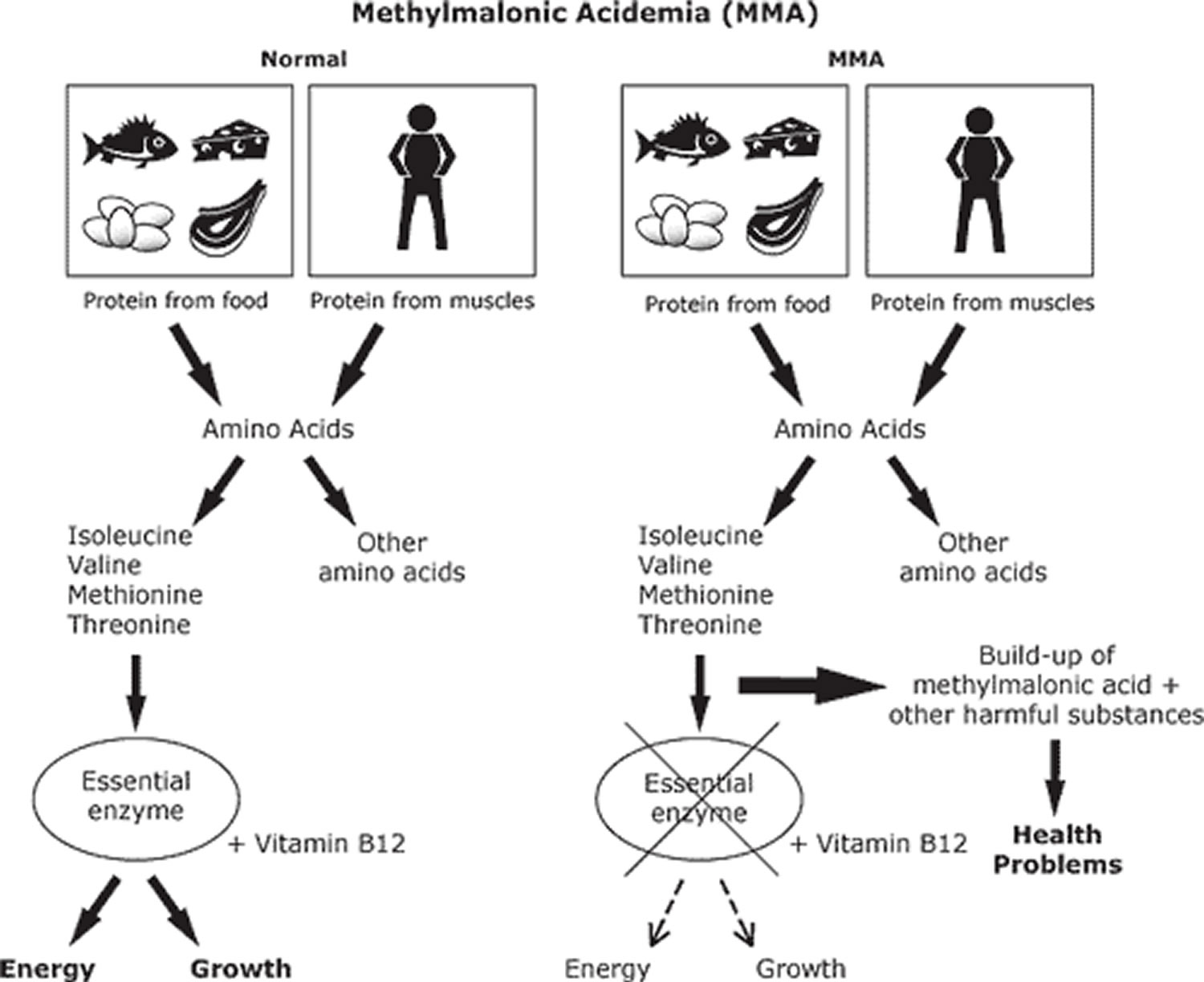
Methylmalonic acidemia
Methylmalonic acidemia also called methylmalonic aciduria (MMA), is an inherited autosomal recessive disorder of amino acid metabolism in which the body is unable to process certain proteins (methylmalonyl-coenzyme A (CoA) to succinyl-CoA) and fats (lipids) properly. Isolated methylmalonic acidemia or methylmalonic aciduria is caused by complete (mut 0 enzymatic subtype) or partial deficiency of the enzyme methylmalonyl-CoA mutase (mut– enzymatic subtype), a defect in the transport or synthesis of its cofactor, adenosyl-cobalamin (cblA, cblB, or cblD-MMA), or deficiency of the enzyme methylmalonyl-CoA epimerase 1. Vitamin B12 deficiency states that are not due to genetic causes, such as vitamin B12 deficiency, can also cause methylmalonic acid to build up in the body.
