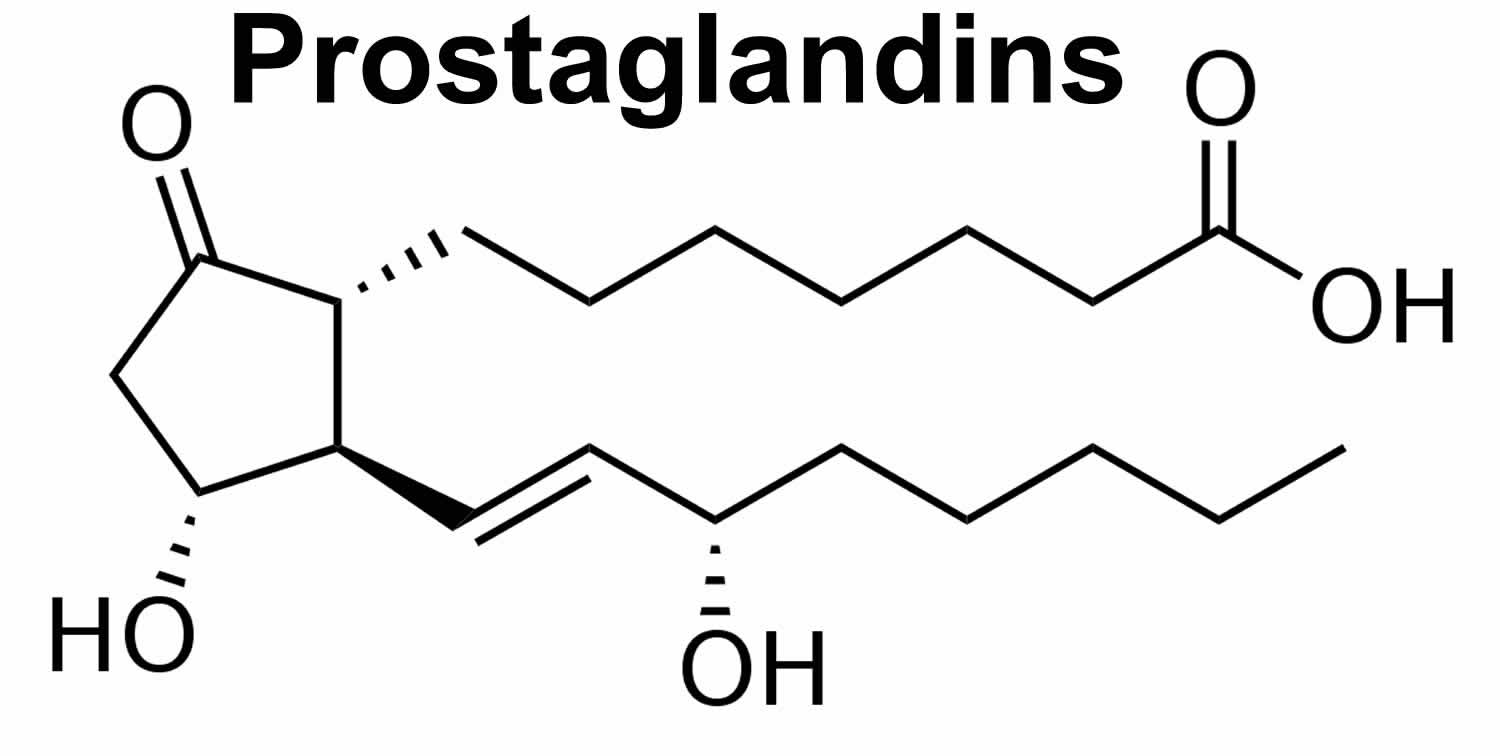
What are prostaglandins
Prostaglandins are a family of lipid compounds that are derived enzymatically from arachidonic acid acting locally as autocrine or paracrine mediators with a wide range of effects throughout the body. The name prostaglandin derives from the prostate gland. When prostaglandin was first isolated from seminal fluid in 1935 by the Swedish physiologist Ulf von Euler, and independently by M.W. Goldblatt, it was believed to be part of the prostatic secretions 1. In fact, prostaglandins are produced by the seminal vesicles. It was later shown that many other tissues secrete prostaglandins for various functions. The first total syntheses of prostaglandin F2α and prostaglandin E2 were reported by E. J. Corey in 1969 2.
