MELAS syndrome
MELAS syndrome is short for Mitochondrial encephalomyopathy, Lactic acidosis and Stroke-like episodes syndrome, is a mitochondrial inherited genetic disorder primarily affecting the nervous system and muscles 1. MELAS syndrome is a multisystem and progressive neurodegenerative disorder that is maternally inherited (see Figure 2 below), that presents in children or young adults as recurrent episodes of encephalopathy with seizures and/or dementia, myopathy with muscle weakness and exercise intolerance, recurrent headaches, recurrent vomiting, hearing impairment, peripheral neuropathy, learning disability, focal neurological deficits, stroke-like episodes and short stature 2. During the stroke-like episodes neuroimaging shows increased T2-weighted signal areas that do not correspond to the classic vascular distribution (hence the term “stroke-like”) 2. Lactic acidemia is very common and muscle biopsies typically show ragged red fibers 2. MELAS syndrome is relentlessly progressive, resulting in neurological impairment by adolescence or early adulthood 1.
Mitochondrial genetic disorders are the result of mutations causing impaired mitochondrial function, including oxidative phosphorylation and energy production. In MELAS syndrome, mutations in transfer RNAs (tRNAs) are believed to cause impairment of protein assembly into respiratory chain complexes, though the exact mechanisms have yet to be elucidated. Mitochondria are the powerhouse of cells. Any mitochondrial disorder will affect the most metabolically active organs of the body especially the brain and muscles.
Many different transfer RNA (tRNA) mutations can cause MELAS syndrome. The most common mutation is in the MTTL1 mitochondrial gene. A single base pair mutation, m.3243A>G, is found in 80% patients, and a second common mutation, m.3271T>C, is found in 10% 3. However, many other genes are being identified with similar phenotypic syndromes such as POLG and BCS1L.
Mitochondrial disorders including MELAS affect mainly tissues with the highest metabolic activities. These include skeletal and heart muscles, brain, eye, and the inner ear. Due to the impaired mitochondrial respiratory chain function and oxidative phosphorylation, the increased anaerobic glycolytic activities will lead to increased lactic acid during acute attacks.
The neurological symptoms of MELAS syndrome are believed to result from a combination of impaired mitochondrial energy production, microvascular angiopathy, and nitric oxide deficiency, which may cause impaired cerebral vasodilation 4.
The exact incidence of MELAS syndrome is unknown 5. MELAS syndrome is one of the more common conditions in a group known as mitochondrial diseases. Together, mitochondrial diseases occur in about 1 in 4,000 people 5.
Both genders are equally affected, but only women can pass the condition on as mitochondria are carried in the tails of sperm cells and therefore shed outside the zygote during fertilization.
The hallmark of mitochondrial disease is an accumulation of mitochondria in muscle fibers visualized on Gomori trichrome stain. The diseased mitochondria proliferate in an attempt to compensate for poor energy production and appear bright red compared to the blue myofibers, therefore called “ragged red fibers.” MELAS syndrome also shows strongly succinate dehydrogenase-reactive blood vessels due to mitochondrial proliferation in perivascular smooth muscle and endothelial cells. However, genetic evaluation should be done first, which eliminates the need for muscle biopsy in most cases.
Clinical testing may include measurement of lactate and pyruvate concentrations and CSF protein which are elevated in MELAS syndrome. Brain imaging techniques such as magnetic resonance imaging (MRI) may be used to look for stroke-like lesions and magnetic resonance spectroscopy (MRS) may be used to look for a lactate peak in the brain. Electrocardiogram may be used to diagnose heart rhythm abnormalities and echocardiogram may be used to diagnose cardiomyopathy. Muscle biopsy will usually show ragged red fibers.
The mitochondrial DNA (mtDNA) mutations associated with MELAS syndrome can usually be detected in white blood cells, but due to heteroplasmy, other tissue samples may be necessary such as skin, hair follicles, urinary sediment and skeletal muscle. Urinary sediment has the best yield for detecting the mutation when compared to blood, skin, and hair follicles.
