What is myelin
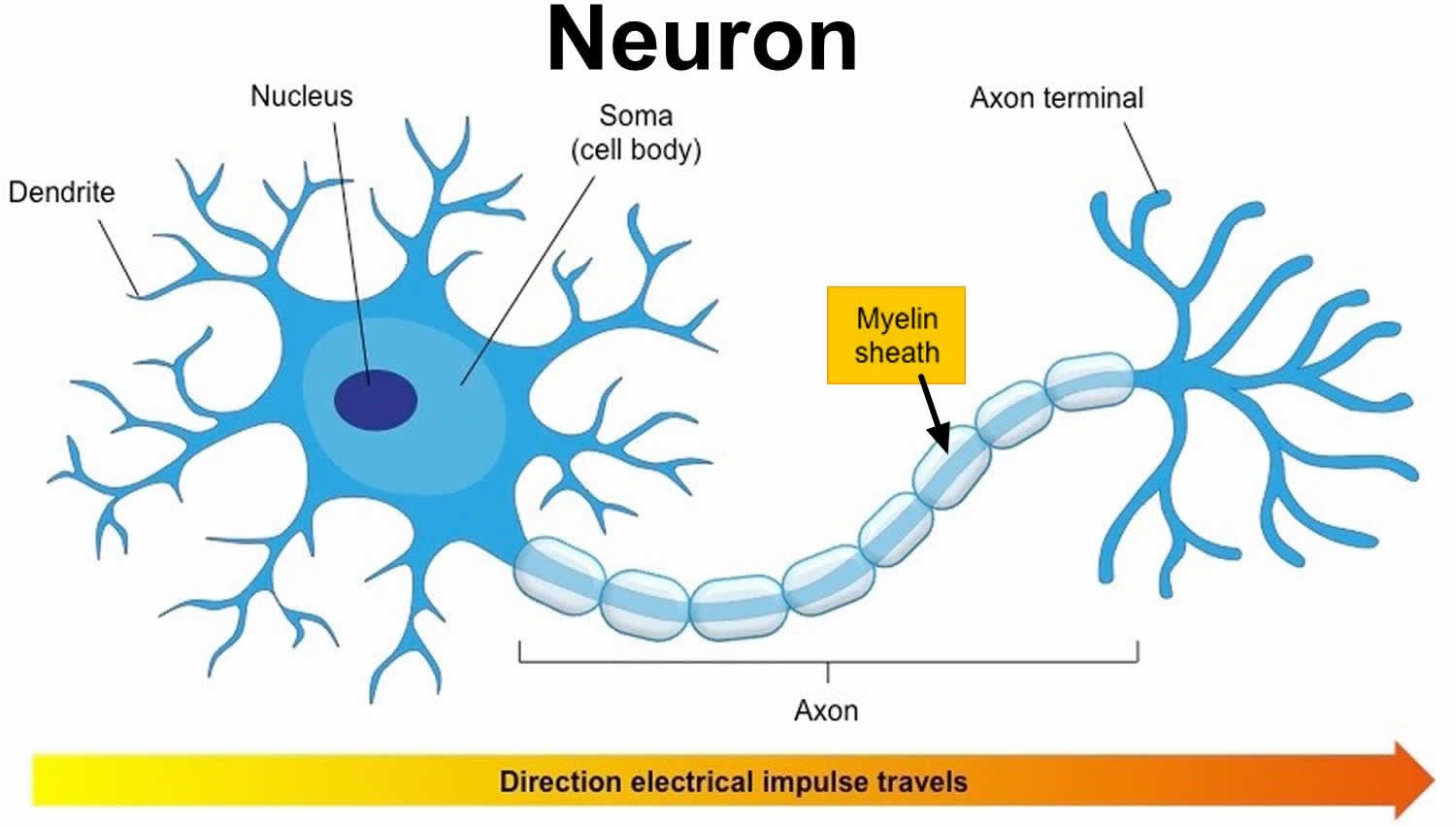
Myelin is a lipid-rich (fatty) substance and the myelin membranes originate from and are a part of the Schwann cells in the peripheral nervous system and the glial cells called oligodendrocytes in the central nervous system (CNS). Myelin is an electrical insulator that insulates nerve cell axons to increase the speed at which information (encoded as an electrical signal) travels from one nerve cell body to another (as in the central nervous system) or from a nerve cell body to a muscle (as in the peripheral nervous system). The myelinated axon can be likened to an electrical wire (the axon) with insulating material (myelin) around it. However, unlike the plastic covering on an electrical wire, myelin does not form a single long sheath over the entire length of the axon. Rather, each myelin sheath insulates the axon over a single section and in general, each axon comprises multiple long myelinated sections separated from each other by short gaps called the nodes of Ranvier. Nodes of Ranvier are the short (~1 micron) unmyelinated regions of the axon between adjacent long (~0.2 mm – >1 mm) myelinated internodes 1. Each myelin sheath is formed by the concentric wrapping of an oligodendrocyte or Schwann cell process around the axon. Each myelin-generating cell (oligodendrocyte in the CNS or Schwann cell in the peripheral nervous system) furnishes myelin for only one segment of any given axon. The periodic interruptions where short portions of the axon are left uncovered by myelin, the nodes of Ranvier, are critical to the functioning of myelin.
In myelinated axons, the excitable axonal membrane is exposed to the extracellular space only at the nodes of Ranvier; this is the location of sodium channels 2. When the membrane at the node of Ranvier is excited, the local circuit generated cannot flow through the high-resistance sheath and, therefore, flows out through and depolarizes the membrane at the next node, which might be 1 mm or farther away (Figure 3). The low capacitance of the myelin sheath means that little energy is required to depolarize the remaining membrane between the nodes of Ranvier, which results in local circuit spreading at an increased speed. Active excitation of the axonal membrane jumps from node to node; this form of impulse propagation is called saltatory conduction (Latin saltare, “to jump”). This saltatory conduction whereby the action potential “jumps” from one node of Ranvier, over a long myelinated stretch of the axon called the internode, before ‘recharging’ at the next node of Ranvier, and so on, until it reaches the axon terminal. Once it reaches the axon terminal, this electrical signal provokes the release of a chemical message or neurotransmitter that binds to receptors on the adjacent post-synaptic cell (e.g. nerve cell in the CNS or muscle cell in the peripheral nervous system) at specialized regions called synapses.
Furthermore, such movement of the wave of depolarization is much more rapid in myelinated nerve fibers than in unmyelinated fibers, because only the nodes of Ranvier are excited during conduction in myelinated fibers, Na+ (sodium) flux into the nerve is much less than in unmyelinated fibers, where the entire membrane is involved. An example of the advantage of myelination is obtained by comparison of two different nerve fibers, both of which conduct at 25 m/sec at 20°C. The 500-mm diameter unmyelinated giant axon of the squid requires 5,000 times as much energy and occupies about 1,500 times as much space as the 12-mm diameter myelinated nerve in the frog.
In another word, myelin speeds the transmission of electrical impulses called action potentials along myelinated axons by insulating the axon and reducing axonal membrane capacitance. Conduction velocity in myelinated fibers is proportional to the diameter, while in unmyelinated fibers it is proportional to the square root of the diameter 3. Thus, differences in energy and space requirements between the two types of fiber are exaggerated at higher conduction velocities. If nerves were not myelinated and equivalent conduction velocities were maintained, the human spinal cord would need to be as large as a good-sized tree trunk. Myelin, then, facilitates conduction while conserving space and energy 4.
This “insulating” role for myelin is essential for normal motor function (i.e. movement such as walking), sensory function (e.g. hearing, seeing or feeling the sensation of pain) and cognition (e.g. acquiring and recalling knowledge), as demonstrated by the consequences of disorders that affect it, such as the genetically determined leukodystrophies 5, the acquired inflammatory demyelinating disorder, multiple sclerosis 6 and the inflammatory demyelinating peripheral neuropathies 7. Due to its high prevalence, multiple sclerosis, which specifically affects the central nervous system (brain, spinal cord and optic nerve), is the best known disorder of myelin.
