Pyruvate dehydrogenase deficiency
Pyruvate dehydrogenase deficiency also called pyruvate dehydrogenase complex deficiency, is a rare inherited disorder of carbohydrate metabolism caused by a deficiency of one of the three enzymes in the pyruvate dehydrogenase complex 1. Pyruvate dehydrogenase deficiency is characterized by the buildup of a chemical called lactic acid in the body and a variety of neurological problems. Pyruvate dehydrogenase deficiency prevalence is unknown 2. Several hundred cases of pyruvate dehydrogenase deficiency have been reported. More males than females have severe disease and early death and progressive neurological symptoms are observed more often in females, although some females have severe symptoms. The age of onset and severity of disease depends on the activity level of the pyruvate dehydrogenase complex enzymes. Individuals with pyruvate dehydrogenase deficiency beginning prenatally or in infancy usually die in early childhood. Those who develop pyruvate dehydrogenase deficiency later in childhood may have mental retardation and other neurological symptoms and usually survive into adulthood. Most individuals with pyruvate dehydrogenase deficiency have an abnormality in the PDHA1 gene located on the X chromosome. Some affected individuals have rarer forms of the disorder that follow autosomal recessive inheritance.Some individuals have a thiamine responsive form of this disorder.
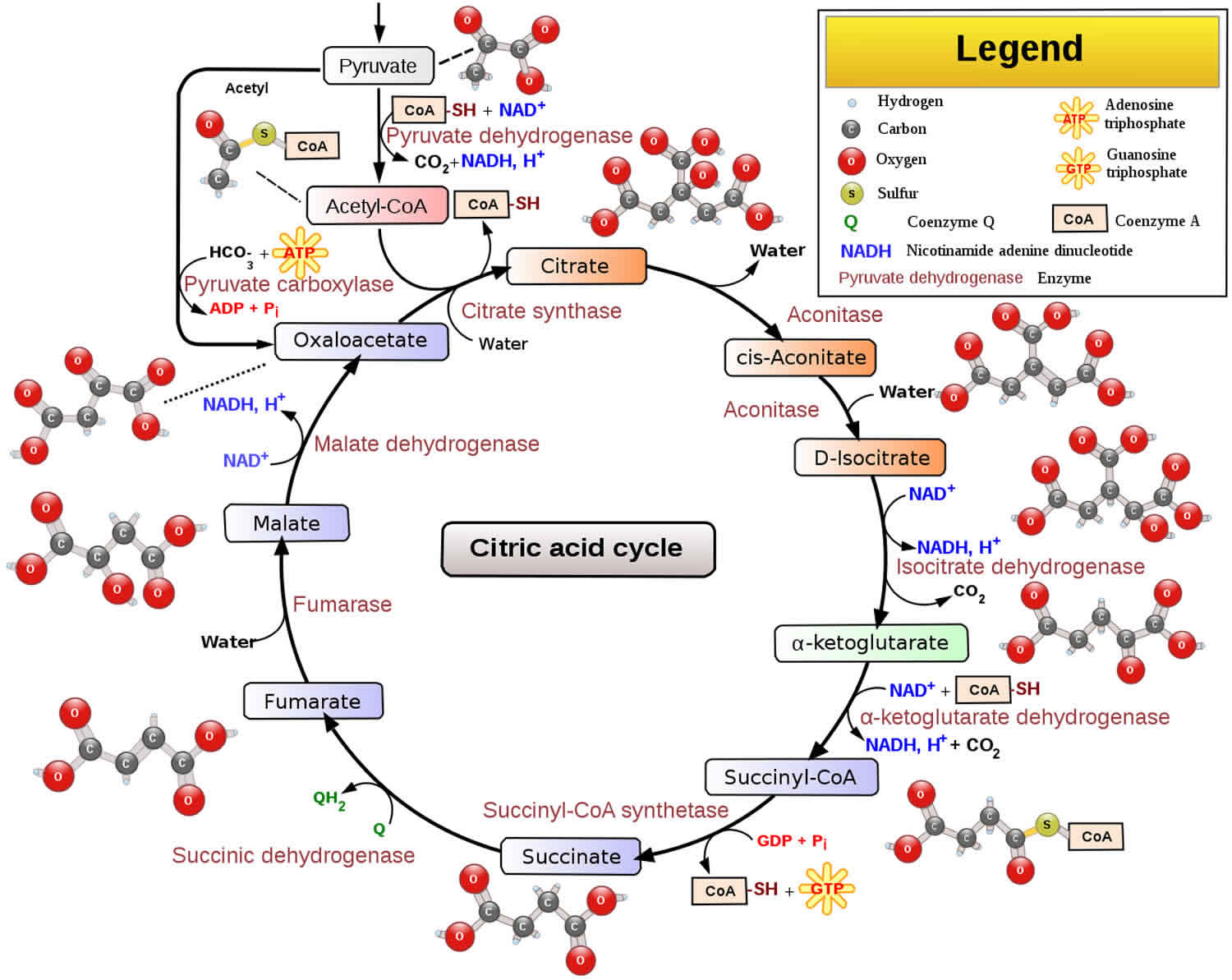