No specific treatment is available for MELAS syndrome. Anti-convulsant drugs are used to help prevent and control seizures associated with MELAS syndrome. Valproic acid should not be used as an anticonvulsant. Cochlear implants have been used to treat sensorineural deafness. Therapies are sometimes used to increase energy production by the mitochondria and slow the effects of the condition. Coenzyme Q10 and L-carnitine have been beneficial in some patients. In patients with mitochondrial myopathies in general, moderate treadmill training may result in improvement of aerobic capacity and drop in resting lactate levels. The use of intravenous L-arginine has been reported to improve the symptoms of disease during the acute stroke-like episodes. The use of oral arginine has been reported to decrease the recurrence of stroke-like episodes when used during the asymptomatic period.
Genetic counseling is recommended for affected individuals and their families.
Figure 1. MELAS syndrome
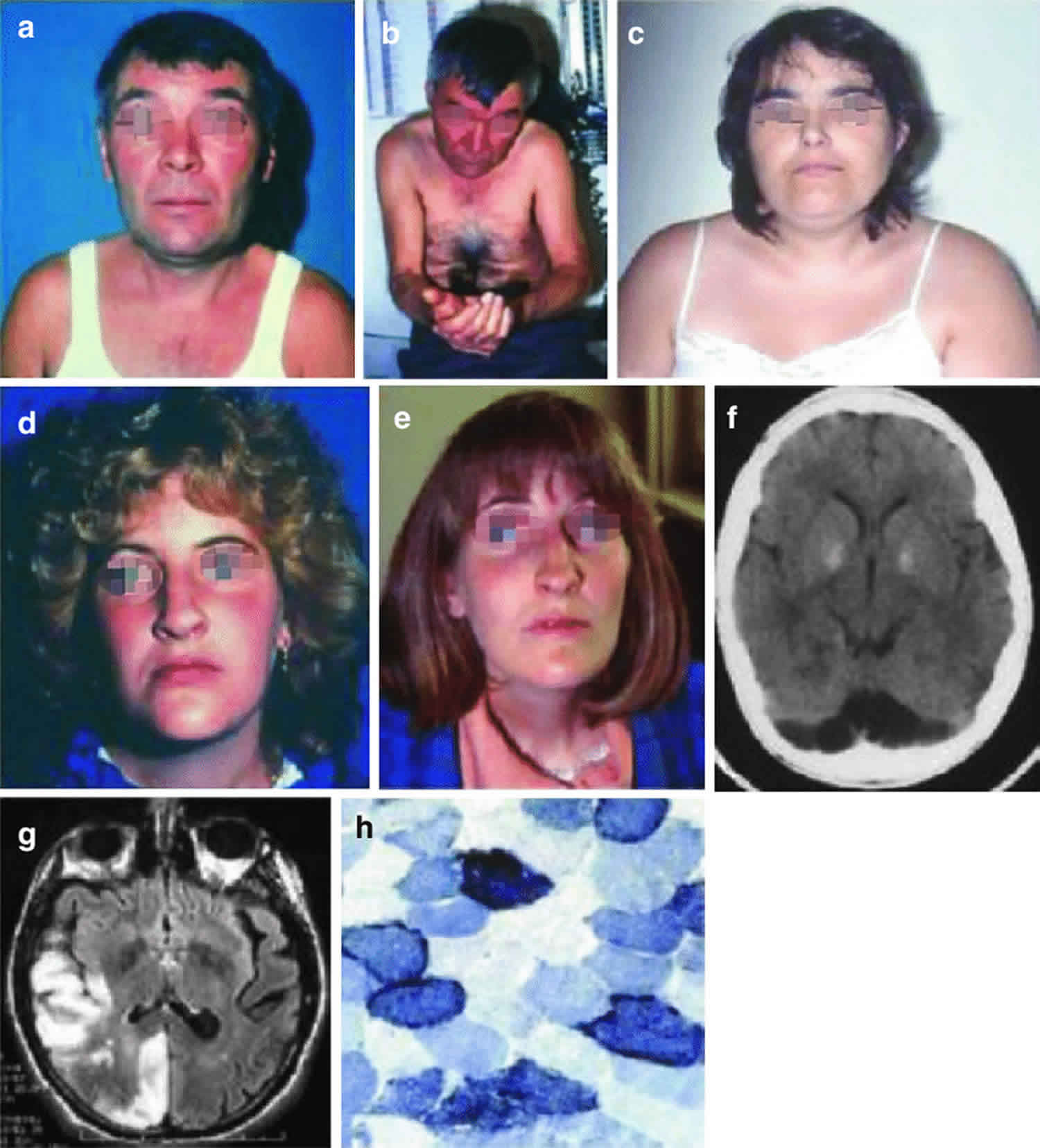
Footnote: Three patients with MELAS syndrome. Patient 1 (a, b) had round face and palpebral ptosis, and after multiple strokes, he developed dementia and apraxia. His brain CT scan showed many lesions with nonvascular distribution (f). His niece (patient 4) (d, e) started with an ocular myopathy and died of dilated cardiomyopathy. Patient 3 (c) had round face, short stature, and limb weakness and developed an untreatable epileptic status. Muscle biopsy showed ragged-red fibers which strongly reacted with succinate dehydrogenase reaction (h). Brain MRI showed multiple stroke-like lesions (g) in occipital and temporoparietal areas.