The effects of methylmalonic acidemia vary from mild to life-threatening, usually appear in early infancy (age of 1 month to 1 year). Affected infants can experience vomiting, dehydration, weak muscle tone (hypotonia), developmental delay, excessive tiredness (lethargy), an enlarged liver (hepatomegaly), and failure to gain weight and grow at the expected rate (failure to thrive). Long-term complications can include feeding problems, neurologic manifestations, such as seizure, encephalopathy, stroke and intellectual disability, chronic kidney disease, and inflammation of the pancreas (pancreatitis). Several cases have involved stroke in the bilateral globus pallidi as a result of methylmalonic acidemia.
Reduced blood flow or faulty oxidative metabolism may cause strokes in methylmalonic acidemia. The sequence of events in reduced blood flow may be acidosis, hypocapnia, and vasoconstriction. Several magnetic resonance spectroscopic studies have shown that lactate accumulates in areas of the brain that are damaged in methylmalonic acidemia.
Some authors suggest that the accumulation of methylmalonic acid and odd-chain fatty acids may be directly toxic to neuronal and glial cells. This toxic effect may impair oxidative metabolism, leading to infarctions. An alternate hypothesis suggests that toxic metabolites may result from treatment with cyanocobalamin, which metabolizes to cyanide, a known central nervous system toxin.
Based on reports of liver transplantation reports meant to address the issue of metabolic derangement in methylmalonic acidemia, the neurologic consequences of methylmalonic acidemia may not be a result of metabolic abnormalities in the liver; rather, they may be a local metabolic disturbance in the brain. Liver transplantation did not prevent further neurologic worsening or occurrence of strokelike episodes 2.
Without treatment, methylmalonic acidemia can lead to coma and death in some cases.
Methylmalonic acidemia occurs in an estimated 1 in 50,000 to 100,000 people 3.
If the patient’s family or sibling history suggests a diagnosis of acidemia, prenatal and neonatal diagnosis must be pursued aggressively. Early diagnosis and treatment may delay the progression of symptoms.
The diet of children with methylmalonic acidemias must be carefully controlled. Treatment includes a low-protein diet and avoidance of the amino acids isoleucine, valine, threonine and methionine because these substances can turn into methylmalonic acid in an affected patient. To assure a balanced diet, certain medical foods must be fed to affected children. Most patients also need to take a special formula missing certain amino acids but containing others to make sure they are getting enough protein for growth. Each patient needs an individually adjusted diet and medication regimen. Massive doses of vitamin B12 (cobalamin) are indicated in the B12-responsive variants. In the disorders of cobalamin (vitamin B12) metabolism, administration of intramuscular and/or oral hydroxycobalamin (man-made injectable form of vitamin B12) may correct the defect and restore normal metabolism.
Unfortunately, there is no cure for methylmalonic acidemia. Genetic counseling is recommended for the families of children with methylmalonic acidemias.