Figure 1. Neuron with myelin sheath
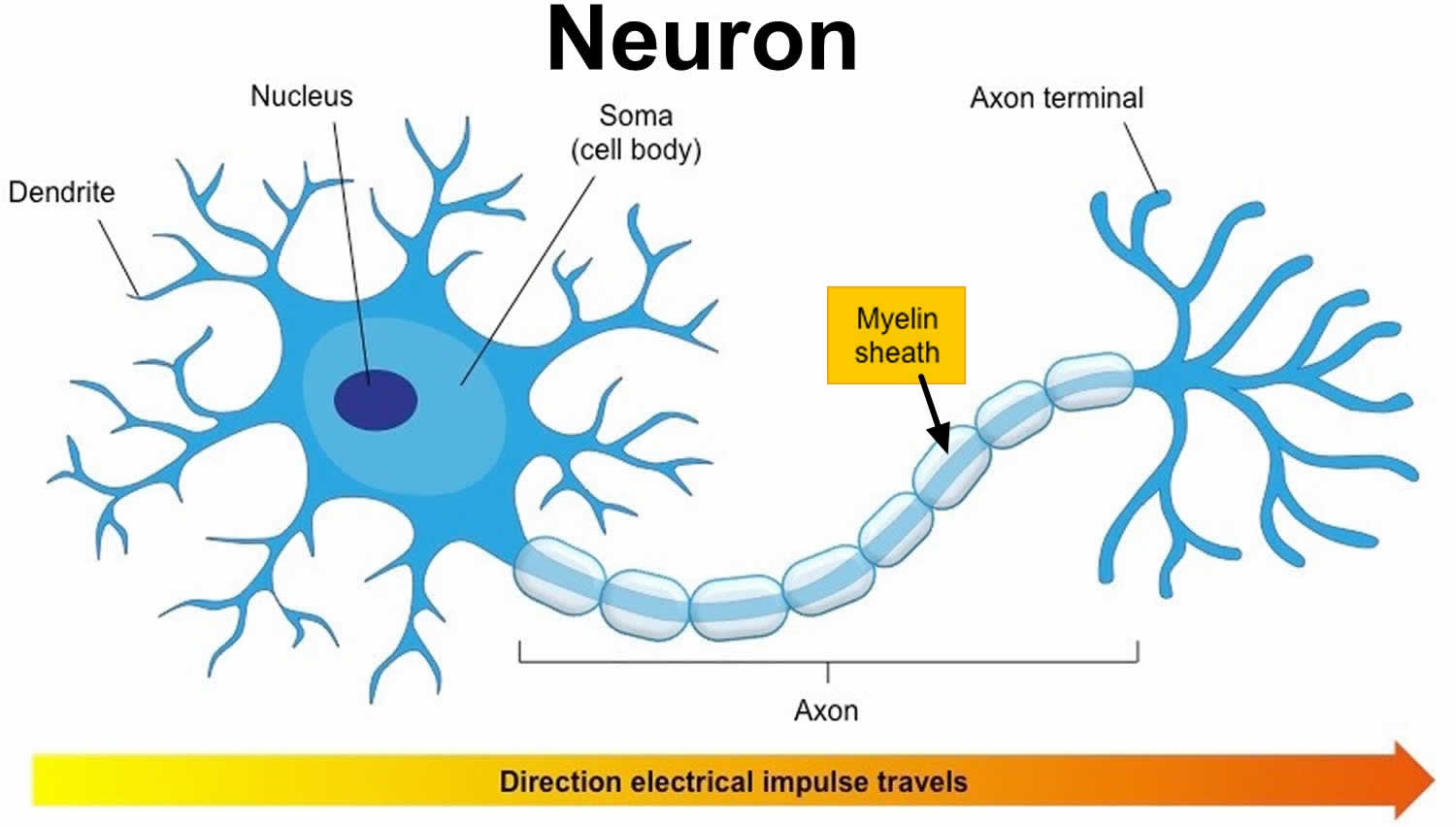
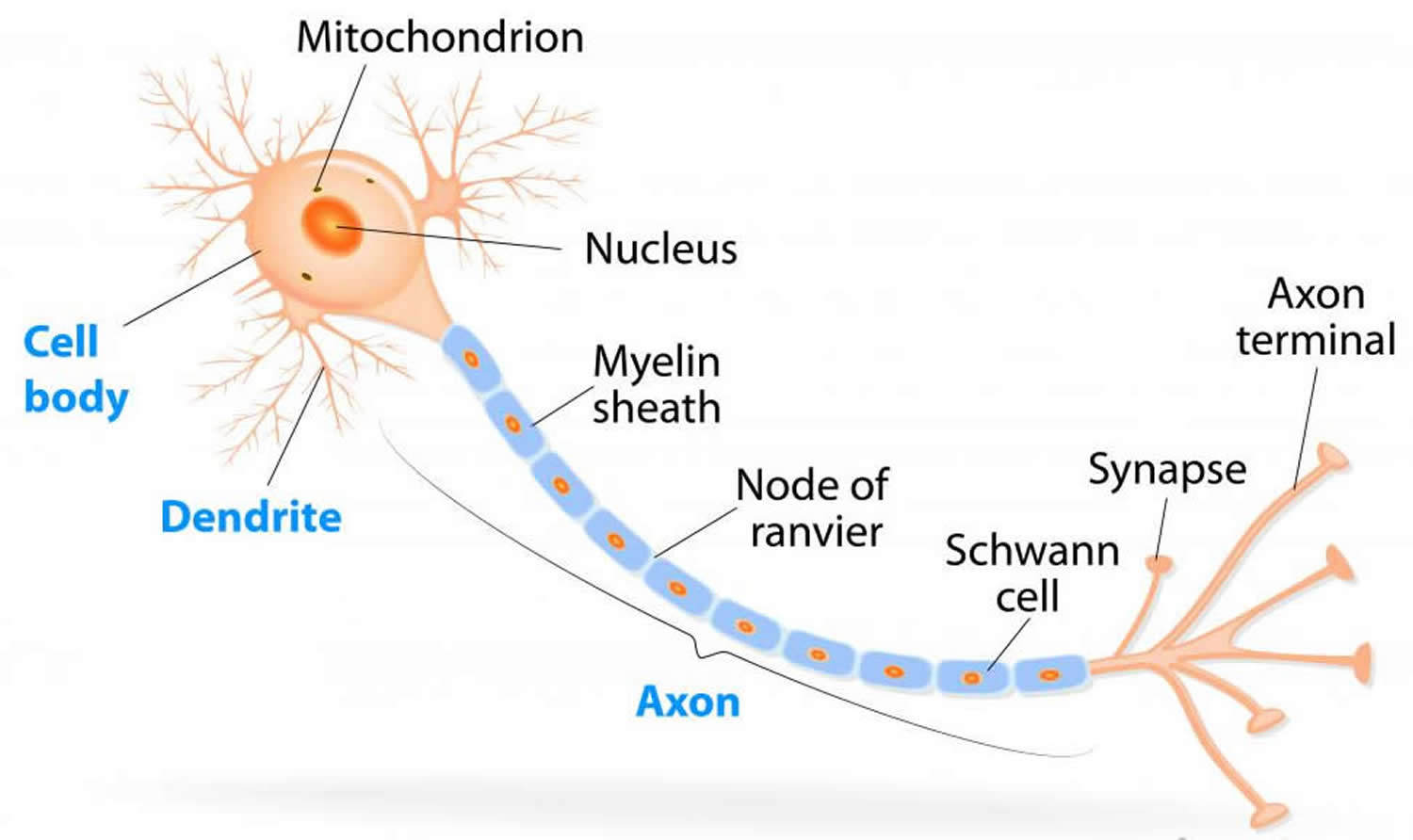
Figure 2. How electrical impulses travel down a neuron (myelinated and unmyelinated)
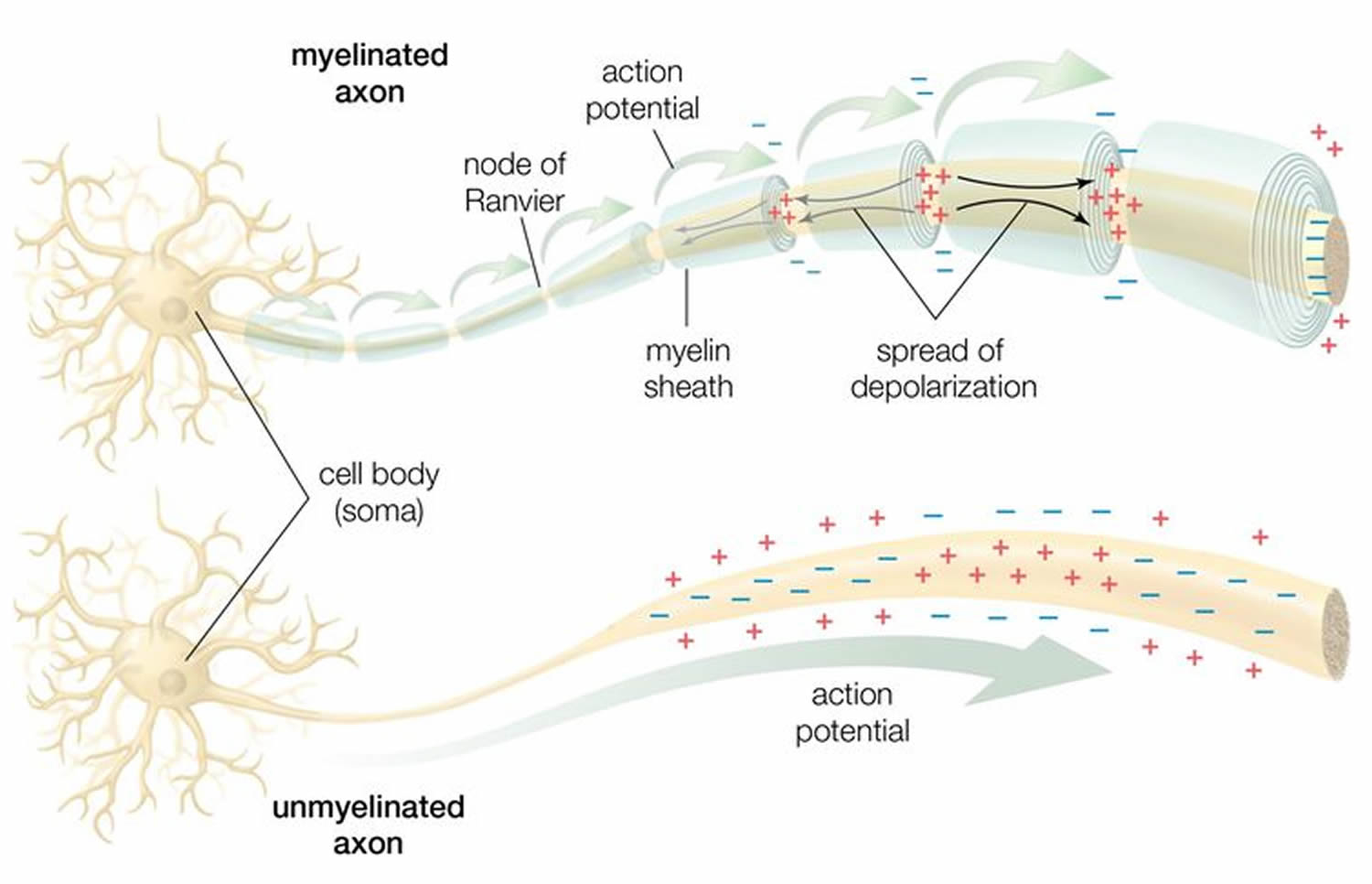
Myelin sheath formation
In the peripheral nervous system (PNS), myelination is preceded by invasion of the nerve bundle by Schwann cells, rapid multiplication of these cells and segregation of the individual axons by Schwann cell processes. Smaller axons (≤1 μm), which will remain unmyelinated, are segregated; several may be enclosed in one cell, each within its own pocket. Large axons (≥1 μm) destined for myelination are enclosed singly, one cell per axon per internode. These cells line up along the axons with intervals between them; the intervals become the nodes of Ranvier.