[Source 6 ]What is the chance that a sibling of an individual with MELAS syndrome will be affected?
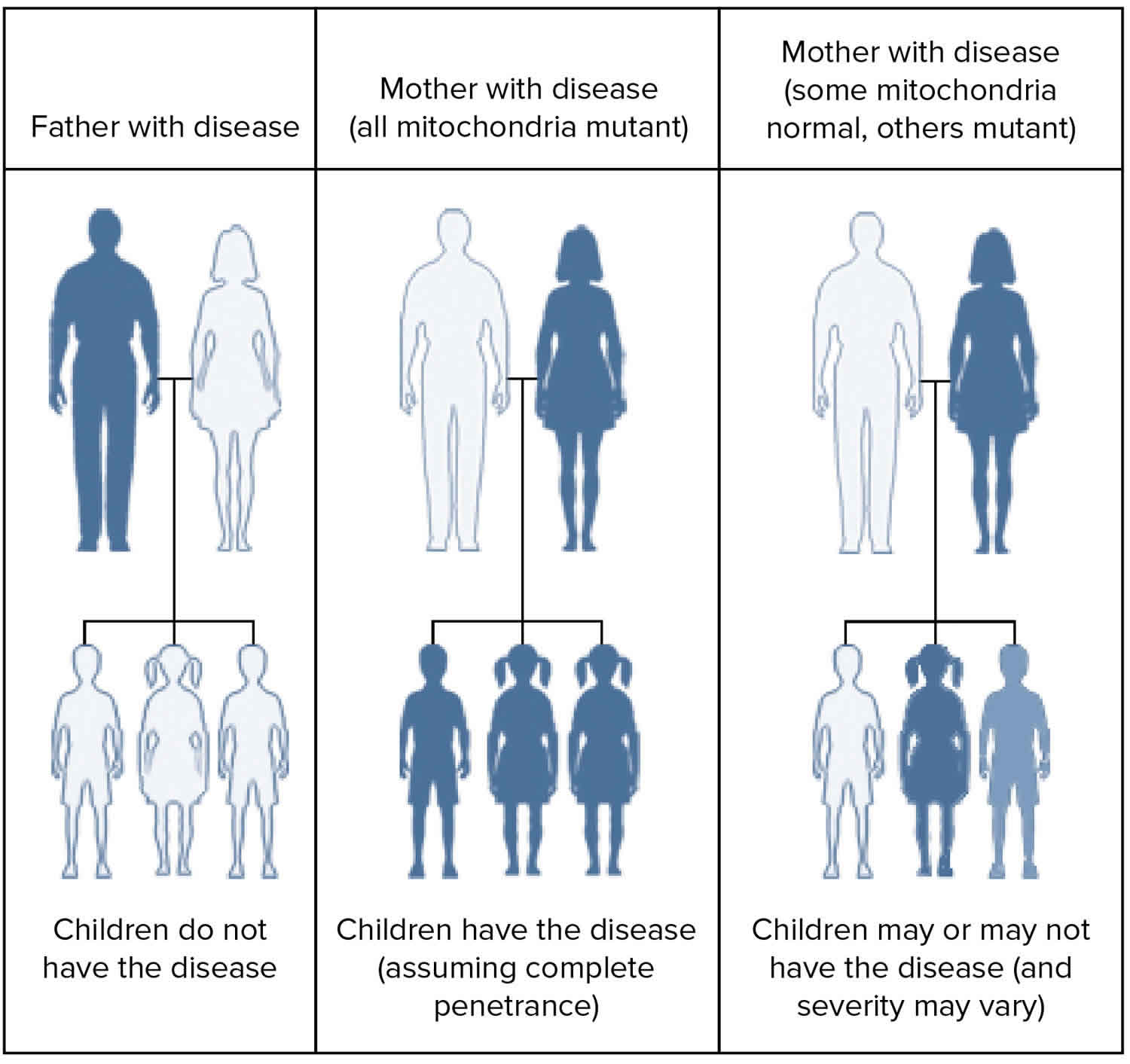
The risk to the siblings of an individual affected with MELAS syndrome depends on the genetic status of their mother. If their mother has the mitochondrial DNA (mtDNA) mutation, all of the siblings of the affected individual will inherit the disease-causing mitochondrial DNA (mtDNA) mutation; however, the siblings may or may not have symptoms. One study reported that women with higher levels of the mutated mtDNA in their blood have a greater likelihood of having affected offspring (i.e. children with symptoms of the condition) 2.
Unfortunately, it is not possible to predict whether an individual with an mitochondrial DNA (mtDNA) mutation will have specific symptoms. The possible effects of an mtDNA mutation depend on a combination of factors, including the severity of the mutation, the percentage of mitochondria that have the mutation, and the organs and tissues in which the mutated mitochondria are located. Different family members often inherit different percentages of mutated mtDNA and therefore can have a wide range of clinical symptoms. Test results of at-risk family members who don’t currently have symptoms are very hard to interpret 2.