There are four principal bioactive prostaglandins (PG) generated in your body: prostaglandin E2 (PGE2), prostacyclin (PGI2), prostaglandin D2 (PGD2) and prostaglandin F2α (PGF2α). Prostaglandins are ubiquitously produced – usually each cell type generates one or two dominant products and act as autacrine and paracrine lipid mediators to maintain local homeostasis in the body. During an inflammatory response, both the level and the profile of prostaglandin production changes dramatically. Prostaglandin production is generally very low in uninflamed tissues, but increases immediately in acute inflammation prior to the recruitment of leukocytes and the infiltration of immune cells.
Prostaglandins play a key role in the generation of the inflammatory response. Their biosynthesis is significantly increased in inflamed tissue and they contribute to the development of the cardinal signs of acute inflammation. For example, prostaglandin E2 (PGE2) and prostacyclin (PGI2) have significant vasodilatory properties, whereas platelet-released thromboxane A2 (TXA2) is known as a vasoconstrictor that increases platelet aggregation. While the pro-inflammatory properties of individual prostaglandins during the acute inflammatory response are well established, their role in the resolution of inflammation is more controversial.
Prostaglandin synthesis
Prostaglandins are found in most tissues and organs. Prostaglandins are produced by almost all nucleated cells. Prostaglandins are autocrine and paracrine lipid mediators that act upon platelets, endothelium, uterine and mast cells. They are synthesized in the cell from the essential fatty acids. Arachidonic acid (AA) transformation is regulated by two cyclooxygenase (COX) isoenzymes, COX-1 and COX-2. Due to subsequent cyclooxygenation and peroxygenation, unstable prostaglandins PGG2 and PGH2 are formed. Next, these prostaglandins undergo further transformation in the presence of specific prostanoid synthases. The final transformation substrates are the prostaglandins PGE2, PGF2 and PGD2; prostacyclin (PGI2); and thromboxane A2 (TXA2) 3. The prostanoid synthesis profile is dependent on differences in enzymatic expression in cells under inflammation. Macrophages, for instance, release PGE2 and TXA2, whereas mast cells release prostaglandin PGD2 4.
Prostaglandins, prostacyclin (PGI2) and thromboxane A2 (TXA2), collectively termed prostanoids, are formed when arachidonic acid (AA), a 20-carbon unsaturated fatty acid, is released from the plasma membrane by phospholipases (PLAs) and metabolized by the sequential actions of prostaglandin G/H synthase also known as cyclooxygenase (COX), and respective synthases. Arachidonic acid metabolites are released from cells under the influence of many factors that act though specific membrane receptors. Growth factors (e.g, FGF, IFG and PDGF), hormones (vasopressin) and neurotransmitters (norepinephrine and acetylcholine) are among those factors 5.
Prostaglandin production (Figure 1) depends on the activity of COXs (cyclooxygenases), bifunctional enzymes that contain both cyclooxygenase and peroxidase activity and which exist as distinct isoforms referred to as COX-1 (cyclooxygenase-1) and COX-2 (cyclooxygenase-2) 6.
- Cyclooxygenase-1 (COX-1), expressed constitutively in most cells, is the dominant source of prostanoids that subserve housekeeping functions, such as gastric epithelial cytoprotection and homeostasis 7.
- Cyclooxygenase-2 (COX-2), induced by inflammatory stimuli, hormones and growth factors, is the more important source of prostanoid formation in inflammation and in proliferative diseases, such as cancer 7.
However, both enzymes COX-1 (cyclooxygenase-1) and COX-2 (cyclooxygenase-2) contribute to the generation of autoregulatory and homeostatic prostanoids, and both can contribute to prostanoid release during inflammation.
PGH2 (prostaglandin H2) is produced by both COX (cyclooxygenase) isoforms and it is the common substrate for a series of specific isomerase and synthase enzymes that produce PGE2, PGI2, PGD2, PGF2α and thromboxane A2 (TXA2). COX-1 (cyclooxygenase-1) couples preferentially, but not exclusively, with thromboxane synthase (TXS), prostaglandin F synthase, and the cytosolic (c) prostaglandin E synthase (PGES) isozymes 8. COX-2 (cyclooxygenase-2) prefers prostaglandin I synthase (PGIS) and the microsomal (m) PGES isozymes, both of which are often coinduced along with COX-2 by cytokines and tumor promoters 8.