Methylmalonic aciduria and homocystinuria
Methylmalonic acidemia with homocystinuria is an inherited disorder in which the body is unable to properly process certain nutrients from food including amino acids, lipids and cholesterol. People with this disorder have a combination of features from two separate conditions: methylmalonic acidemia and homocystinuria. When the condition begins early in life, babies have difficulty gaining weight (failure to thrive), feeding difficulties, and a pale appearance. Babies may also have weak muscle tone (hypotonia) and seizures. Most babies and children with this condition have an unusually small head size (microcephaly), intellectual disability and developmental delay. Less common features of the condition include eye problems and a blood disorder called megaloblastic anemia 4. When methylmalonic acidemia with homocystinuria begins in adolescence or adulthood, the signs and symptoms usually include behavior and personality changes and cognitive problems (issues with learning, memory, perception etc). In some cases, abilities are lost, resulting in a decline of performance, memory and speech problems, dementia and lethargy 5.
Methylmalonic acidemia with homocystinuria can be caused by mutations in one of several genes: MMACHC, MMADHC, LMBRD1, ABCD4, or HCFC1. Mutations in these genes account for the different types of the disorder, cblC, cblD, cblF, cblJ, and cblX, respectively 4. Although there is no cure for this conditions, treatment may include intramuscular injections of hydroxycobalamin, oral betaine, and folic acid 6.
Methylmalonic acidemia causes
Mutations in the MMUT, MMAA, MMAB, MMADHC, and MCEE genes cause methylmalonic acidemia. The long term effects of methylmalonic acidemia depend on which gene is mutated and the severity of the mutation.
About 60 percent of methylmalonic acidemia cases are caused by mutations in the MMUT gene. This gene provides instructions for making an enzyme called methylmalonyl CoA mutase. This enzyme works with vitamin B12 (also called cobalamin) to break down several protein building blocks (amino acids), certain lipids, and cholesterol. Mutations in the MMUT gene alter the enzyme’s structure or reduce the amount of the enzyme, which prevents these molecules from being broken down properly. As a result, a substance called methylmalonyl CoA and other potentially toxic compounds can accumulate in the body’s organs and tissues, causing the signs and symptoms of methylmalonic acidemia.
Mutations in the MMUT gene that prevent the production of any functional enzyme result in a form of the condition designated mut0. Mut0 is the most severe form of methylmalonic acidemia and has the poorest outcome. Mutations that change the structure of methylmalonyl CoA mutase but do not eliminate its activity cause a form of the condition designated mut-. The mut- form is typically less severe, with more variable symptoms than the mut0 form.
Some cases of methylmalonic acidemia are caused by mutations in the MMAA, MMAB, or MMADHC gene. Proteins produced from the MMAA, MMAB, and MMADHC genes are needed for the proper function of methylmalonyl CoA mutase. Mutations that affect proteins produced from these three genes can impair the activity of methylmalonyl CoA mutase, leading to methylmalonic acidemia.
A few other cases of methylmalonic acidemia are caused by mutations in the MCEE gene. This gene provides instructions for producing an enzyme called methylmalonyl CoA epimerase. Like methylmalonyl CoA mutase, this enzyme also plays a role in the breakdown of amino acids, certain lipids, and cholesterol. Disruption in the function of methylmalonyl CoA epimerase leads to a mild form of methylmalonic acidemia.
It is likely that mutations in other, unidentified genes also cause methylmalonic acidemia.