Individuals concerned about specific risks to themselves or family members should speak with a genetics professional.
MELAS syndrome causes
MELAS syndrome can result from mutations in one of several genes, including MT-ND1, MT-ND5, MT-TH, MT-TL1, and MT-TV 5. These genes are found in the DNA of cellular structures called mitochondria, which convert the energy from food into a form that cells can use. Although most DNA is packaged in chromosomes within the nucleus, mitochondria also have a small amount of their own DNA, known as mitochondrial DNA or mtDNA.
Some of the genes related to MELAS syndrome provide instructions for making proteins involved in normal mitochondrial function. These proteins are part of a large enzyme complex in mitochondria that helps convert oxygen, fats, and simple sugars to energy. Other genes associated with this disorder provide instructions for making molecules called transfer RNAs (tRNAs), which are chemical cousins of DNA. These molecules help assemble protein building blocks called amino acids into full-length, functioning proteins within mitochondria.
Mutations in a particular transfer RNA gene, MT-TL1, cause more than 80 percent of all cases of MELAS syndrome. These mutations impair the ability of mitochondria to make proteins, use oxygen, and produce energy. Researchers have not determined how changes in mitochondrial DNA (mtDNA) lead to the specific signs and symptoms of MELAS syndrome. They continue to investigate the effects of mitochondrial gene mutations in different tissues, particularly in the brain.