Pyruvate dehydrogenase deficiency metabolically converts pyruvate into acetyl-coenzyme A (ACoA), one of the first steps in the citric acid cycle (see Figure 1). The citric acid cycle is a major biochemical process in the mitochondrial matrix that derives energy from several metabolic substrates, including carbohydrates, fatty acids, and amino acids. Malfunction of this cycle deprives the body of energy. In pyruvate dehydrogenase deficiency, pyruvate, which is derived from the breakdown of carbohydrates, is not converted into ACoA. This decreases the major substrate necessary for the CAC to function and results in an abnormal buildup of lactate and alanine, which are alternative metabolic products of pyruvate. Without function of the CAC, the mitochondria cannot produce energy for the cells to function. Lack of energy product and the buildup of unusable metabolites results in nonspecific symptoms (eg, severe lethargy, poor feeding, tachypnea), especially during times of illness, stress, or high carbohydrate intake.
In pyruvate dehydrogenase deficiency, progressive neurological symptoms usually start in infancy but may be evident at birth or in later childhood and very rarely in adulthood. Symptoms may include developmental delay, intermittent ataxia, poor muscle tone, abnormal eye movements, or seizures. Childhood- and adult-onset forms of this disorder are often associated with intermittent periods of decompensation but normal or mildly delayed neurological development.
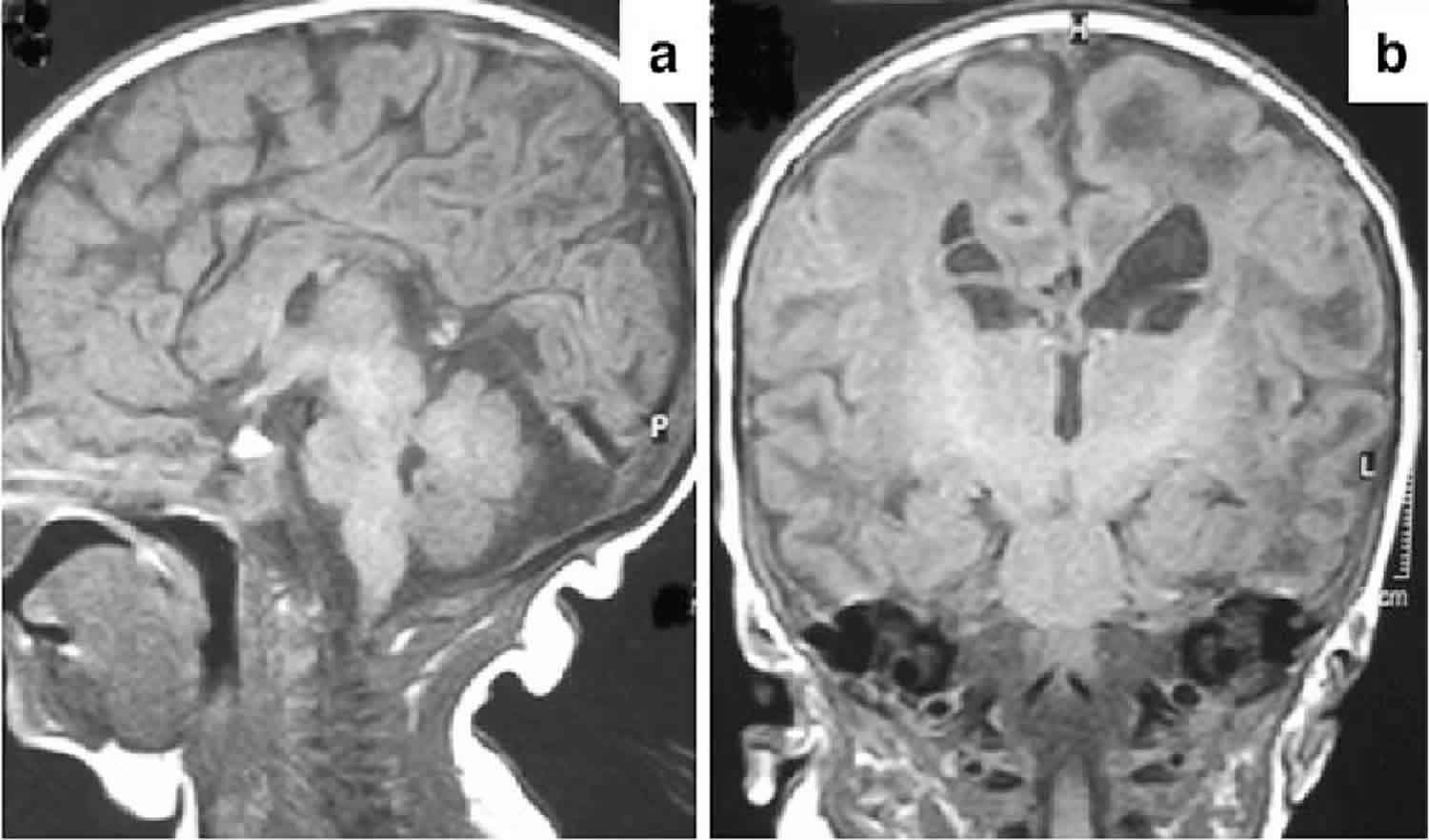