Methylmalonic acidemia inheritance pattern
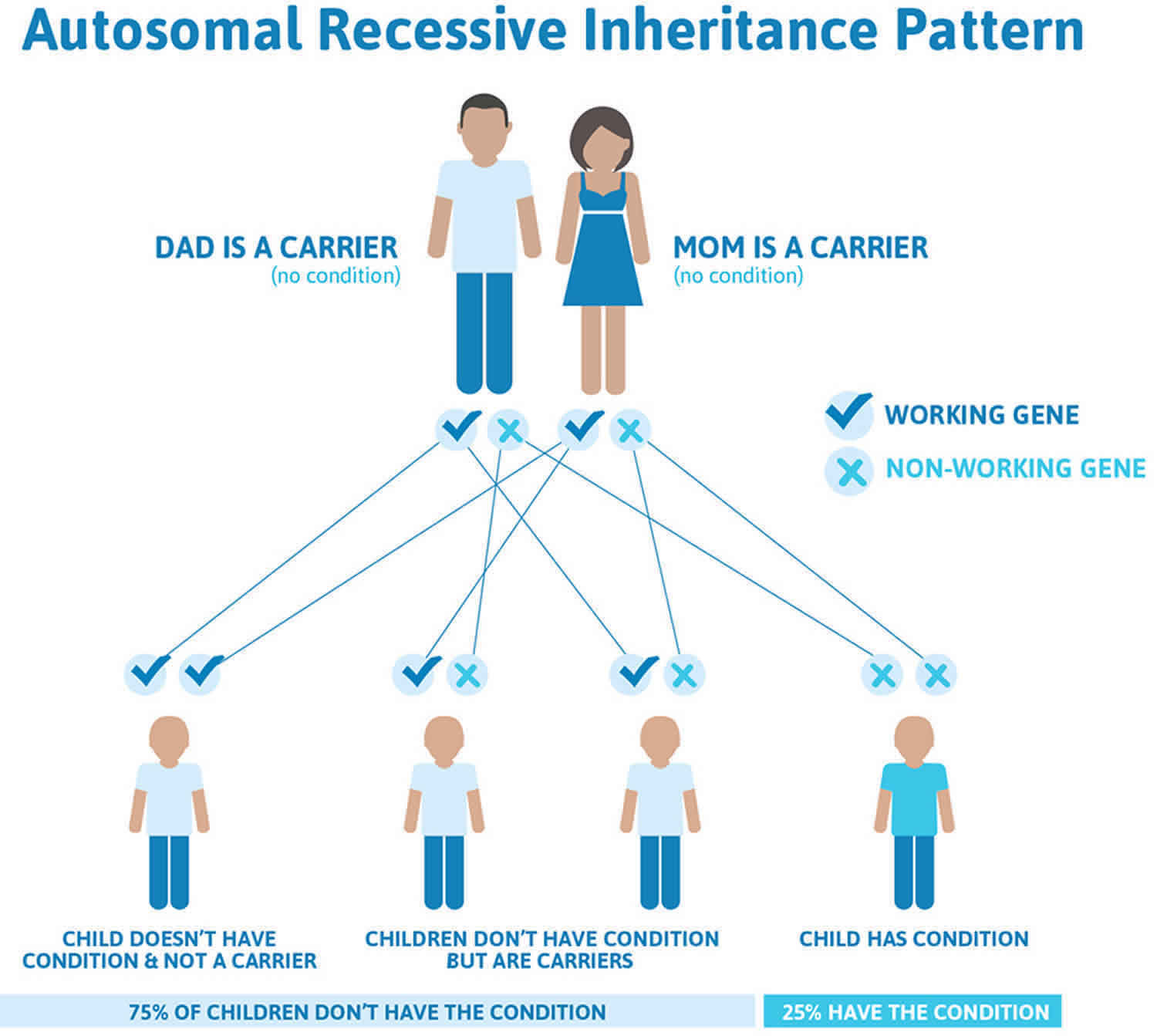
Methylmalonic acidemia is inherited in an autosomal recessive pattern, which means both copies of the MMUT, MMAA, MMAB, MMADHC, or MCEE gene in each cell have mutations. Most often, the parents of an individual with an autosomal recessive condition are carriers of one copy of the mutated gene but do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 1 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 1. Methylmalonic acidemia autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Methylmalonic acidemia symptoms
Children with methylmalonic acidemia may be healthy at birth and develop symptoms soon after starting protein intake. The onset of the methylmalonic aciduria usually occurs during the first few months of life although onset to late childhoods has been described. The patient’s family history may be positive for methylmalonic acidemia (eg, siblings with similar episodes of recurrent illnesses or with acidopathy). Symptoms may include lethargy, failure to thrive, recurrent vomiting, acidosis, dehydration, respiratory distress, diminished muscle tone, developmental retardation, seizures and/or an enlarged liver.