The profile of prostanoid production is determined by the differential expression of these enzymes within cells present at sites of inflammation. For example, mast cells predominantly generate PGD2 (prostaglandin D2) while macrophages produce PGE2 (prostaglandin E2) and thromboxane A2 (TXA2) 9. In addition, alterations in the profile of prostanoid synthesis can occur upon cellular activation. While resting macrophages produce TXA2 in excess of PGE2 (prostaglandin E2), this ratio changes to favor PGE2 (prostaglandin E2) production after bacterial lipopolysaccharide (LPS) activation 9.
Figure 1. Prostaglandin synthesis
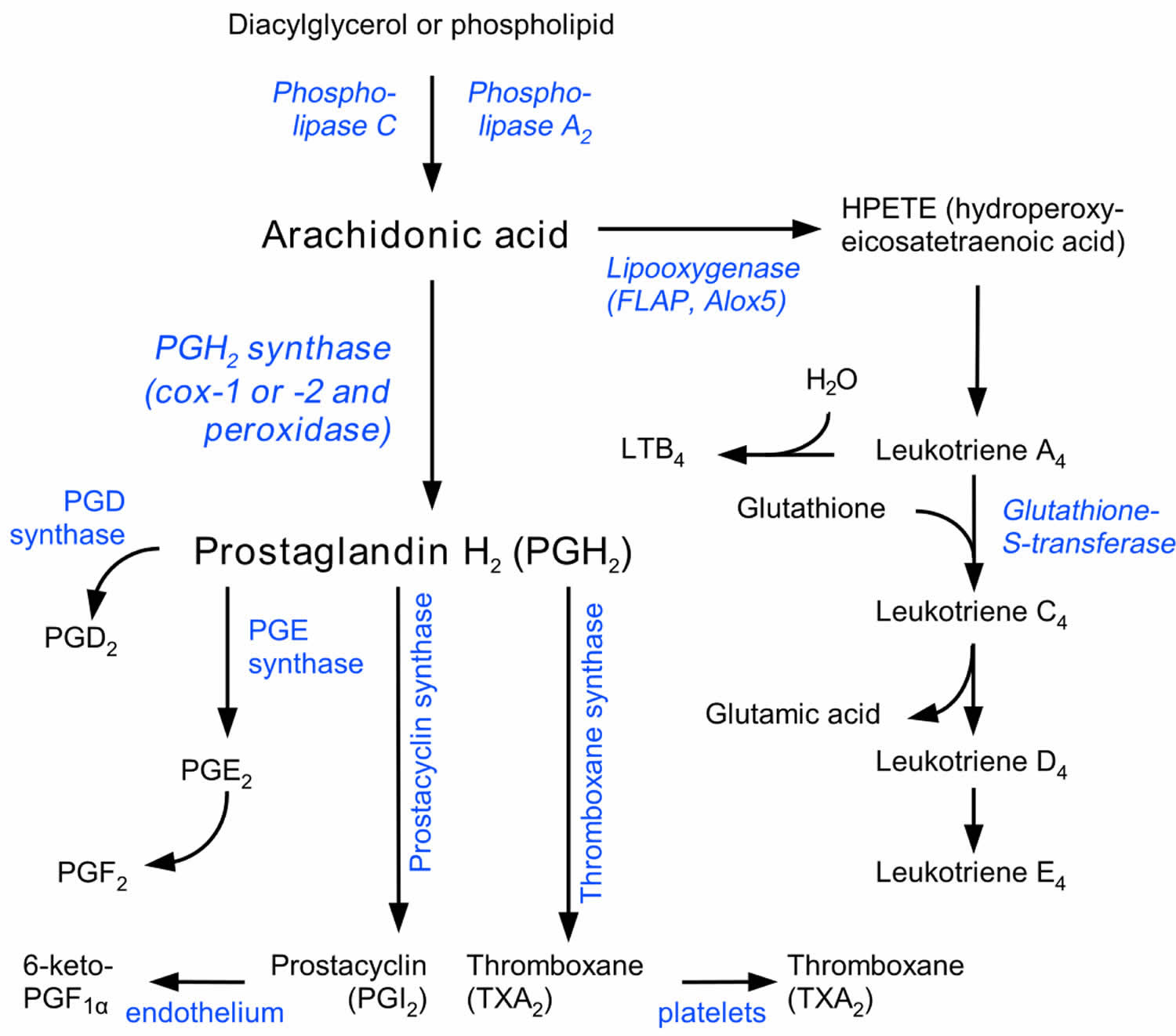
Footnotes: Prostaglandin synthesis pathway. The COX enzymes COX1 and COX2, which reduce arachidonic acid to prostaglandin H2 (PGH2), are the main targets of COX2-specific inhibitors and NSAIDs. PGH2 becomes a substrate for thromboxane, prostacyclin and prostaglandin synthases, which are converted into thromboxane A2 (TXA2), PGI2, PGE2 and prostaglandin D2 (PGD2). Arachidonic lipoxygenesases and hydroperoxides (HPETEs) convert arachidonic acid into leukotrienes and other inflammation-mediating eicosanoids.