The key feature of this condition is gray matter degeneration with foci of necrosis and capillary proliferation in the brainstem in many but not all patients. The group of disorders that result in this pathology are termed Leigh syndrome. Defects in one of many of the mitochondrial enzymes involved in energy metabolism may demonstrate similar brain pathology. Leigh syndrome may be caused by mitochondrial defects other than pyruvate dehydrogenase deficiency.
Therapies are suboptimal for most forms of pyruvate dehydrogenase complex deficiency; although resolution of the lactic acidosis may occur, cessation of the underlying progressive neurological damage is rare. Some affected individuals respond to treatment with thiamine (vitamin B1), carnitine or lipoic acid. Thiamine may need to be given in high doses to be effective. A diet low in carbohydrates and high in fat (ketogenic diet) has been used to treat the symptoms of pyruvate dehydrogenase deficiency but is not always successful. Oral citrate is often used to treat acidosis
Genetic counseling is recommended for families of children with pyruvate dehydrogenase complex deficiency.
Figure 1. Citric acid cycle
Pyruvate dehydrogenase deficiency causes
The genes involved in pyruvate dehydrogenase deficiency each provide instructions for making a protein that is a component of a group of proteins called the pyruvate dehydrogenase complex. The pyruvate dehydrogenase complex plays an important role in the pathways that convert the energy from food into a form that cells can use. The pyruvate dehydrogenase complex converts a molecule called pyruvate, which is formed from the breakdown of carbohydrates, into another molecule called acetyl-coenzyme A (acetyl-CoA). This conversion is essential to begin the series of chemical reactions that produce energy for cells.