MELAS syndrome inheritance pattern
MELAS syndrome is inherited in a mitochondrial pattern, which is also known as maternal inheritance. This pattern of inheritance applies to genes contained in mitochondrial DNA (mtDNA). Mitochondria are structures in each cell that turn molecules into energy, and each contain a small amount of DNA. Because egg cells, but not sperm cells, contribute mitochondria to the developing embryo, children can only inherit disorders resulting from mtDNA mutations from their mother. These disorders can appear in every generation of a family and can affect both males and females, but fathers do not pass traits associated with changes in mtDNA to their children.
In most cases, people with MELAS syndrome inherit an altered mitochondrial gene from their mother.
In rare instances, MELAS syndrome results from a sporadic mutation in a mitochondrial gene and occurs in people with no family history of MELAS syndrome 1.
Figure 2. MELAS syndrome mitochondrial inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
MELAS syndrome symptoms
The signs and symptoms of MELAS syndrome most often appear in childhood following a period of normal development, although they can begin at any age. Children with MELAS syndrome often have normal early psychomotor development until the onset of symptoms between 2 and 10 years old. Though less common, infantile onset may occur and may present as failure to thrive, growth retardation and progressive deafness. Onset in older children typically presents as recurrent attacks of a migraine-like headache, anorexia, vomiting, and seizures. Children with MELAS syndrome are also frequently found to have short stature.
Early symptoms may include muscle weakness and pain, recurrent headaches, loss of appetite, vomiting, and seizures. Myopathy in MELAS syndrome initially presents as exercise intolerance or proximal muscle weakness. Due to the severity of the other neurological features, the myopathy is often under-recognized. The myopathy in MELAS syndrome often does not cause a significant functional disturbance. Seizures in MELAS syndrome may be focal or generalized and have no recognized syndrome-specific semiology or EEG findings. Children with younger age of onset tend to have higher rates of drug-resistance epilepsy and therefore more severe clinical dysfunction.
Seizures are often accompanied by sudden focal neurological deficits which are termed “stroke-like episodes.” Most affected individuals experience stroke-like episodes beginning before age 40. These “stroke-like episodes” often involve temporary muscle weakness on one side of the body (hemiparesis), altered consciousness, vision abnormalities (hemianopia), seizures, aphasia and severe headaches resembling migraines. Repeated stroke-like episodes can progressively damage the brain, by adolescence or young adulthood, leading to vision loss, sensorineural hearing loss (deafness), problems with movement, and a loss of intellectual function (dementia).
These “stroke-like episodes” differ from a typical stroke in that they do not conform to any vascular territory, the MRI findings may fluctuate or resolve more quickly than typical stroke, and the apparent diffusion coefficient on MRI is not always decreased like typical infarct, and may instead be increased or mixed.
Encephalopathy is another key feature. Altered level of consciousness is often associated with the stroke-like episodes. Mental deterioration begins during childhood and is slowly progressive.
Other organ systems may also be involved in MELAS syndrome. Patients are reported to have cardiac conduction disorders, diabetes, and chronic fatigue.
Most people with MELAS have a buildup of lactic acid in their bodies, a condition called lactic acidosis. Increased acidity in the blood can lead to vomiting, abdominal pain, extreme tiredness (fatigue), muscle weakness, and difficulty breathing. Less commonly, people with MELAS may experience involuntary muscle spasms (myoclonus), impaired muscle coordination (ataxia), hearing loss, heart and kidney problems, diabetes, and hormonal imbalances.
MELAS syndrome diagnosis
MELAS syndrome is diagnosed based on clinical findings and molecular genetic testing 1:
- Stroke-like episodes before 40 years old
- Encephalopathy with seizures or dementia
- Blood lactic acidosis* or ragged red fibers on muscle biopsy
*Due to mitochondrial heteroplasmy, urine and blood testing is preferable to blood alone.