Before myelination, the axon lies in an invagination of the Schwann cell (Figure 3). The plasmalemma of the Schwann cell then surrounds the axon and joins to form a double-membrane structure that communicates with the cell surface. This structure, called the mesaxon, elongates around the axon in a spiral fashion. Thus, formation of myelin topologically resembles rolling up a sleeping bag; the mesaxon winds about the axon, and the cytoplasmic surfaces condense into a compact myelin sheath and form the major dense line. The two external surfaces form the myelin intraperiod line.
Myelin deposition in the peripheral nervous system (PNS) may result in a single axon having up to 100 myelin layers; therefore, it is improbable that myelin is laid down by a simple rotation of the Schwann cell nucleus around the axon. In the central nervous system (CNS), such a postulate is precluded by the fact that one glial cell can myelinate several axons. During myelination, there are increases in the length of the internode, the diameter of the axon and the number of myelin layers. Myelin, therefore, expands in all planes at once. Any mechanism to account for this growth must assume that the membrane system is able to expand and contract and that layers slip over each other.
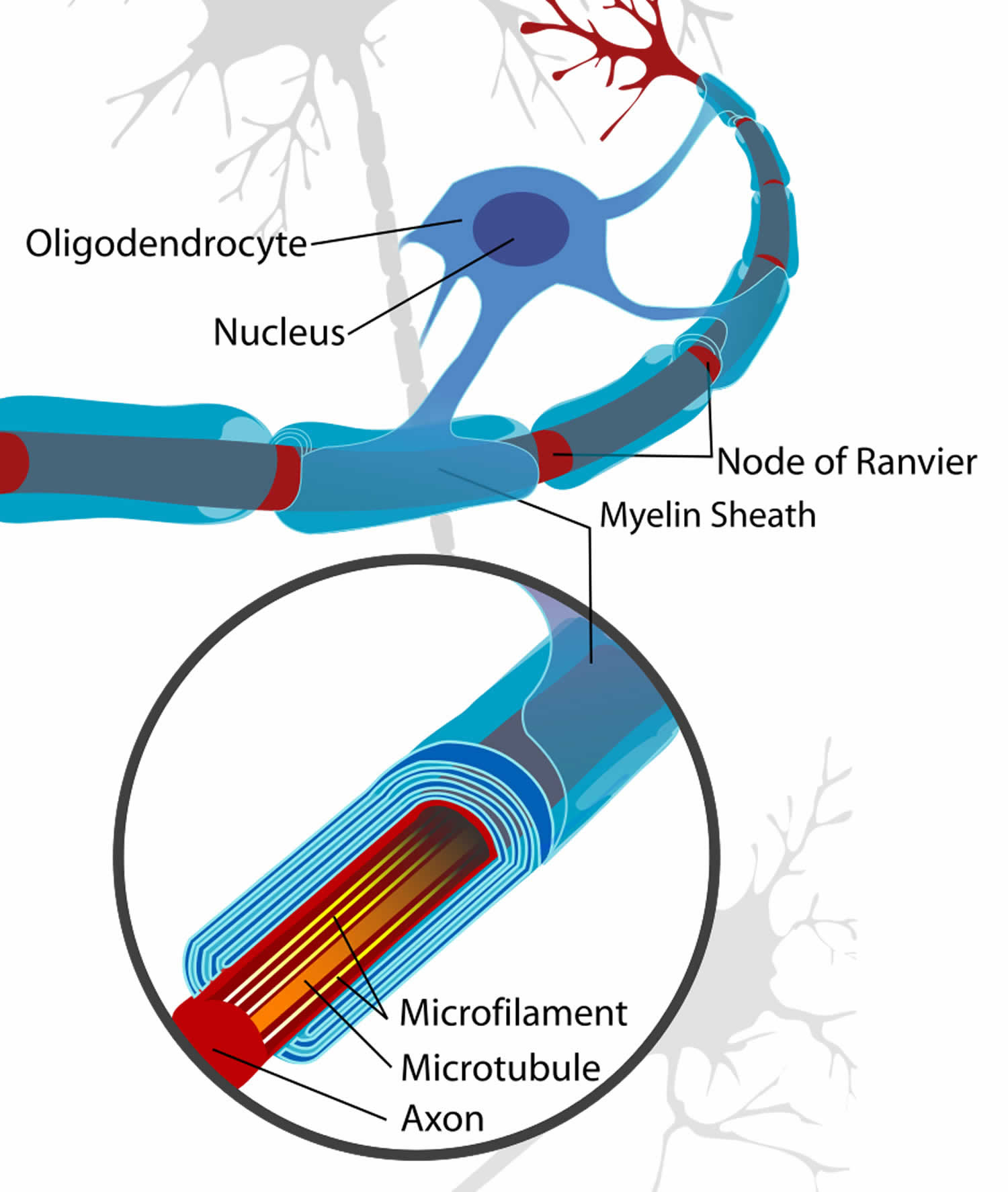
In the central nervous system (CNS), the structures of myelin are formed by the oligodendroglial cell 8. This has many similarities but also points of difference with respect to myelination in the PNS. Central nervous system (CNS) nerve fibers are not separated by connective tissue, nor are they surrounded by cell cytoplasm, and specific glial nuclei are not obviously associated with particular myelinated fibers. Central nervous system (CNS) myelin is a spiral structure similar to peripheral nervous system (PNS) myelin; it has an inner mesaxon and an outer mesaxon that ends in a loop, or tongue, of glial cytoplasm. Unlike the peripheral nerve, where the sheath is surrounded by Schwann cell cytoplasm, the cytoplasmic tongue in the central nervous system is restricted to a small portion of the sheath. This glial tongue is continuous with the plasma membrane of the oligodendroglial cell through slender processes. One glial cell can myelinate 40 or more separate axons 9.
Figure 3. Formation of myelin sheath in the peripheral nervous system (PNS) – note that Schwann cell cytoplasm forms a ring both inside and outside of the myelin sheath.