The pyruvate dehydrogenase complex is made up of multiple copies of several enzymes called E1, E2, and E3, each of which performs part of the chemical reaction that converts pyruvate to acetyl-CoA. In addition, other proteins included in the complex ensure its proper function. One of these proteins, E3 binding protein, attaches E3 to the complex and provides the correct structure for the complex to perform its function. Other associated proteins control the activity of the complex: pyruvate dehydrogenase phosphatase turns on (activates) the complex, while pyruvate dehydrogenase kinase turns off (inhibits) the complex.
The E1 enzyme, also called pyruvate dehydrogenase, is composed of four parts (subunits): two alpha subunits (called E1 alpha) and two beta subunits (called E1 beta). Mutations in the gene that provides instructions for making E1 alpha, the PDHA1 gene, are the most common cause of pyruvate dehydrogenase deficiency, accounting for approximately 80 percent of cases. These mutations lead to a shortage of E1 alpha protein or result in an abnormal protein that cannot function properly. A decrease in functional E1 alpha leads to reduced activity of the pyruvate dehydrogenase complex. The gene responsible for this form of pyruvate dehydrogenase deficiency has been located on the short arm of the X chromosome (Xp22.2-Xp22.1) and is known as the E1-alpha subunit pyruvate dehydrogenase gene (PDHA1). There are many different variations (allelic variants) of this gene that can cause pyruvate dehydrogenase deficiency. Most individuals with a PDHA1 gene mutation have a new gene mutation that is not inherited.
Other components of the pyruvate dehydrogenase complex are also involved in pyruvate dehydrogenase deficiency. Mutations in the genes that provide instructions for E1 beta (the PDHB gene), the E2 enzyme (the DLAT gene), E3 binding protein (the PDHX gene), and pyruvate dehydrogenase phosphatase (the PDP1 gene) have been identified in people with this condition. Although it is unclear how mutations in each of these genes affect the complex, reduced functioning of one component of the complex appears to impair the activity of the whole complex. As with PDHA1 gene mutations, changes in these other genes lead to a reduction of pyruvate dehydrogenase complex activity.
With decreased function of this complex, pyruvate builds up and is converted in another chemical reaction to lactic acid. The excess lactic acid causes lactic acidosis in affected individuals. In addition, the production of cellular energy is diminished. The brain, which requires especially large amounts of energy, is severely affected, resulting in the neurological problems associated with pyruvate dehydrogenase deficiency.
Pyruvate dehydrogenase deficiency inheritance pattern
Pyruvate dehydrogenase deficiency can have different inheritance patterns. When the condition is caused by mutations in the PDHA1 gene, it is inherited in an X-linked pattern. The PDHA1 gene is located on the X chromosome, which is one of the two sex chromosomes. In males, who have only one X chromosome, a mutation in the only copy of the gene in each cell is sufficient to cause the condition. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
In females, who have two copies of the X chromosome, one altered copy of the PDHA1 gene in each cell can lead to less severe features of the condition or may cause no signs or symptoms at all. However, many females with one altered copy of this gene have pyruvate dehydrogenase deficiency similar to affected males because the X chromosome with the normal copy of the PDHA1 gene is turned off through a process called X-inactivation. Early in embryonic development in females, one of the two X chromosomes is permanently inactivated in somatic cells (cells other than egg and sperm cells). X-inactivation ensures that females, like males, have only one active copy of the X chromosome in each body cell. Usually X-inactivation occurs randomly, such that each X chromosome is active in about half of the body cells. Sometimes X-inactivation is not random, and one X chromosome is active in more than half of cells. When X-inactivation does not occur randomly, it is called skewed X-inactivation.