Signs may include hypotonia, lethargy, failure to thrive, hepatosplenomegaly, and monilial infections are some classic findings. In patients with methylmalonic acidemia, acute onset of choreoathetosis, dystonia, dysphagia, or dysarthria should alert the physician to the possibility of stroke. Neurologic manifestations may be present, even in the absence of more traditional findings.
The majority of methylmalonic acidemia onset from neurological damage, but some cases may first onset with unusal presentations such as mimicked diabetic ketoacidosis, late-onset diffuse lung disease, and juvenile gout 7.
Onset of the manifestations of isolated methylmalonic acidemia or methylmalonic acidemia ranges from the neonatal period to adulthood. All phenotypes are characterized by periods of relative health and intermittent metabolic decompensation, usually associated with intercurrent infections and stress.
- In the neonatal period the disease can present with lethargy, vomiting, hypotonia, hypothermia, respiratory distress, severe ketoacidosis, hyperammonemia, neutropenia, and thrombocytopenia and can result in death within the first four weeks of life.
- In the infantile/non-B12-responsive phenotype, infants are normal at birth, but develop lethargy, vomiting, dehydration, failure to thrive, hepatomegaly, hypotonia, and encephalopathy within a few weeks to months of age.
- An intermediate B12-responsive phenotype can occasionally be observed in neonates, but is usually observed in the first months or years of life; affected children exhibit anorexia, failure to thrive, hypotonia, and developmental delay, and sometimes have protein aversion and/or vomiting and lethargy after protein intake.
- Atypical and “benign”/adult methylmalonic acidemia phenotypes are associated with increased, albeit mild, urinary excretion of methylmalonate.
In most children, the disease is diagnosed in the middle of an episode of metabolic decompensation 8. Vomiting, dehydration, lethargy, seizures, recurrent infections, and progressive encephalopathy are some features of methylmalonic acidemia. These metabolic perturbations can be caused by an infection or a change in feeding habit. Some children may present with strokes during a metabolic crisis.
Methylmalonic acidemia due to derangement of adenosylcobalamin synthesis (cblA, cblB, cblH) and cobalamin catabolism (cblC, cblD, cblF) may have features not shared by pure methylmalonyl-CoA mutase disorders.
A case study of a 4-year-old girl with methylmalonic acidemia with partial clbC subtype showed that onset of disease can be following aHUS as opposed to neurological damage which is more common 7.
There have been cases of late-onset cblC form of disease reported. Most presented with neurological symptoms including cognitive decline, hypertensive encephalopathy, unsteady gait, myelopathy, and behavioral abnormalities. One study reports two siblings with late-onset cblC which presented with manic-depressive psychosis at the first onset. As this late-onset form of methylmalonic acidemia is rare, inclusion of psychiatric symptoms as a first symptom may help yield higher diagnostic results 9.
Major secondary complications of methylmalonic acidemia include: intellectual impairment (variable); tubulointerstitial nephritis with progressive renal failure; “metabolic stroke” (acute and chronic basal ganglia injury) causing a disabling movement disorder with choreoathetosis, dystonia, and para/quadriparesis; pancreatitis; growth failure; functional immune impairment; and optic nerve atrophy.
Signs, symptoms, and nonspecific presentation generally making the diagnosis of methylmalonic acidemia difficult.
Laboratory findings include an abnormally high amount of methylmalonic acid in the blood and urine. Metabolic acidosis also occurs. Elevated levels of ketone bodies such as acetone in the blood (ketonemia) or in the urine (ketonuria) may develop. An elevated level of ammonia in the blood (hyperammonemia) may also be present. Excessive levels of the amino acid, glycine in the blood (hyperglycinemia) and in the urine (hyperglycinuria) is found. The concentration of white blood cells, blood platelets and red blood cells may be lower than normal. Low blood sugar (hypoglycemia) may also occur.