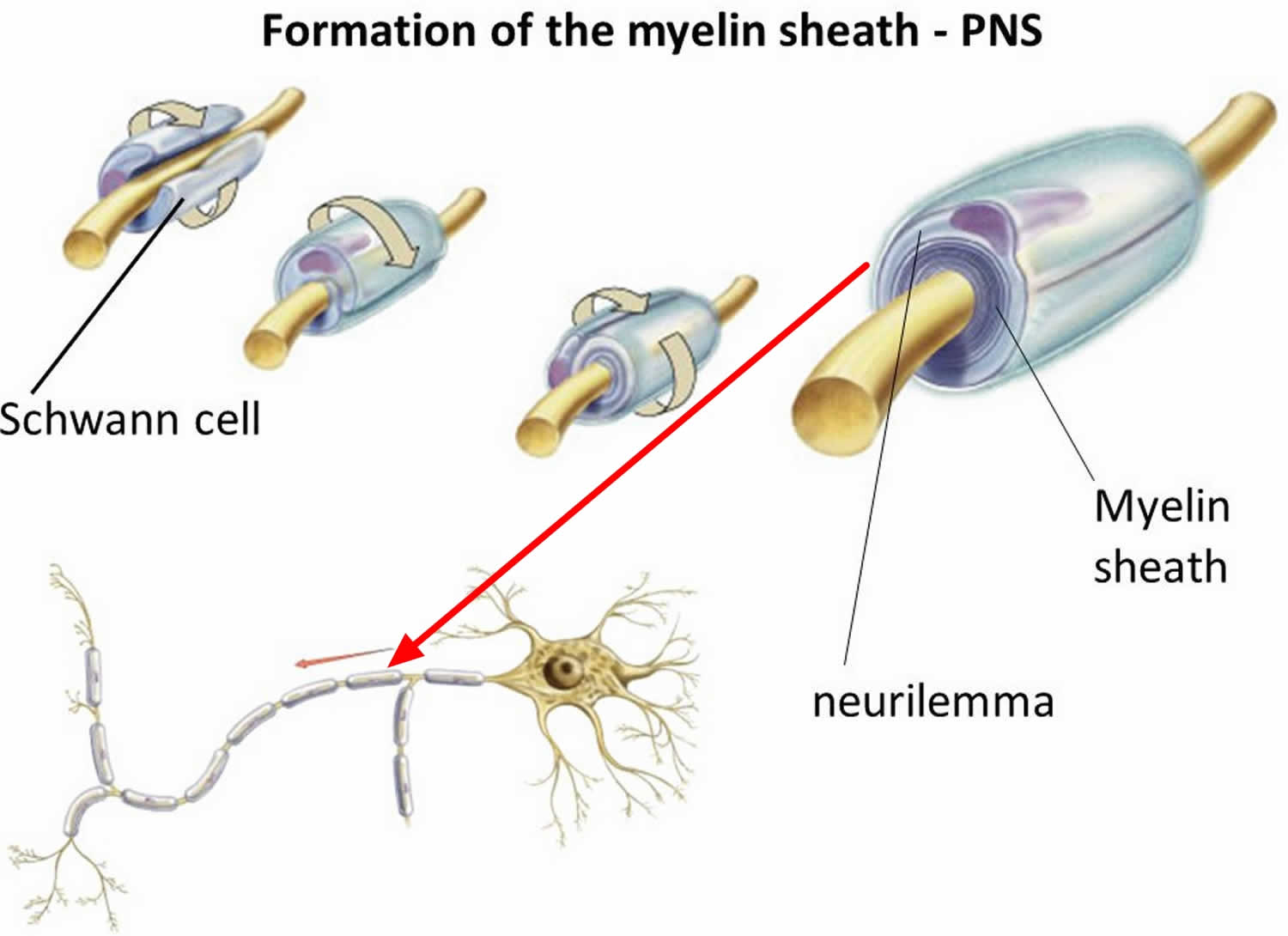
Figure 4. Myelin sheath (the Schwann cell has surrounded the nerve axon)
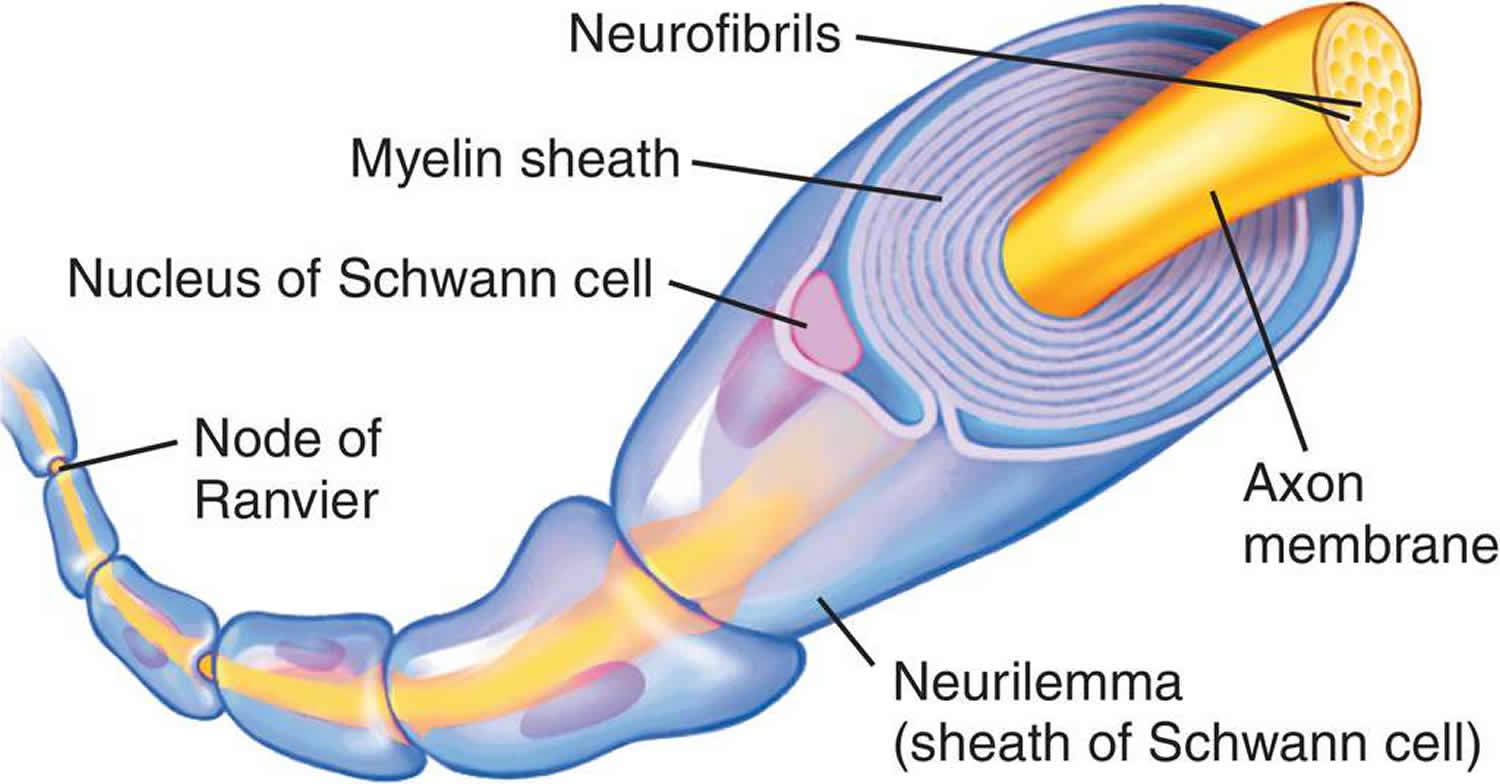
Figure 5. Formation of myelin sheath in the central nervous system (CNS)
What is the function of myelin
The main purpose of myelin is to increase the speed at which electrical impulses travel along the myelinated fiber. In unmyelinated fibers, electrical impulses (action potentials) travel as continuous waves, but in myelinated fibers, they “hop” or propagate by saltatory conduction. The latter is markedly faster than the former, at least for axons over a certain diameter. Myelin decreases capacitance and increases electrical resistance across the axonal membrane (the axolemma). It has been suggested that myelin permits larger body size by maintaining agile communication between distant body parts 10.
Myelinated fibers lack voltage-gated sodium channels along the myelinated internodes, exposing them only at the nodes of Ranvier. Here, they are highly abundant and densely packed. Positively charged sodium ions can enter the axon through these voltage-gated channels, leading to depolarisation of the membrane potential at the node of Ranvier. The resting membrane potential is then rapidly restored due to positively charged potassium ions leaving the axon through potassium channels. The sodium ions inside the axon then diffuse rapidly through the axoplasm (axonal cytoplasm), to the adjacent myelinated internode and ultimately to the next (distal) node of Ranvier, triggering the opening of the voltage gated sodium channels and entry of sodium ions at this site. Although, the sodium ions diffuse through the axoplasm rapidly, diffusion is decremental by nature, thus nodes of Ranvier have to be (relatively) closely spaced, to secure action potential propagation 11. The action potential “recharges” at consecutive nodes of Ranvier as the axolemmal membrane potential depolarises to approximately +35 mV 12. Along the myelinated internode, energy-dependent sodium/potassium pumps, pump the sodium ions back out of the axon and potassium ions back into the axon, to restore the balance of ions between the intracellular (inside the cell i.e. axon in this case) and extracellular (outwith the cell) fluids.
Whilst the role of myelin as an “axonal insulator” is well-established, other functions of myelinating cells are less well known or only recently established. The myelinating cell “sculpts” the underlying axon by promoting the phosphorylation of neurofilaments, thus increasing the diameter or thickness of the axon at the internodal regions; helps cluster molecules on the axolemma (such as voltage-gated sodium channels) at the node of Ranvier 13; and modulates the transport of cytoskeletal structures and organelles such as mitochondria, along the axon 1. Recently, evidence came to light to support a role for the myelinating cell in “feeding” the axon 14. In other words, the myelinating cell seems to act as a local ‘fueling station’ for the axon, which uses a great deal of energy to restore the normal balance of ions between it and its environment 15, following the generation of action potentials.