Clinical diagnostic criteria for MELAS syndrome have been published 7. MELAS syndrome should be suspected in individuals with the following features:
Clinical features
- Stroke-like episodes before the age of 40 years
- Acquired encephalopathy with seizures and/or dementia
- Recurrent headaches
- Muscle weakness and exercise intolerance
- Cortical vision loss
- Hemiparesis
- Recurrent vomiting
- Short stature
- Hearing impairment
- Normal early psychomotor development
- Peripheral neuropathy
- Learning disability
Brain Imaging
Brain MRI
- During the stroke-like episodes, the affected areas:
- Have increased T2 signal;
- Do not correspond to the classic vascular distribution (hence the term “stroke-like”);
- Are asymmetric;
- Typically involve predominantly the posterior cerebrum (temporal, parietal, and occipital lobes);
- Can be restricted to cortical areas or involve subcortical white matter 8.
- Slow spreading of the stroke-like lesions occurs in the weeks following the first symptoms, typically documented by T2-weighted MRI 9.
- Diffusion-weighted MRI shows increased apparent diffusion coefficient (ADC) in the stroke-like lesions of MELAS, in contrast to the decreased apparent diffusion coefficient seen in ischemic strokes 10.
- MR angiography is usually normal and MR spectroscopy shows decreased N-acetylaspartate signals and accumulation of lactate 8.
Head CT
- Basal ganglia calcifications are occasionally seen.
Electromyography and Nerve Conduction Studies
Findings are consistent with a myopathic process, but neuropathy may coexist. Neuropathy can be axonal or mixed axonal and demyelinating 11.
Laboratory Findings
Lactic acidosis both in blood and Cerebrospinal fluid (CSF). Lactic acidemia is very common. CSF lactate is also elevated in most affected individuals.
Lactic acidemia is not specific for MELAS syndrome as it can occur in other mitochondrial diseases, metabolic diseases, and systemic illness. Other situations (unrelated to the diagnosis of MELAS) in which lactate can be elevated are acute neurologic events such as seizure or stroke. On the other hand, lactate level can be normal in a minority of individuals with MELAS syndrome 12.
Elevated CSF protein rarely surpasses 100 mg/dL.
Muscle biopsy
- Ragged red fibers with the modified Gomori trichrome stain, which represent mitochondrial proliferation below the plasma membrane of the muscular fibers causing the contour of the muscle fiber to become irregular. These proliferated mitochondria also stain strongly with the succinate dehydrogenase stain giving the appearance of ragged blue fibers.
- Although ragged red fibers are present in many other mitochondrial diseases e.g., MERRF (myoclonic epilepsy with ragged red fibers), most of the ragged red fibers in MELAS stain positively with the cytochrome c oxidase (COX) histochemical stain, unlike other mitochondrial diseases where ragged red fibers do not react with COX.
- An overabundance of mitochondria in smooth muscle and endothelial cells of intramuscular blood vessels, best revealed with the succinate dehydrogenase stain (“strongly succinate dehydrogenase-reactive blood vessels”)
- Respiratory chain studies on muscle tissue: typically multiple partial defects, especially involving complex I and/or complex IV. However, biochemical results can also be normal.
Note: Muscle biopsy is not required to make this diagnosis; molecular genetic testing is frequently used in lieu of muscle biopsy to establish the diagnosis.
Establishing the MELAS syndrome diagnosis
Two sets of clinical diagnostic criteria for MELAS syndrome have been published:
1. A clinical diagnosis of MELAS syndrome can be made if the following three criteria are met 13:
- Stroke-like episodes before age 40 years
- Encephalopathy characterized by seizures and/or dementia
- Mitochondrial myopathy is evident by the presence of lactic acidosis and/or ragged-red fibers on muscle biopsy.