Research shows that females with pyruvate dehydrogenase deficiency caused by mutation of the PDHA1 gene often have skewed X-inactivation, which results in the inactivation of the X chromosome with the normal copy of the PDHA1 gene in most cells of the body. This skewed X-inactivation causes the chromosome with the mutated PDHA1 gene to be expressed in more than half of cells.
Some pyruvate dehydrogenase deficiency cases are caused by a mutation in a gene in another subunit of the pyruvate dehydrogenase complex. One of the genes responsible for this form of pyruvate dehydrogenase deficiency has been mapped to the short arm of the 11th chromosome (11p13) and has been termed PDHX. Others include the PDHB located on the short arm of the 3rd chromosome (3p21.1-p14.2), DLAT on the long arm of the 11th chromosome (11q23.1), PDP1 located on the long arm of the 8th chromosome (8q22.1), and DLD located on the long arm of the 7th chromosome (7q31-q32). The gene mutations in these subunits are usually inherited and follow autosomal recessive inheritance, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. The risk for two carrier parents to both pass the defective gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Pyruvate dehydrogenase deficiency symptoms
Signs and symptoms of pyruvate dehydrogenase complex deficiency usually first appear shortly after birth, and they can vary widely among affected individuals. The most common feature is a potentially life-threatening buildup of lactic acid (lactic acidosis), which can cause poor feeding, nausea, vomiting, lethargy, severe breathing problems and rapid breathing (tachypnea) and an abnormal heartbeat in an infant. People with pyruvate dehydrogenase deficiency usually have neurological problems as well. Neurologic symptoms are progressive and usually start in infancy but may even be apparent at birth. Most have delayed development of mental abilities and motor skills such as sitting and walking. Other neurological problems can include intellectual disability, seizures, poor muscle tone (hypotonia), poor coordination (ataxia), difficulty walking, abnormal eye movements and poor visual tracking. Some affected individuals have abnormal brain structures, such as underdevelopment of the tissue connecting the left and right halves of the brain (corpus callosum), wasting away (atrophy) of the exterior part of the brain known as the cerebral cortex, or patches of damaged tissue (lesions) on some parts of the brain. Because of the severe health effects, many individuals with pyruvate dehydrogenase deficiency do not survive past childhood, although some may live into adolescence or adulthood.
Individuals with the early childhood-onset form of pyruvate dehydrogenase deficiency may have normal neurologic development with intermittent periods of ataxia, often associated with upper respiratory infection or other minor stress. Varying degrees of neurologic deficits and mental retardation may occur in individuals with pyruvate dehydrogenase deficiency.
Pyruvate dehydrogenase deficiency diagnosis
Pyruvate dehydrogenase complex deficiency is suspected in people who have lactic acidosis or signs of early-onset neurological disease such as seizures, lethargy, and poor feeding.
Biochemical abnormalities may vary from severe acidosis due to abnormally high levels of lactic acid appearing shortly after birth to a mildly elevated level which usually follows a meal high in carbohydrates. In some cases elevation of blood lactate levels is seen only during the acute episodes. Excretion of abnormally large amounts of the amino acid alanine (alaninuria) may occur only during acute episodes.
A doctor may wish to order more tests including:
- Brain magnetic resonance imaging (MRI) and magnetic resonance spectroscopy (MRS) to check for brain damage
- Blood test to measure levels of lactic acid or pyruvate
- Tests to measure levels of lactic acid or pyruvate in the fluid surrounding the brain and spinal cord (cerebrospinal fluid)
- Blood test or urine test to analyze levels of the amino acid alanine
A diagnosis of pyruvate dehydrogenase complex deficiency can be confirmed by testing the activity of the pyruvate dehydrogenase complex and the activity of all of the specific enzymes within the complex. This can be completed by sampling the blood leukocytes, skin (fibroblast), or muscle biopsy 3. Genetic testing may be used to confirm the diagnosis 4.