Methylmalonic acidemia diagnosis
Signs, symptoms, and nonspecific presentation generally making the diagnosis of methylmalonic acidemia difficult. Methylmalonic acidemias can usually be diagnosed before birth (prenatally) by measuring the concentration of methylmalonic acid in amniotic fluid or activity of the deficient enzyme in fluid or tissue samples obtained from the fetus or uterus during pregnancy (amniocentesis or chorionic villus sampling [CVS]). During amniocentesis, a sample of fluid surrounding the developing fetus is removed and analyzed. Chorionic villus sampling (CVS) involves the removal and examination of tissue from a portion of the placenta. The disorder can be identified at birth through expanded newborn screening with tandem mass spectrometry.
In most affected infants, methylmalonic aciduria is diagnosed or confirmed in the first weeks of life, based upon a thorough clinical evaluation, a detailed patient and family history, and a variety of specialized tests. Laboratory studies (assays) are typically conducted on certain white blood cells (leukocytes) or cultured skin cells (fibroblasts) to confirm deficient activity of the deficient enzyme. Additional laboratory studies may reveal excessive levels of acids and increased accumulations of ketone bodies in bodily tissues and fluids (ketoacidosis), increased levels of glycine in the blood and urine (hyperglycinemia and hyperglycinuria), high levels of ammonia in the blood (hyperammonemia), and/or decreased levels of circulating platelets and white blood cells (thrombocytopenia and neutropenia).
Diagnosis of isolated methylmalonic acidemia relies on analysis of organic acids in plasma and/or urine by gas-liquid chromatography and mass spectrometry. Establishing the specific subtype of methylmalonic acidemia requires cellular biochemical studies (including C 14 propionate incorporation and B12 responsiveness, complementation analysis, and cobalamin distribution assays) and molecular genetic testing. The finding of biallelic pathogenic variants in one of the five genes (MMUT, MMAA, MMAB, MCEE, and MMADHC) associated with isolated methylmalonic acidemia – with confirmation of carrier status in the parents – can establish the diagnosis.
Methylmalonic acidemia treatment
Unfortunately, there is no cure for methylmalonic acidemia. Critically ill individuals are stabilized by restoring volume status and acid-base balance; reducing or eliminating protein intake (decreases the key amino acids eg, isoleucine, valine, threonine, methionine); providing increased calories via high glucose-containing fluids and insulin to arrest catabolism; and monitoring serum electrolytes and ammonia, venous or arterial blood gases, and urine output. Management includes a high-calorie diet low in propiogenic amino acid precursors; hydroxocobalamin intramuscular injections; carnitine supplementation; antibiotics such as neomycin or metronidazole to reduce propionate production from gut flora; gastrostomy tube placement as needed; and aggressive treatment of infections. Other therapies used in a limited number of patients include N-carbamylglutamate for the treatment of acute hyperammonemic episodes; liver, kidney, or combined liver and kidney transplantation; and antioxidants for the treatment of optic nerve atrophy.
Patients with methylmalonic acidemia are prone to anorexia and often need a temporary or permanent method for enteral feeding. When appropriate, tube feeding should be used 10.
Levo-carnitine (L-carnitine) is a dietary supplement that is also used to treat all patients with methylmalonic acidemia, who apparently have a relative carnitine deficiency. The D-isomer of carnitine may not be therapeutic.
Medical management of methylmalonic acidemia
Treatment strategies for methylmalonic acidemia are designed to reduce metabolic poisons and/or to accelerate their clearance 7.
Implement a protein-restricted diet (0.5-1.5 g/kg/d) with L-carnitine and cobalamin supplementation.
Cobalamin supplementation may help because cobalamin is a cofactor in the enzymatic conversion of methylmalonyl-coenzyme A (CoA) to succinyl-CoA. This therapy can be started while the diagnosis is being confirmed. If cobalamin supplementation is not helpful, restrict the patient’s isoleucine, threonine, methionine, and valine intake. The typical formulation and starting dosage is hydroxocobalamin 1000 μg intramuscular once daily.