AND at least two of the following criteria are present:
- Normal early psychomotor development
- Recurrent headaches
- Recurrent vomiting episodes
2. A clinical diagnosis of MELAS syndrome can also be made in an individual with at least two category A and two category B criteria 7:
Category A criteria
- Headaches with vomiting
- Seizures
- Hemiplegia
- Cortical blindness
- Acute focal lesions on neuroimaging
Category B criteria
- High plasma or cerebrospinal fluid (CSF) lactate
- Mitochondrial abnormalities on muscle biopsy.
- A MELAS-related pathogenic variant
The diagnosis of MELAS is established in a proband who meets the clinical diagnostic criteria discussed above and who has a pathogenic variant in one of the genes listed in Table 1 identified by molecular genetic testing.
Table 1. Genetic causes of MELAS syndrome
| Gene | % of MELAS Attributed to Pathogenic Variants in This Gene | Proportion of Pathogenic Variants Detectable by Sequence Analysis |
|---|---|---|
| MT-TL1 | >80% | 100% |
| MT-ND5 | <10% | 100% |
| MT-TC MT-TF MT-TH MT-TK MT-TL2 MT-TQ MT-TV MT-TW MT-TS1 MT-TS2 MT-ND1 MT-ND6 MT-CO2 MT-CO3 MT-CYB | Rare | 100% |
MELAS syndrome treatment
There is no treatment to stop disease progression. Treatment for MELAS syndrome is generally supportive. During the acute stroke-like episode, a bolus of intravenous arginine (500 mg/kg for children or 10 g/m² body surface area for adults) within three hours of symptom onset is recommended followed by the administration of a similar dosage of intravenous arginine as a continuous infusion over 24 hours for the next three to five days. Coenzyme Q10, L-carnitine, and creatine have been beneficial in some individuals. Sensorineural hearing loss has been treated with cochlear implantation; seizures respond to traditional anticonvulsant therapy (although valproic acid should be avoided). Ptosis, cardiomyopathy, cardiac conduction defects, nephropathy, and migraine headache are treated in the standard manner. Diabetes mellitus is managed by dietary modification, oral hypoglycemic agents, or insulin therapy. Exercise intolerance and weakness may respond to aerobic exercise.
Once an individual with MELAS has the first stroke-like episode, arginine should be administered prophylactically to reduce the risk of recurrent stroke-like episodes. A daily dose of 150 to 300 mg/kg body weight per day oral arginine in three divided doses is recommended.
Because febrile illnesses may trigger acute exacerbations, individuals with MELAS should receive standard childhood vaccinations, flu vaccine, and pneumococcal vaccine.
Surveillance
Affected individuals and their at-risk relatives should be followed at regular intervals to monitor progression and the appearance of new symptoms. Annual ophthalmologic, audiology, and cardiologic (electrocardiogram and echocardiogram) evaluations are recommended. Annual urinalysis and fasting blood glucose level are also recommended.
Agents/circumstances to avoid
Mitochondrial toxins, including aminoglycoside antibiotics, linezolid, cigarettes, and alcohol; valproic acid for seizure treatment; metformin because of its propensity to cause lactic acidosis; dichloroacetate because of increased risk for peripheral neuropathy.
Pregnancy management
Affected or at-risk pregnant women should be monitored for diabetes mellitus and respiratory insufficiency, which may require therapeutic interventions.
Investigational therapies
Medications including carnitine, coenzyme Q10, menadione, ascorbic acid, riboflavin, thamine, nicotinamide, creatine monohydrate, idebenone, succinate and dichloroacetate have been helpful in individual patients but further studies are needed to prove their efficacy. Arginine and citrulline are being investigated as potential therapies to reduce brain damage from stroke-like episodes.
