cases. Br. J. Dermatol. 2014 Nov;171(5):1138-46.
- Kempf W, Pfaltz K, Vermeer MH, Cozzio A, Ortiz-Romero PL, Bagot M, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011 Oct 13. 118(15):4024-35.
- Kempf W, Levi E, Kamarashev J, et al. Fascin expression in CD30-positive cutaneous lymphoproliferative disorders. J Cutan Pathol. 2002 May. 29(5):295-300.
- Liu HL, Hoppe RT, Kohler S, Harvell JD, Reddy S, Kim YH. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003 Dec. 49 (6):1049-58.





{kind=link}