What does prostaglandin do
The structural differences between prostaglandins account for their different biological activities. A given prostaglandin may have different and even opposite effects in different tissues in some cases. The ability of the same prostaglandin to stimulate a reaction in one tissue and inhibit the same reaction in another tissue is determined by the type of receptor to which the prostaglandin binds. They act as autocrine or paracrine factors with their target cells present in the immediate vicinity of the site of their secretion. Prostaglandins differ from endocrine hormones in that they are not produced at a specific site but in many places throughout the human body.
Prostaglandins (PG) are powerful locally acting vasodilators and inhibit the aggregation of blood platelets. Through their role in vasodilation, prostaglandins are also involved in inflammation. They are synthesized in the walls of blood vessels and serve the physiological function of preventing needless clot formation, as well as regulating the contraction of smooth muscle tissue 11. Conversely, thromboxanes (TXAs) (produced by platelet cells) are vasoconstrictors and facilitate platelet aggregation. Their name comes from their role in clot formation (thrombosis).
Specific prostaglandins are named with a letter (which indicates the type of ring structure) followed by a number (which indicates the number of double bonds in the hydrocarbon structure). For example, prostaglandin E1 is abbreviated PGE1 or PGE1, and prostaglandin I2 is abbreviated PGI2 or PGI2.
There are currently ten known prostaglandin receptors on various cell types. Prostaglandins ligate a sub-family of cell surface seven-transmembrane receptors, G-protein-coupled receptors. These receptors are termed DP1-2, EP1-4, FP, IP1-2, and TP, corresponding to the receptor that ligates the corresponding prostaglandin (e.g., DP1-2 receptors bind to PGD2).
The diversity of receptors means that prostaglandins act on an array of cells and have a wide variety of effects such as:
- cause constriction or dilation in vascular smooth muscle cells
- cause aggregation or disaggregation of platelets
- sensitize spinal neurons to pain
- induce labor
- decrease intraocular pressure
- regulate inflammation
- regulate calcium movement
- regulate hormones
- control cell growth
- acts on thermoregulatory center of hypothalamus to produce fever
- acts on mesangial cells (specialised smooth muscle cells) in the glomerulus of the kidney to increase glomerular filtration rate
- acts on parietal cells in the stomach wall to inhibit acid secretion
- increase mucus production and bicarbonate secretion
- brain masculinization (in rats) 12
- increases mating behaviors in goldfish 13
- Prostaglandins are released during menstruation, due to the destruction of the endometrial cells, and the resultant release of their contents 14. Release of prostaglandins and other inflammatory mediators in the uterus cause the uterus to contract. These substances are thought to be a major factor in primary dysmenorrhea 15.
- Cyclooxygenases blocking by lornoxicam in acute stage of inflammation reduced the frequency of membrane formation by 43% in the dispase model of proliferative vitreoretinopathy and by 31% in the concanavalin one. Lornoxicam not only normalized the expression of cyclooxygenases in both models of proliferative vitreoretinopathy, but also neutralized the changes of the retina and the choroid thickness caused by the injection of pro-inflammatory agents. These facts underline the importance of prostaglandins in the development of proliferative vitreoretinopathy 16.
Prostaglandins are potent but have a short half-life before being inactivated and excreted. Therefore, they send only paracrine (locally active) or autocrine (acting on the same cell from which it is synthesized) signals.
The following is a comparison of different types of prostaglandin, including prostaglandin I2 (prostacyclin; PGI2), prostacyclin D2 (PGD2), prostaglandin E2 (PGE2), and prostaglandin F2α (PGF2α).