When a peripheral fiber is severed, the myelin sheath provides a track along which regrowth can occur. However, the myelin layer does not ensure a perfect regeneration of the nerve fiber. Some regenerated nerve fibers do not find the correct muscle fibers, and some damaged motor neurons of the peripheral nervous system die without regrowth. Damage to the myelin sheath and nerve fiber is often associated with increased functional insufficiency.
Unmyelinated fibers and myelinated axons of the mammalian central nervous system do not regenerate.
Some studies have revealed that optic nerve fibers can be regenerated in postnatal rats. This regeneration depends upon two conditions: axonal die-back has to be prevented with appropriate neurotrophic factors, and neurite growth inhibitory components have to be inactivated. These studies may lead to further understanding of nerve fiber regeneration in the central nervous system.
Myelin sheath disorders
Demyelination is the loss of the myelin sheath insulating the nerves, and is the hallmark of some neurodegenerative autoimmune diseases, including multiple sclerosis, acute disseminated encephalomyelitis, neuromyelitis optica, transverse myelitis, chronic inflammatory demyelinating polyneuropathy, Guillain–Barré syndrome, central pontine myelinosis, inherited demyelinating diseases such as leukodystrophy, and Charcot-Marie-Tooth disease. Sufferers of pernicious anaemia can also suffer nerve damage if the condition is not diagnosed quickly. Subacute combined degeneration of spinal cord secondary to pernicious anaemia can lead to slight peripheral nerve damage to severe damage to the central nervous system, affecting speech, balance, and cognitive awareness. When myelin degrades, conduction of signals along the nerve can be impaired or lost, and the nerve eventually withers. A more serious case of myelin deterioration is called Canavan disease.
The immune system may play a role in demyelination associated with such diseases, including inflammation causing demyelination by overproduction of cytokines via upregulation of tumor necrosis factor 16 or interferon.
Symptoms of Demyelination
Demyelination results in diverse symptoms determined by the functions of the affected neurons. It disrupts signals between the brain and other parts of the body; symptoms differ from patient to patient, and have different presentations upon clinical observation and in laboratory studies.
Typical symptoms include:
- blurriness in the central visual field that affects only one eye, may be accompanied by pain upon eye movement
- double vision
- loss of vision/hearing
- odd sensation in legs, arms, chest, or face, such as tingling or numbness (neuropathy)
- weakness of arms or legs
- cognitive disruption, including speech impairment and memory loss
- heat sensitivity (symptoms worsen or reappear upon exposure to heat, such as a hot shower)
- loss of dexterity
- difficulty coordinating movement or balance disorder
- difficulty controlling bowel movements or urination
- fatigue
- tinnitus
Central Nervous System Myelin Sheath Diseases
Demyelinating diseases of the CNS can be classified according to their pathogenesis into several categories: demyelination due to inflammatory processes, viral demyelination, demyelination caused by acquired metabolic derangements, hypoxic–ischaemic forms of demyelination and demyelination caused by focal compression. Some of these distinctions are rather simplistic in that there is overlap in pathogenesis between the entities in the different categories, but the classification provides a conceptual framework that may be useful in accurate diagnosis.
Inflammatory demyelination
Three diseases fall into this category: multiple sclerosis, acute‐disseminated encephalomyelitis and acute hemorrhagic leucoencephalitis. The commonest of these, multiple sclerosis, is pathologically and pathogenetically heterogeneous, and has been divided according to clinical and pathological features into four main subtypes (classical, acute, neuromyelitis optica and concentric sclerosis) with further subdivision of plaque types on the basis of a combination of morphological and immunohistochemical findings.
Multiple sclerosis
Multiple sclerosis (MS) is the commonest of the demyelinating diseases 17. Multiple sclerosis can affect the brain and/or spinal cord, causing a wide range of potential symptoms, including problems with vision, arm or leg movement, sensation or balance. Multiple sclerosis (MS) a lifelong condition that can sometimes cause serious disability, although it can occasionally be mild. In many cases, it’s possible to treat symptoms. MS itself is rarely fatal, but complications may arise from severe MS, such as chest or bladder infections, or swallowing difficulties.
The average life expectancy for people with MS is around 5 to 10 years lower than average, and this gap appears to be getting smaller all the time.
Multiple sclerosis (MS) is most commonly diagnosed in people in their 20s and 30s, although it can develop at any age. It’s about two to three times more common in women than men.
Multiple sclerosis (MS) is thought to be caused by the interaction of multiple genetic and environmental factors. The risk of developing multiple sclerosis is increased 100‐fold to 190‐fold if an identical twin has the disease, 20‐fold to 40‐fold in a full sibling, 7‐fold to 13‐fold in a half‐sibling and 5.5‐fold in an affected parent 18. The concordance rate is 25–30% among monozygotic twins compared with 2–5% between dizygotic twins with multiple sclerosis 18. The strongest evidence for the involvement of environmental factors comes from prevalence and migration studies. The prevalence of multiple sclerosis varies markedly geographically, from below 5/100 000 in many areas of Africa, South America and Asia, to over 100/100 000 in Scotland and parts of Scandinavia and Canada 19. Migration before 15 years of age from a high‐prevalence to a lower‐prevalence area reduces the likelihood of developing multiple sclerosis. Migration to a higher‐prevalence area between 11 and 45 years of age increases the risk of developing multiple sclerosis, even above that of the natives 20. Genetic studies have shown linkage of multiple sclerosis to the region of the major histocompatibility complex on chromosome 6p21; among northern European populations, this seems to be largely attributable to association with the human leucocyte antigen‐DR2 allele 21.
Symptoms of multiple sclerosis
The symptoms of multiple sclerosis (MS) vary widely from person to person and can affect any part of the body.
The main symptoms include:
- fatigue
- difficulty walking
- vision problems, such as blurred vision
- problems controlling the bladder
- numbness or tingling in different parts of the body
- muscle stiffness and spasms
- problems with balance and co-ordination
- problems with thinking, learning and planning
Depending on the type of MS you have, your symptoms may come and go in phases, or get steadily worse over time (progress).
Types of multiple sclerosis (MS)
MS starts in one of two general ways: with individual relapses (attacks or exacerbations) or with gradual progression.
- Relapsing remitting multiple sclerosis (MS)
More than 8 out of every 10 people with MS are diagnosed with the “relapsing remitting” type.
Someone with relapsing remitting MS will have episodes of new or worsening symptoms, known as “relapses”. These typically worsen over a few days, last for days to weeks to months, then slowly improve over a similar time period.
Relapses often occur without warning, but are sometimes associated with a period of illness or stress.
The symptoms of a relapse may disappear altogether, with or without treatment, although some symptoms often persist, with repeated attacks happening over several years.
Periods between attacks are known as periods of “remission”. These can last for years at a time.
After many years (usually decades), many, but not all people, with relapsing remitting MS go on to develop secondary progressive MS. In this type of MS, symptoms gradually worsen over time without obvious attacks. Some people continue to have infrequent relapses during this stage.
Around half of people with relapsing remitting MS will develop secondary progressive MS within 15-20 years, and the risk of this happening increases the longer you have the condition.
- Primary progressive multiple sclerosis (MS)
Just over 1 in 10 people with the condition start their MS with a gradual worsening of symptoms.
In primary progressive MS, symptoms gradually worsen and accumulate over several years, and there are no periods of remission, though people often have periods where their condition appears to stabilize.
Causes of multiple sclerosis
Exactly why someone develops multiple sclerosis (MS) isn’t known. It’s not caused by anything you’ve done and it’s not clear whether it can be prevented.
What is known so far suggests it’s caused by a combination of genetic and environmental factors.
MS is an autoimmune condition, which means your immune system mistakes part of your body for a foreign substance and attacks it.
In the case of MS, it attacks the myelin sheath in the brain and/or spinal cord. This is the layer that surrounds your nerves, protecting them and helping electrical signals travel from the brain to the rest of the body.
The attacks cause the myelin sheath to become inflamed in small patches (plaques or lesions), which can be seen on a magnetic resonance imaging (MRI) scan.
These patches of inflammation can disrupt the messages traveling along the nerves. It can slow them down, jumble them, send them the wrong way, or stop them from getting through completely. This disruption leads to the symptoms and signs of MS.
When the inflammation goes away, it can leave behind scarring of the myelin sheath (sclerosis). These attacks, particularly if frequent and repeated, can eventually lead to permanent damage to the underlying nerves.
Why do people get MS?
It’s not clear what causes the immune system to attack the myelin sheath.
It seems likely that it’s partly caused by genes you inherit from your parents and partly by outside factors that may trigger the condition.