The following studies are indicated in pyruvate dehydrogenase complex deficiency:
Lactate and pyruvate levels
High blood lactate and pyruvate levels with or without lactic acidemia suggest an inborn error of metabolism at the mitochondrial level.
Cerebrospinal fluid also shows elevation of lactate and pyruvate (at times even in the absence of elevated blood levels).
In mild cases of pyruvate dehydrogenase complex deficiency, blood lactate and pyruvate levels may be elevated only slightly or not at all under normal conditions; elevated levels are usually found during periods of crisis.
A recent study suggests that the lactate-to-pyruvate ratio is only diagnostically useful to differentiate pyruvate dehydrogenase complex deficiency from other forms of congenital lactic acidosis at higher lactate levels (>5 mmol/L) 5.
Serum and urine analysis
Serum and urine amino acid analyses reveal hyperalaninemia.
Deficiency of the E3 enzyme also causes an elevation in branched-chain amino acids in the serum and alpha-ketoglutarate in the serum and urine.
Amino acid levels vary with the general metabolic state of the patient; a catabolic state, in which gluconeogenesis is activated and proteins are degraded, elevates many amino acids, leading to a nonspecific amino acid profile.
Hyperammonemia and nonspecific amino acid elevation are associated with E2 enzyme deficiency, which is more common during acute illnesses.
Thiamine pyrophosphate-adenosine triphosphate phosphoryl transferase inhibitor can be detected in urine or blood by a specific assay.
Other studies
Definitive diagnosis is made by showing abnormal enzyme function.
Functional assays can be performed on leukocytes, fibroblasts, or properly preserved tissue samples. Pyruvate dehydrogenase complex activity should be measured with and without thiamine in order to detect cases of thiamine-responsive pyruvate dehydrogenase complex deficiency.
Blood and fibroblast enzyme tests are the easiest to obtain, but mosaicism can cause normal enzymatic activity in leukocytes and fibroblasts, requiring a tissue biopsy if the diagnosis is strongly suspected.
A skin sample grows if obtained within 2 days of death.
Imaging studies
MRI
MRI shortly after birth may reveal ventricular dilation, cerebral atrophy, hydranencephaly, partial or complete absence of the corpus callosum, absence of the medullary pyramids, or abnormal and ectopic inferior olives.
MRI of infants with progressive neurological symptoms may reveal symmetric cystic lesions and gliosis in the cortex, basal ganglia, brainstem, or cerebellum, or generalized hypomyelination.
Individuals with a deficiency in the E2 subunit may reveal discrete lesions restricted to the globus pallidus.
Magnetic resonance spectroscopy
Magnetic resonance spectroscopy (MRS) of the brain reveals high lactate levels in individuals with pyruvate dehydrogenase complex deficiency.
N -acetylaspartate and choline levels are consistent with hypomyelination.
Pyruvate dehydrogenase deficiency treatment
The goal of the treatment for pyruvate dehydrogenase complex deficiency is to stimulate the pyruvate dehydrogenase complex to produce as much energy as possible. This can prevent immediate worsening of the disease 6. Treatment options typically include supplementing cofactors including carnitine, thiamine, and lipoic acid. These are substances in the body that help the chemical reactions in the cells occur 4. Administration of thiamine and lipoic acid cofactors to all patients with pyruvate dehydrogenase complex deficiency is done to optimize pyruvate dehydrogenase complex function, and carnitine is given to facilitate fatty acid transport into mitochondria and to potentially increase cellular ATP production. Certain genetic changes (mutations or pathogenic variants) that cause pyruvate dehydrogenase complex deficiency may be more responsive to thiamine treatment than others 7. Thiamine-responsive cases are more likely in children who are diagnosed at older than 1 year, and high-dose thiamine (400 mg/day) should be continued in patients who are responsive clinically and chemically 8.
Doctors may also recommend a ketogenic diet with restricted carbohydrate intake that is high in fats and low in carbohydrates. This can help prevent lactic acidosis but typically does not stop neurological symptoms 4. Medications to help prevent seizures may be recommended for some people with pyruvate dehydrogenase complex deficiency 9.