L-carnitine, an enzyme involved in the metabolism of long-chain fatty acids, buffers the acyl-CoA metabolites that accumulate with protein-restricted diets. Acyl-carnitine produced by this buffering action is excreted in the urine.
Response to cobalamin supplementation and dietary changes may be monitored in terms of clinical and laboratory improvement. Quantitative measurement of methylmalonic acid in the urine can monitor the success of therapy.
Medical dietary changes are often critical for therapy in inborn errors of metabolism, however, their optimal composition is under review by researchers. The combination of protein restriction and high leucine content in medical foods may have unintended effects such as imbalanced branched-chain amino acid (BCAA) content, which could lead to decreased plasma levels of valine and isoleucine and predicts impairment of brain uptake of essential amino acids 11.
Candidal infection may be the first sign that treatment adjustments are necessary.
Liver transplantation alone or in conjunction with kidney transplantation has been attempted. Organ transplantation may not prevent future neurologic damage or reverse old damage 12. Since the liver is the site of most of the metabolic conversion of propionate, replacing the liver may contribute enough enzyme activity to avert metabolic decompensation. Although liver transplant may help to protect against metabolic instability, it is not a cure and methylmalonic acidemia patients still remain at risk for long-term complications 13.
Low sodium bicarbonate levels should be treated aggressively. This formula can be used to calculate the mmol of base to provide for half-correction, which is an appropriate goal for the first 24 hours of treatment. [Desired bicarbonate level (mmol/L) – current bicarbonate level (mmol/L)] × [volume of distribution (0.7 in neonate; 0.6 in older child and adult) × body weight (kg)] × [0.5] 10.
Hemodialysis is indicated when initial ammonia levels are greater than 500μmol/L. Nephrology should be consulted in this case 10.
Regular medical screenings for known complications of methylmalonic acidemia are recommended for optimal management of disease.
Methylmalonic acidemia prognosis
Outcomes are better in patients with cobalamin-responsive disease than in those with the cobalamin-nonresponsive disease, in association with dietary changes and supplementation of carnitine and cobalamin 14. Over the last 3 decades, observations of patients have revealed that their response to treatment is correlated with their prognosis. Of the 9 recognized defects in methylmalonate metabolism, cblA has the best prognosis; mut0, the worst. The remaining classes (cblB, cblC, cblD, cblE, cblF, clbG) have intermediate prognoses. cblH is a newly identified variant of cblA.
Patients with cobalamin-responsive disease may reach some early developmental milestones, and they may have long-term prognoses better than those of the other group. However, this group remains at risk for acute decompensation, which may result in clinical signs and symptoms of globus pallidal lesions.
While transplantation may help improve metabolic stability, the central nervous system (CNS) remains at high ongoing risk regardless 15. Early transplantation may help to maintain neurological development and improve growth of patients and in one study of 13 Japanese methylmalonic acidemia patients, 10 were free of acidosis attacks following transplantation 16.
In a cross-sectional study of 35 patients from the United Kingdom, early-onset cobalamin-nonresponders had the worst outcomes, with a median survival of approximately 6 years 17. Neurologic outcomes remained unchanged despite dietary modifications and management of infections.
In a case study of 4 children with combined methylmalonic acidemia and homocysteinemia, all developed diffuse lung diseases. This study indicates that the combination of methylmalonic acidemia and homocysteinemia may be the primary cause of diffuse lung diseases in this cohort of patients. It also presents justification for combined methylmalonic acidemia and homocysteinemia to be considered in the differential diagnosis of diffuse lung diseases.
In a study of 35 families affected by methylmalonic acidemia, health-related quality of life was evaluated using the Pediatric Quality of Life Inventory (PedsQL™) parent version 18. Children with methylmalonic acidemia mut 0 enzymatic subtype had a lower mean score on the Generic Core Scales than healthy children with the most impaired domains in social and school functioning. This indicates that children living with methylmalonic acidemia have substantial impairments in quality of life, and their families also showed significant impairments in cognitive functioning, worry, family relationships, and daily activities 18.