Some of the factors that have been suggested as possible causes of MS include:
- your genes – MS isn’t directly inherited, but people who are related to someone with the condition are more likely to develop it; the chance of a sibling or child of someone with MS also developing it is estimated to be around 2-3%
- lack of sunlight and vitamin D – MS is more common in countries far from the equator, which could mean that a lack of sunlight and low vitamin D levels may play a role in the condition, although it’s not clear whether vitamin D supplements can help prevent MS
- smoking – people who smoke are about twice as likely to develop MS compared to those who don’t smoke
- viral infections – it has been suggested that infections, particularly those caused by Epstein-Barr virus (responsible for glandular fever), might trigger the
- immune system, leading to MS in some people
Further research is needed to understand more about why MS occurs and whether anything can be done to prevent it.
Treatment for multiple sclerosis
There’s currently no cure for multiple sclerosis (MS), but it’s possible to treat the symptoms with medications and other treatments.
Treatment for MS depends on the specific symptoms and difficulties the person has.
It may include:
- treating relapses of MS symptoms (with steroid medication)
- treating specific MS symptoms
- treatment to reduce the number of relapses (disease-modifying therapies)
You’ll be supported by a team of different healthcare professionals working together.
This may include a neurologist (specialist in treating conditions of the nervous system), a physiotherapist, a speech and language therapist, and a number of other professionals.
Your team will also include a specialist MS nurse, who will usually serve as your main point of contact.
Treatment for MS relapses
Contact your specialist MS nurse or doctor if you think you’re having a relapse.
A flare-up of symptoms can sometimes be caused by something other than a relapse, such as an infection, so your nurse or GP needs to check for other possible causes.
Treatment for a relapse usually involves either:
- a 5-day course of steroid tablets taken at home
- injections of steroid medication given in hospital for 3 to 5 days
Steroids can help speed up your recovery from a relapse, but they don’t prevent further relapses or stop MS getting worse over time.
They’re only given for a short period of time to avoid possible steroid side effects, such as osteoporosis (weak bones), weight gain and diabetes, although some people will still experience problems.
Not using steroids more than 3 times a year (if possible) will also help to reduce the risk of side effects.
Disease-modifying therapies
Although MS can’t currently be cured, there are medicines that can help reduce the number and severity of relapses in some people. These are called disease-modifying therapies.
These aim to reduce the amount of damage and scarring to the myelin sheath (a layer surrounding your nerves), which is associated with MS relapses.
These treatments may also help to slow worsening disability in MS, although definitive research into their long-term benefits is limited.
Disease-modifying therapies aren’t suitable for everyone with MS. They’re only prescribed to those with relapsing-remitting MS or secondary progressive MS who meet certain criteria, such as the number of relapses they have had.
People without relapses are very unlikely to benefit from the treatments and could still experience side effects from them.
Beta interferon
- The types of beta interferon licensed for use in the US are interferon beta-1a (Avonex, Rebif and Plegridy) and interferon beta-1b (Betaferon and Extavia). These are all given by injection.
- You may be offered treatment with one of these medicines if either:
- you have relapsing-remitting MS and have had a recent relapse and/or MRI scans show that your MS is active
- you have secondary progressive MS and still have significant relapses
- All beta interferons often cause mild side effects, particularly flu-like symptoms (headaches, chills and mild fever), for 24 to 48 hours after they’re injected, and temporary pain or redness at the injection site.
Glatiramer acetate
- One brand of glatiramer acetate, called Copaxone, is licensed for use in the US. It’s injected under the skin either every day or at a higher dose 3 times a week.
- You may be offered treatment with glatiramer acetate if you have relapsing-remitting MS and have had a recent relapse, and/or MRI scans show that your MS is active.
- Common side effects of glatiramer acetate include problems with redness or hardening of the skin at the injection sites, and occasionally palpitations or flushing after the injection.
Teriflunomide
- Teriflunomide, branded as Aubagio, is a tablet taken once a day.
- You may be offered treatment with teriflunomide if you have relapsing-remitting MS and have had a recent relapse, and/or MRI scans show that your MS is active.
- Common side effects of teriflunomide include headaches, feeling sick, diarrhoea and hair thinning or hair loss.
- You’ll also need blood tests frequently in the early months of treatment to check for problems with liver function.
Natalizumab
- Natalizumab, branded as Tysabri, is injected into a vein (known as an infusion) once every 28 days.
- You may be offered treatment with natalizumab if you have severe relapsing-remitting MS that’s getting worse quite quickly. For example, if you have had 2 or more severe relapses within a year and MRI scans show that your MS is getting worse.
- Side effects of natalizumab are rare, but can include an increased risk of an itch or rash (hives), headaches, dizziness, joint pain, and feeling or being sick at the time of the infusions.
- The main concern of treatment with natalizumab is a risk of a brain infection called progressive multifocal leukoencephalopathy (PML).
- This is uncommon, but can become a significant problem with long-term treatment in some people.
Fingolimod
- Fingolimod, branded as Gilenya, is taken as a capsule once a day.
- You may be offered treatment with fingolimod if you have relapsing-remitting MS and you experience the same or an increased number of relapses, despite treatment with other medications, such as beta interferons.
- The medication doesn’t usually cause significant side effects, but some people experience an increased risk of infections, headaches, diarrhoea, liver problems and visual problems.
Alemtuzumab and ocrelizumab
- Alemtuzumab, branded as Lemtrada, is initially given into a vein once a day for 5 consecutive days.
- This is followed by a second course of treatment a year later, which lasts for 3 consecutive days.
- You may be offered treatment with alemtuzumab if you have relapsing-remitting MS and have had a relapse in the past year, and/or MRI scans show that your MS is active.
- Common side effects of alemtuzumab include an increased risk of infections, headaches, rashes and fever. Regular blood and urine tests will be carried out to monitor treatment.
- If you can’t take alemtuzumab, you may be offered a similar drug called ocrelizumab, branded as Ocrevus. It’s given into a vein and again 2 weeks later. Further doses are given every 6 months.
Dimethyl fumarate
- Dimethyl fumarate, branded as Tecfidera, is a tablet taken twice a day.
- You may be offered treatment with dimethyl fumarate if you have relapsing-remitting MS and have had a recent relapse, and/or MRI scans show that your MS is active.
- Common side effects of dimethyl fumarate include hot flushes, diarrhoea, nausea, abdominal (tummy) pain and headaches.
- Stomach upsets usually settle after a month or so. The flushes can continue, but aren’t normally a major issue.
Cladribine
- Cladribine, branded as Mavenclad, is a tablet taken as a course of treatment over 2 years.
- You take a tablet once a day for 4 or 5 days in the first month that you start treatment and then again a month later for another 4 or 5 days. This cycle is then repeated a year later.
- You may be offered treatment with cladribine if you have very active relapsin- remitting MS. This means you have severe MS that’s getting worse quite quickly. For example, you have had at least 2 relapses in the past year and MRI scans show that your MS is getting worse.
- You may also be offered cladribine if you have had a relapse in the past year and MRI scans show that your MS is active, even if you have been taking a different disease-modifying therapy.
- Common side effects of cladribine include a reduced white blood cell count, cold sores and shingles, rashes and hair loss. Regular blood tests will be carried out to monitor treatment.
Treatment for specific MS symptoms
MS can cause a range of symptoms that can be treated individually.
Treatments for some of the main symptoms are discussed in the following sections.
Fatigue
Many people with MS experience fatigue.
You may be prescribed amantadine for fatigue caused by MS, although this medication may only have a limited effect.
You should also be given general advice on ways to manage fatigue, such as:
- exercise
- keeping healthy sleep patterns
- energy-saving techniques
- avoiding medications that can worsen fatigue (including some painkillers)
Specialist fatigue management courses or therapy, such as cognitive behavioural therapy (CBT), can also help some people with MS cope with their fatigue.
Visual problems
MS-related visual problems will often improve on their own, usually within a few weeks, so you may not need any treatment.
If your symptoms are particularly severe, you may be prescribed steroids to help speed up recovery.
If you have problems with involuntary eye movements, medication such as gabapentin can sometimes help.
Some people with double vision need help from ophthalmologists (eye specialists).
Muscle spasms and stiffness
Muscle spasms and stiffness (spasticity) can be improved with physiotherapy.
Techniques such as stretching exercises can help if your movement is restricted.
If your muscle spasms are more severe, you may be prescribed a medicine that can relax your muscles.
This will usually be either baclofen or gabapentin, although there are alternative medicines, such as tizanidine, diazepam, clonazepam and dantrolene.
These medicines all have side effects, such as dizziness, weakness, nausea and diarrhoea, so discuss which of these would be best for you with your specialist MS nurse or doctor.
Mobility problems
Mobility problems are often the result of muscle spasms and spasticity, but they can also be caused by muscle weakness, or problems with balance or dizziness.
If you have problems with mobility, you might benefit from:
- an exercise programme supervised by a physiotherapist
- special exercises called vestibular rehabilitation if you have problems with balance
- medication for dizziness or tremors
- mobility aids, such as a walking stick, or occasionally a wheelchair
- home adaptations, such as stair lifts or railings
An occupational therapist can carry out an assessment of your home and suggest adaptations that may be of help.