A medication called dichloroacetate may help treat some people with pyruvate dehydrogenase complex deficiency. Dichloroacetate reduces the inhibitory phosphorylation of pyruvate dehydrogenase complex. Resolution of lactic acidosis is observed in patients with E1 alpha enzyme subunit mutations that reduce enzyme stability. Oral dichloroacetate administered for 6 months was found to be well tolerated and blunted the postprandial increase in circulating lactate but did not improve neurologic or other clinical measures 10.
Studies with human fibroblast have demonstrated that certain gene deletions are more response to dichloroacetate than others.
Other lactic acidemias have been treated successfully with this compound.
Long-term use is associated with reversible peripheral neuropathy and elevation in liver transaminases.
Coadministration of thiamine appears to protect against neuropathy in animals.
Because of the largely unknown benefit of this compound, it remains an investigational drug.
Oral citrate is often used to treat acidosis.
Genetic counseling for the parents of the individual with pyruvate dehydrogenase complex deficiency is important in order to estimate the recurrence risk for future pregnancies.
Progressive renal failure is common in pyruvate dehydrogenase complex deficiency. A nephrologist should be consulted if signs of renal failure are evident.
Pyruvate dehydrogenase deficiency diet
Limit carbohydrate administration to 3-4 mg per kg body weight to prevent lactate buildup. The appropriate oral carbohydrate intake depends on the residual enzyme activity and must be individually treated. It may be as low as 10-20 carbohydrate calories per kg carbohydrate.
A ketogenic diet may be indicated. Ketogenic diets minimize the carbohydrate content and maximize the daily intake of fat content.
Fat intake should account for 65-80% of the caloric intake, with protein accounting for about 10% of the caloric intake and carbohydrate caloric intake making up the balance.
Manipulate the percent of dietary fat and carbohydrate calories to provide an appropriate lactic acid level.
Although the ketogenic diet may reduce the blood lactic acid level and extend lifespan, CNS metabolic abnormalities persist, as evidenced by high lactic acid levels in the cerebrospinal fluid and progressive neurological degeneration.
The vulnerability of the CNS is a result of its dependence on glucose as a fuel. However, the brain will change glucose to lipid energy sources after a few days of a ketogenic diet.
Pyruvate dehydrogenase deficiency prognosis
The prognosis of pyruvate dehydrogenase deficiency is proportional to residual pyruvate dehydrogenase complex activity. In general, the lower the pyruvate dehydrogenase complex activity, the earlier the onset and the more severe the disease progression. Since the most common form of the pyruvate dehydrogenase deficiency is X-linked recessive, carrier females can have a milder form of the disease, depending on variable X inactivation, whereas males with the X-linked form are more affected. Prognosis also significantly related to whether the form of pyruvate dehydrogenase deficiency is responsive to thiamine or lipoic acid.
Unfortunately, the long-term outlook for people with pyruvate dehydrogenase complex deficiency is poor. People who show signs and symptoms early in life may pass away from complications of the disease in the first years of life. If people with pyruvate dehydrogenase deficiency survive, they may be affected with complications including intellectual disability 6. People who have symptoms of PDC deficiency beginning in late childhood may survive longer because they have higher levels of functioning enzymes in the body. However, these individuals can still have complications of the disease 4.
Individuals with mild deficiencies in the E1 enzyme of the pyruvate dehydrogenase complex have a better prognosis than those with deficiencies in the E2 and E3 pyruvate dehydrogenase complex enzymes.
Prediction of prognosis is unclear because of the small number of children with pyruvate dehydrogenase complex deficiency studied and the large number of mutations involved. Complications include Leigh syndrome, seizures, and central nervous system (CNS) deterioration in many patients with pyruvate dehydrogenase deficiency, and there is a high morbidity and mortality rate 11.