Columbia University in New York City is seeking study participants for a double-blind, placebo controlled clinical trial of idebenone in MELAS (Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like Episodes) (https://clinicaltrials.gov/ct2/show/NCT00887562). The Phase IIa study will compare two different doses of an experimental medication, idebenone, administered over a one month period to determine the efficacy of the drug. People with MELAS and the 3243 mutation, aged 8-65 years, may be eligible. The main goal of the clinical trial is to determine if idebenone has an effect on brain lactate as measured by magnetic resonance spectroscopy (MRS). Magnetic resonance spectroscopy is done in an MRI scanner, and is safe and typically well tolerated. An additional goal is to study the safety and tolerability of idebenone in people with MELAS.
MELAS syndrome prognosis
MELAS syndrome widely varies in presentation; however, patients in general tend to have a poor prognosis and outcome. The encephalomyopathy tends to be severe and progressive to dementia. The patient with MELAS syndrome may end up in a state of cachexia. Currently, no therapies have proven efficacy 14.
References- Pia S, Lui F. Melas Syndrome. [Updated 2019 Jan 16]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK532959
- El-Hattab AW, Almannai M, Scaglia F. MELAS. 2001 Feb 27 [Updated 2018 Nov 29]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1233
- Niedermayr K, Pölzl G, Scholl-Bürgi S, Fauth C, Schweigmann U, Haberlandt E, Albrecht U, Zlamy M, Sperl W, Mayr JA, Karall D. Mitochondrial DNA mutation “m.3243A>G”-Heterogeneous clinical picture for cardiologists (“m.3243A>G”: A phenotypic chameleon). Congenit Heart Dis. 2018 Sep;13(5):671-677.
- Koga Y, Povalko N, Inoue E, Nakamura H, Ishii A, Suzuki Y, Yoneda M, Kanda F, Kubota M, Okada H, Fujii K. Therapeutic regimen of L-arginine for MELAS: 9-year, prospective, multicenter, clinical research. J. Neurol. 2018 Dec;265(12):2861-2874.
- Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes. https://ghr.nlm.nih.gov/condition/mitochondrial-encephalomyopathy-lactic-acidosis-and-stroke-like-episodes#statistics
- Angelini, Corrado. (2018). Genetic Neuromuscular Disorders: A Case-Based Approach. 10.1007/978-3-319-56454-8. Angelini, Corrado. (2018). Genetic Neuromuscular Disorders: A Case-Based Approach. 10.1007/978-3-319-56454-8.
- Yatsuga S, Povalko N, Nishioka J, Katayama K, Kakimoto N, Matsuishi T, Kakuma T, Koga Y, Matsuoka T, et al. MELAS: a nationwide prospective cohort study of 96 patients in Japan. Biochim Biophys Acta. 2012;1820:619–24.
- Sproule DM, Kaufmann P. Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Ann NY Acad Sci. 2008;1142:133–58.
- Iizuka T, Sakai F, Kan S, Suzuki N. Slowly progressive spread of the stroke-like lesions in MELAS. Neurology. 2003;61:1238–44.
- Kolb SJ, Costello F, Lee AG, White M, Wong S, Schwartz ED, Messé SR, Ellenbogen J, Kasner SE, Galetta SL. Distinguishing ischemic stroke from the stroke-like lesions of MELAS using apparent diffusion coefficient mapping. J Neurol Sci. 2003;216:11–5.
- Kaufmann P, Pascual JM, Anziska Y, Gooch CL, Engelstad K, Jhung S, DiMauro S, De Vivo DC. Nerve conduction abnormalities in patients with MELAS and the A3243G mutation. Arch Neurol. 2006b;63:746–8.
- Hirano M, Pavlakis SG. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS): current concepts. J. Child Neurol. 1994;9:4–13.
- Hirano M, Ricci E, Koenigsberger MR, Defendini R, Pavlakis SG, DeVivo DC, DiMauro S, Rowland LP. MELAS: an original case and clinical criteria for diagnosis. Neuromuscul Disord. 1992;2:125–35.
- MELAS syndrome. https://emedicine.medscape.com/article/946864-overview





{kind=link}