Neuropathic pain
Neuropathic pain is caused by damage to your nerves, and is usually sharp and stabbing.
It can also occur in the form of extreme skin sensitivity or a burning sensation.
This type of pain can be treated using the medicines gabapentin or carbamazepine, or with a medication called amitriptyline.
This is an older type of antidepressant, but these days it’s mainly used for pain control.
Musculoskeletal pain
Living with MS can cause stresses and strains to the muscles and joints in your body.
A physiotherapist may be able to help with this pain by suggesting exercise techniques or better seating positions.
If your pain is more severe, you may be prescribed painkillers. Alternatively, you may have a device that stimulates your nerves called a transcutaneous electrical nerve stimulation (TENS) machine.
Problems with thinking, learning and memory
If you experience problems with thinking and memory, any treatment you receive will be fully explained and recorded so it’s clear to you.
You should be referred to a clinical psychologist, who will assess your problems and suggest ways to manage them.
Emotional problems
If you experience emotional outbursts, such as laughing or crying for no apparent reason, you should be assessed by a specialist like a clinical psychologist. They may suggest treatment with an antidepressant.
People with MS who have depression can also be treated with antidepressants or therapy, such as CBT.
If you often feel anxious or worried, you may be prescribed antidepressants or benzodiazepines, which are a type of tranquilliser that have a calming effect.
Sexual problems
Men with MS who find it hard to obtain or maintain an erection (erectile dysfunction) may be prescribed medication to temporarily increase the blood flow to the penis, such as sildenafil (Viagra). This is provided by the NHS if you have MS.
Relationship counselling or seeing a sex therapist may also help both men and women with MS who are having problems with reduced interest in sex or difficulty reaching orgasm.
Bladder problems
Various medications are available if you have an overactive bladder or need to pee frequently during the night.
If you find it difficult to empty your bladder, advice from a continence nurse or physiotherapist can help.
Handheld external stimulators can also help some people start peeing or empty the bladder.
Occasionally, a catheter can be used to empty the bladder when needed.
In rare cases, people with MS may need a long-term catheter to keep the bladder emptying safely.
You may be referred to a continence adviser or urologist, who can offer specialist treatment and advice, such as botulinim toxin injections, bladder exercises or electrical treatment for your bladder muscles.
Bowel problems
It may be possible to treat mild to moderate constipation by changing your diet or taking laxatives.
More severe constipation may need to be treated with suppositories, which are inserted into your bottom, or an enema.
An enema involves having a liquid medication rinsed through your bottom and large bowel, which softens and flushes out your stools.
Bowel incontinence can sometimes be treated with anti-diarrhoea medication or by doing pelvic floor exercises to strengthen your rectal muscles.
Speech and swallowing difficulties
A speech and language therapist can help you find ways to overcome problems with speech and swallowing.
For example, they can offer advice about foods that are easy to swallow and recommend exercises to strengthen the muscles used in speech and swallowing.
If swallowing problems become very severe, some people need to be fed using a tube, which is fitted into the stomach through the skin.
Acute‐disseminated encephalomyelitis
This inflammatory demyelinating disease mainly affects children. It typically occurs within 3 weeks of infection, vaccination or giving drugs, and is thought to be due to a T cell hypersensitivity reaction 22. The infections most often responsible are viral (usually measles, mumps, varicella, rubella or infectious mononucleosis), but Mycoplasma pneumoniae, Campylobacter jejuni, group A streptococci or other bacteria are sometimes the causative agents. Vaccination is a less common antecedent, and drug‐induced acute‐disseminated encephalomyelitis is rare. In some patients, no preceding illness or other initiating event is identified. The incidence of recorded acute‐disseminated encephalomyelitis (ADEM) is about 0.4/100 000/year, but this is probably an underestimate as in children with encephalopathy the diagnosis is readily missed unless MRI is carried out 22.
Clinical features
The most prominent clinical features are usually ataxia, headache and weakness 23. Other manifestations can include vomiting, slurring or impairment of speech, extraocular or other cranial nerve nerve palsies, agitation, seizures, lethargy, delirium and stupor. Approximately 80% of patients make a full recovery. Although acute‐disseminated encephalomyelitis (ADEM) is classically a monophasic disease, relapses have been reported in 5–10% of cases (multiphasic disseminated encephalomyelitis); if relapse occurs on more than one occasion, a diagnosis of multiple sclerosis rather than multiphasic disseminated encephalomyelitis is probably appropriate. A recurrent form of demyelinating perivenous encephalomyelitis can occur in patients with familial erythrophagocytic lymphohistiocytosis 24.
Acute hemorrhagic leucoencephalitis
This rare, usually fatal, disease is thought to be a hyperacute variant of acute‐disseminated encephalomyelitis (ADEM). Like acute‐disseminated encephalomyelitis, acute hemorrhagic leucoencephalitis may be preceded by viral or Mycoplasma pneumoniae infection. Other rare associations include ulcerative colitis, Crohn’s disease, septicaemia and some drugs. In many patients there is no obvious precipitant.
Clinical features
A typical presentation is pyrexia, headache, vomiting, multifocal neurological deficits and seizures, progressing within 2 or 3 days through drowsiness and coma to death. The outcome is usually death or severe disability, but good recovery has been documented after aggressive medical and surgical reduction of raised intracranial pressure and after giving intravenous immunoglobulin 25.
Viral demyelination
Progressive multifocal leucoencephalopathy
The principal viral demyelinating disease in humans is progressive multifocal leucoencephalopathy caused by the papovavirus, human polyomavirus JC virus (JohnCunningham virus). Approximately 50% of adolescents and 75% of adults have serological evidence of JC virus infection, but it is usually asymptomatic. The virus establishes latent infection in B cells, kidney and possibly CNS 26. Reactivation occurs under conditions of impaired cell‐mediated immunity—for example, after organ transplantation, in patients with leukaemia or lymphoma or in those with AIDS 27.
Clinical features
Patients usually present with insidious onset of neurological deficits that often affect motor function, speech, vision, personality and cognition. Conventional examination of the CSF is usually normal, but human polyomavirus JC virus nucleic acids can be shown by polymerase chain reaction (PCR) 28. MRI typically shows multiple small lesions in the white matter, but these may increase rapidly in size, and occasionally cause mass effect 29. Until a few years ago, progressive multifocal leucoencephalopathy was usually a rapidly progressive fatal disease. However, successful reversal of impairment of cell‐mediated immunity—for example, by administration of antiretroviral drugs to patients with AIDS, can lead to remission of progressive multifocal leucoencephalopathy.
HIV infection can cause a range of white matter abnormalities, including microglial nodule or multinucleated giant cell encephalitis, diffuse leucoencephalopathy and vacuolar myelopathy. In addition, patients with AIDS receiving highly active antiretroviral treatment may occasionally develop severe inflammatory demyelination that is not due to progressive multifocal leucoencephalopathy 30. There is an intense perivascular inflammatory infiltrate of lymphocytes and macrophages. At least occasional macrophages (many, in some reported cases) are immunopositive for HIV. Viral RNA is usually abundant.
Subacute sclerosing panencephalitis, due to measles virus, causes perivascular inflammation and gliosis of white matter (as well as grey matter). This may be associated with patchy or extensive loss of myelinated fibres. Axonal loss is usually commensurate with that of myelin but, particularly towards the edges of severely affected white matter, axons are sometimes relatively preserved. Measles virus intranuclear inclusion bodies may be seen, but tend to be sparse. In most cases the clinical and serological findings are diagnostic, and viral RNA can be detected by PCR.
Rarely, in patients with AIDS, varicella‐zoster virus causes multifocal lesions in the cerebral white matter that are partly demyelinating 31. Viral inclusions and antigen can be seen in oligodendrocytes.
Acquired metabolic demyelination
The most common diseases in this category are central pontine myelinolysis and extrapontine myelinolysis. Very rarely, demyelination occurs in association with chronic alcoholism and malnourishment (Marchiafava–Bignami disease) 32.
Central pontine and extrapontine myelinolysis
Central pontine myelinolysis is a monophasic demyelinating disease of the pons and lower midbrain. It most often occurs in association with alcoholic liver disease or correction of hyponatraemia (especially if the hyponatraemia is marked and the correction rapid) 33. Another clinical context in which central pontine myelinolysis is increasingly common is in patients with liver transplant, in whom high ciclosporin levels may have a role 34. Central pontine myelinolysis rarely occurs in patients who are normonatraemic or hypernatraemic. About 25–50% of patients with central pontine myelinolysis also have extrapontine myelinolysis; this usually affects the cerebellum but may also affect parts of the cerebrum. In up to 25% of patients the demyelination is exclusively extrapontine.
The presentation is usually rapid onset of confusion, limb weakness (often progressing to spastic tetraparesis) and mutism. Other frequent manifestations include ataxia, dysphagia and hypotension. Movement disorders (dystonia, choreoathetosis and parkinsonism) occur in some patients and are probably related to extrapontine myelinolysis. With good supporting care, most patients with central pontine myelinolysis and extrapontine myelinolysis now survive the acute disease, but many have residual neurological deficits.