In most cases of neonatal-onset and infantile-onset of pyruvate dehydrogenase complex deficiency, a poor prognosis remains, even when the lactic acidosis is treated successfully. Although lactic acidosis appears to be controlled by thiamine supplementation in individuals who respond to thiamine, the neurological outcome may be poor.
One case report describes cessation of neurological demyelination with the ketogenic diet; however, the ketogenic diet has not been reported to be of significant neurologic benefit to other patients with pyruvate dehydrogenase complex deficiency 12.
Dichloroacetate appears to produce biochemical correction of pyruvate dehydrogenase complex deficiency in many cases, but resolution of neurologic symptoms is exceptional. Structural CNS abnormalities likely cannot be reversed with successful biochemical treatment.
Dichloroacetate may have greater efficacy with particular mutations of the E1 subunit.
In general, treatment of individuals with pyruvate dehydrogenase complex deficiency is most beneficial if started early. Although successful treatment is rare, some cases have been reported.
Although the recurrence rate for subsequent pregnancies is low, test future gestations for pyruvate dehydrogenase complex deficiency because of the possibility of germline mosaicism. Enzyme activity of cultured chorionic villus cells can be determined in time to make an early diagnosis. Inaccuracies in the diagnosis of the female fetus arise from X chromosome inactivation.
Individuals with an E2 subunit deficiency may have a mild phenotype.
References- Pyruvate Dehydrogenase Complex Deficiency. https://rarediseases.org/rare-diseases/pyruvate-dehydrogenase-complex-deficiency/
- Pyruvate dehydrogenase deficiency. https://ghr.nlm.nih.gov/condition/pyruvate-dehydrogenase-deficiency
- Shin HK, Grahame G, McCandless SE, Kerr DS, and Bedoyan JK. Enzymatic testing sensitivity, variability, and practical diagnostic algorithm for pyruvate dehydrogenase complex (PDC) deficiency. Molecular Genetics and Metabolism. November 2017; 122(3):61-66.
- Pyruvate Dehydrogenase Deficiency (PDCD). https://emedicine.medscape.com/article/948360-overview
- Debray FG, Mitchell GA, Allard P, Robinson BH, Hanley JA, Lambert M. Diagnostic accuracy of blood lactate-to-pyruvate molar ratio in the differential diagnosis of congenital lactic acidosis. Clin Chem. 2007 May. 53(5):916-21.
- Brown GK, Otero LJ, LeGris M, and Brown RM. Pyruvate dehydrogenase deficiency. Journal of Medical Genetics. 1994; 31:875-879. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1016663/pdf/jmedgene00001-0059.pdf
- Jauhari P, Sankhyan N, Vyas S, and Singhi P. Thiamine Responsive Pyruvate Dehydrogenase Complex Deficiency: A Potentially Treatable Cause of Leigh’s Disease. Journal of Pediatric Neurosciences. July-September 2017; 12(3):265-267. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5696666
- van Dongen S, Brown RM, Brown GK, Thorburn DR, Boneh A. Thiamine-responsive and non-responsive patients with PDHC-E1 deficiency: a retrospective assessment. JIMD Rep. 2014 Apr 10.
- Pyruvate Dehydrogenase Complex Deficiency (PDCD/PDH). https://www.umdf.org/types/pyruvate-dehydrogenase-complex-deficiency/
- Stacpoole PW, Kerr DS, Barnes C, Bunch ST, Carney PR, Fennell EM. Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics. 2006 May. 117(5):1519-31.
- Patel KP, O’Brien TW, Subramony SH, Shuster J, Stacpoole PW. The spectrum of pyruvate dehydrogenase complex deficiency: clinical, biochemical and genetic features in 371 patients. Mol Genet Metab. 2012 Jan. 105(1):34-43.
- Weber TA, Antognetti MR, Stacpoole PW. Caveats when considering ketogenic diets for the treatment of pyruvate dehydrogenase complex deficiency. J Pediatr. 2001 Mar. 138(3):390-5.





{kind=link}