As more cases are evaluated, better therapies for these patients will emerge.
References- Manoli I, Sloan JL, Venditti CP. Isolated Methylmalonic Acidemia. 2005 Aug 16 [Updated 2016 Dec 1]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1231
- Kasahara M, Horikawa R, Tagawa M, et al. Current role of liver transplantation for methylmalonic acidemia: a review of the literature. Pediatr Transplant. 2006 Dec. 10(8):943-7.
- Methylmalonic acidemia. https://ghr.nlm.nih.gov/condition/methylmalonic-acidemia
- Methylmalonic acidemia with homocystinuria. https://ghr.nlm.nih.gov/condition/methylmalonic-acidemia-with-homocystinuria
- Sloan JL, Carrillo N, Adams D, et al. Disorders of Intracellular Cobalamin Metabolism. 2008 Feb 25 [Updated 2018 Sep 6]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1328
- Methylmalonic acidemia with homocystinuria. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=26
- Chen M, Zhuang J, Yang J, Wang D, Yang Q. Atypical hemolytic uremic syndrome induced by CblC subtype of methylmalonic academia: A case report and literature review. Medicine (Baltimore). 2017 Oct. 96 (43):e8284
- Parini R, Furlan F, Brambilla A, Codazzi D, Vedovati S, Corbetta C, et al. Severe Neonatal Metabolic Decompensation in Methylmalonic Acidemia Caused by CblD Defect. JIMD Rep. 2013. 11:133-7.
- Wu LY, An H, Liu J, Li JY, Han Y, Zhou AH, et al. Manic-depressive Psychosis as the Initial Symptom in Adult Siblings with Late-onset Combined Methylmalonic Aciduria and Homocystinemia, Cobalamin C Type. Chin Med J (Engl). 2017 Feb 20. 130 (4):492-494.
- Aldubayan SH, Rodan LH, Berry GT, Levy HL. Acute Illness Protocol for Organic Acidemias: Methylmalonic Acidemia and Propionic Acidemia. Pediatr Emerg Care. 2017 Feb. 33 (2):142-146.
- Myles JG, Manoli I, Venditti CP. Effects of medical food leucine content in the management of methylmalonic and propionic acidemias. Curr Opin Clin Nutr Metab Care. 2018 Jan. 21 (1):42-48
- Niemi AK, Kim IK, Krueger CE, Cowan TM, Baugh N, et al. Treatment of methylmalonic acidemia by liver or combined liver-kidney transplantation. J Pediatr. 2015 Jun. 166 (6):1455-61.e1
- Manoli I, Sloan JL, Venditti CP, Adam MP, Ardinger HH, Pagon RA, et al. Isolated Methylmalonic Acidemia. 1993.
- Manoli I, Myles JG, Sloan JL, Carrillo-Carrasco N, Morava E, et al. A critical reappraisal of dietary practices in methylmalonic acidemia raises concerns about the safety of medical foods. Part 2: cobalamin C deficiency. Genet Med. 2015 Aug 13.
- Fraser JL, Venditti CP. Methylmalonic and propionic acidemias: clinical management update. Curr Opin Pediatr. 2016 Dec. 28 (6):682-693.
- Sakamoto R, Nakamura K, Kido J, Matsumoto S, Mitsubuchi H, Inomata Y, et al. Improvement in the prognosis and development of patients with methylmalonic acidemia after living donor liver transplant. Pediatr Transplant. 2016 Dec. 20 (8):1081-1086.
- Nicolaides P, Leonard J, Surtees R. Neurological outcome of methylmalonic acidaemia. Arch Dis Child. 1998 Jun. 78(6):508-12.
- Splinter K, Niemi AK, Cox R, Platt J, Shah M, Enns GM, et al. Impaired Health-Related Quality of Life in Children and Families Affected by Methylmalonic Acidemia. J Genet Couns. 2016 Oct. 25 (5):936-44





{kind=link}