Hypoxic–ischemic demyelination
Brain tissue generally undergoes necrosis rather than demyelination in response to hypoxia or ischemia. However, in some circumstances the myelinating oligodendrocyte bears the brunt of the hypoxic or ischemic damage.
In patients with severe small vessel cerebrovascular disease, usually in association with hypertension, the subcortical white matter often shows ischemic damage. Quite often, these patients develop dementia with superimposed focal neurological deficits that are related to lacunar infarcts. Pathology shows marked arteriosclerosis and arteriolosclerosis, particularly within the deep cerebral white matter, which is rarefied and gliotic and usually includes both cavitating and poorly circumscribed demyelinating lesions. These tend to be hypocellular, but may contain small to moderate numbers of foamy macrophages. Lymphocytic inflammation is not a feature.
Rarely, global brain hypoxia due to cardiac arrest, asphyxia or depression of cardiorespiratory function by drug overdose causes a leucoencephalopathy with relative sparing of grey‐matter structures 35. The damage is predominantly necrotic, but there may also be some demyelination.
Exposure to carbon monoxide, which causes hypoxaemia by binding to hemoglobin with 200‐fold greater affinity than oxygen, can cause damage to white matter as well as grey matter structures such as the globus pallidus. White matter damage becomes evident only in patients who survive the acute intoxication. The likelihood of sustaining this damage depends on the level and duration of exposure. The subcortical U fibres tend to be spared. The deep cortical white matter is diffusely rarefied and gliotic, due to a mixture of axonal degeneration and demyelination. As in small vessel cerebrovascular disease, the demyelinated white matter may contain foamy macrophages but shows depletion of oligodendrocytes.
Compression‐induced demyelination
This is rarely encountered in routine diagnostic neuropathology. The best documented example of compression‐induced demyelination is that responsible for many cases of trigeminal neuralgia and due to compression of trigeminal nerve root fibres by an overlying artery or vein, close to the zone of entry of the nerve root into the pons. This produces a focal region of non‐inflammatory demyelination, up to about 2 mm across, in the proximal (CNS) part of the nerve root where the compression has occurred 36. In this there are often groups of demyelinated fibres in close juxtaposition, which has been suggested to allow non‐synaptic, direct (ephaptic) transmission of nerve impulses from one to another.
References- Stassart RM, Möbius W, Nave K-A, Edgar JM. The Axon-Myelin Unit in Development and Degenerative Disease. Frontiers in Neuroscience. 2018;12:467. doi:10.3389/fnins.2018.00467. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6050401/
- Waxman S G , Ritchie J M . Molecular dissection of the myelinated axon. Ann. Neurol. 1993;33:121–136.
- Morell P, Quarles RH. The Myelin Sheath. In: Siegel GJ, Agranoff BW, Albers RW, et al., editors. Basic Neurochemistry: Molecular, Cellular and Medical Aspects. 6th edition. Philadelphia: Lippincott-Raven; 1999. Available from: https://www.ncbi.nlm.nih.gov/books/NBK27954
- Ritchie, J. M. Physiological basis of conduction in myelinated nerve fibers. In P. Morell (ed.), Myelin, 2nd ed. New York: Plenum, 1984, pp. 117–141.
- Van der Knaap MS, Bugiani M. Leukodystrophies: a proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathologica. 2017;134(3):351-382. doi:10.1007/s00401-017-1739-1. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5563342/
- Multiple sclerosis. Lancet. 2008 Oct 25;372(9648):1502-17. doi: 10.1016/S0140-6736(08)61620-7. https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(08)61620-7/fulltext
- Chronic inflammatory demyelinating polyneuropathy. Curr Opin Neurol. 2017 Oct;30(5):508-512. doi: 10.1097/WCO.0000000000000481. https://www.ncbi.nlm.nih.gov/pubmed/28763304
- Bunge R P . Glial cells and the central myelin sheath. Physiol. Rev. 1968;48:197–248.
- Davison, A. N., and Peters, A. Myelination. Springfield, IL: Charles C. Thomas, 1970.
- What is myelin? Neuron Glia Biol. 2008 May;4(2):153-63. doi: 10.1017/S1740925X09990263. https://doi.org/10.1017/S1740925X09990263
- Raine CS (1999). “Characteristics of Neuroglia”. In Siegel GJ, Agranoff BW, Albers RW, Fisher SK, Uhler MD. Basic Neurochemistry: Molecular, Cellular and Medical Aspects (6th ed.). Philadelphia: Lippincott-Raven.
- Saladin KS (2012). Anatomy & physiology: the unity of form and function (6th ed.). New York, NY: McGraw-Hill
- Brivio V, Faivre-Sarrailh C, Peles E, Sherman DL, Brophy PJ. Assembly of CNS Nodes of Ranvier in Myelinated Nerves Is Promoted by the Axon Cytoskeleton. Current Biology. 2017;27(7):1068-1073. doi:10.1016/j.cub.2017.01.025. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5387178/
- Funfschilling U, Supplie LM, Mahad D, et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature. 2012;485(7399):517-521. doi:10.1038/nature11007. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3613737/
- Engl E, Attwell D. Non‐signalling energy use in the brain. The Journal of Physiology. 2015;593(16):3417-3429. doi:10.1113/jphysiol.2014.282517. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4560575/
- Cytokines, signal transduction, and inflammatory demyelination: review and hypothesis. Neurochem Res. 1998 Mar;23(3):277-89. https://www.ncbi.nlm.nih.gov/pubmed/9482240
- Love S. Demyelinating diseases. Journal of Clinical Pathology. 2006;59(11):1151-1159. doi:10.1136/jcp.2005.031195.
- The genetic epidemiology of multiple sclerosis. Kenealy SJ, Pericak-Vance MA, Haines JL. J Neuroimmunol. 2003 Oct; 143(1-2):7-12. https://www.ncbi.nlm.nih.gov/pubmed/14575907/
- Pugliatti M, Sotgiu S, Rosati G. The worldwide prevalence of multiple sclerosis. Clin Neurol Neurosurg 2002104182–191.
- Multiple sclerosis in time and space–geographic clues to cause. J Neurovirol. 2000 May;6 Suppl 2:S134-40. https://www.ncbi.nlm.nih.gov/pubmed/10871801
- Genetic analysis of multiple sclerosis in Europeans. J Neuroimmunol. 2003 Oct;143(1-2):1-6. https://www.ncbi.nlm.nih.gov/pubmed/14575906
- Leake J A, Albani S, Kao A S. et al Acute disseminated encephalomyelitis in childhood: epidemiologic, clinical and laboratory features. Pediatr Infect Dis J 200423756–764.
- Gupte G, Stonehouse M, Wassmer E. et al Acute disseminated encephalomyelitis: a review of 18 cases in childhood. J Paediatr Child Health 200339336–342.
- Martin J J, Cras P. Familial erythrophagocytic lymphohistiocytosis. A neuropathologic study. Acta Neuropathol (Berlin) 198566140–144
- Leake J A, Billman G F, Nespeca M P. et al Pediatric acute hemorrhagic leukoencephalitis: report of a surviving patient and review. Clin Infect Dis 200234699–703
- White FA I I I, Ishaq M, Stoner G L. et al JC virus DNA is present in many human brain samples from patients without progressive multifocal leukoencephalopathy. J Virol 1992665726–5734.
- Sweet T M, Valle L D, Khalili K. Molecular biology and immunoregulation of human neurotropic JC virus in CNS. J Cell Physiol 2002191249–256.
- Sugimoto C, Ito D, Tanaka K. et al Amplification of JC virus regulatory DNA sequences from cerebrospinal fluid: diagnostic value for progressive multifocal leukoencephalopathy. Arch Virol 1998143249–262.
- Thurnher M M, Thurnher S A, Muhlbauer B. et al Progressive multifocal leukoencephalopathy in AIDS: initial and follow‐up CT and MRI. Neuroradiology 199739611–618.
- Langford T D, Letendre S L, Marcotte T D. et al Severe, demyelinating leukoencephalopathy in AIDS patients on antiretroviral therapy. AIDS 2002161019–1029
- Gray F, Belec L, Lescs M C. et al Varicella‐zoster virus infection of the central nervous system in the acquired immune deficiency syndrome. Brain 1994117987–999.
- Love S. Demyelinating diseases. Journal of Clinical Pathology. 2006;59(11):1151-1159. doi:10.1136/jcp.2005.031195. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1860500/
- Karp B I, Laureno R. Pontine and extrapontine myelinolysis: a neurologic disorder following rapid correction of hyponatremia. Medicine (Baltimore) 199372359–373
- Lampl C, Yazdi K. Central pontine myelinolysis. Eur Neurol 2002473–10.
- Ginsberg M D, Hedley‐Whyte E T, Richardson E P., Jr Hypoxic‐ischemic leukoencephalopathy in man. Arch Neurol 1976335–14.
- Love S, Coakham H B. Trigeminal neuralgia: pathology and pathogenesis. Brain 20011242347–2360.





{kind=link}