Table 1. Prostaglandins functions
| Type | Receptor | Receptor type | Function |
|---|---|---|---|
| PGI2 | IP | Gs |
|
| PGD2 | PTGDR (DP1) and CRTH2 (DP2) | GPCR |
|
| PGE2 | EP1 | Gq |
|
| EP2 | Gs |
| |
| EP3 | Gi |
| |
| Unspecified |
| ||
| PGF2α | FP | Gq |
|
Prostaglandin inhibitors
Nonsteroidal anti-inflammatory drugs (NSAIDs) are frequently used for acute and chronic pain control and treatment of inflammation, osteoarthritis and rheumatoid arthritis. Corticosteroids inhibit phospholipase A2 production by boosting production of lipocortin, an inhibitor protein. The two cyclooxygenase isoforms, COX-1 (cyclooxygenase-1) and COX-2 (cyclooxygenase-2), are targets of nonsteroidal anti-inflammatory drugs (NSAIDs). These drugs are competitive active site inhibitors of both COXs. Although both COXs exist as homodimers, only one partner is used at a time for substrate binding 17. COX-1/COX-2 heterodimers may also exist, but their role in biology remains to be established 18. Nonsteroidal anti-inflammatory drugs (NSAIDs) bind to and inactivate the COX site at only one of the monomers of the COX dimer and this is sufficient to shut down prostanoid formation 17. Relatively new drugs, known as COX-2 selective inhibitors or coxibs, are used as specific inhibitors of the COX-2 isoform of cyclooxygenase. The development of these drugs allowed the circumvention of the negative gastrointestinal side effects of NSAIDs while still effectively reducing inflammation.
Both NSAIDs and coxibs have been shown to increase the risk of myocardial infarction when taken on a chronic basis for at least 18 months. One emerging hypothesis that may explain these cardiovascular effects is that coxibs create an imbalance in circulating TxA2 (thromboxane A2) and PGI2 (prostacyclin) levels. An increase in the ratio of TxA2/PGI2 could lead to increased platelet aggregation and dysregulation of platelet homeostasis 19.
The clinical efficacy of structurally distinct NSAIDs, all of which share this capacity for prostanoid inhibition, points to the importance of these mediators in the promotion of pain, fever and inflammation 20. The dramatic increase of COX-2 expression upon provocation of inflammatory cells, its expression in inflamed tissues and the assumption that inhibition of COX-1 derived prostanoids in platelets and gastric epithelium explains NSAID evoked gastrointestinal adverse effects and provided a rationale for development of NSAIDs designed to be selective for inhibition of COX-2 for treating arthritis and other chronic inflammatory diseases 21.
Although COX-2 appears to be the dominant source of prostaglandin formation in inflammation, there is some suggestion that both isoforms of the human enzyme may contribute to the acute inflammatory response. COX-1 is constitutively expressed in resident inflammatory cells and there is evidence for induction of COX-1 during lipopolysaccharide (LPS)-mediated inflammatory response and cellular differentiation 22. Both COX isoforms are co-expressed in circulating inflammatory cells ex vivo and in both inflamed rheumatoid arthritis (RA) synovium and in atherosclerotic plaques obtained from patients 23. Human data are compatible with COX-1-derived products driving the initial phase of an acute inflammation, with COX-2 upregulation occurring within several hours 8. However, controlled clinical trials to test the comparative efficacy of NSAIDs that inhibited both COXs versus COX-2 alone were never performed at scale. Such trials were designed to seek divergence in the incidence of gastrointestinal adverse effects rather than to assess comparative clinical efficacy.
Studies in both COX-1- and COX-2- knockout mice reveal impaired inflammatory responses, although the impacts of gene deletion diverge in time course and intensity. Mice deficient in COX-1, but not COX-2, exhibit a reduction in arachidonic acid (AA)-induced ear edema although arachidonic acid (AA) induces an equivalent inflammatory response in wild-type and COX-2-deficient mice 24. By contrast, the level of edema induced by the tumor promoter tetradecanoyl phorbol acetate was not significantly different between wild-type, COX-1-deficient and COX-2-deficient mice 25. The ear inflammation studies indicate that COX-1, as well as COX-2, may contribute to inflammatory responses and the isoform responsible for the inflammation may depend on the type of inflammatory stimulus and/or the relative levels of each isoform in the target tissue.
Similarly, arthritis models exhibit a significant reliance on either the COX-1 or COX-2 isoforms for the development of clinical synovitis. This varies, depending on the experimental model used. Thus, in a autoantibody-driven K/BxN serum-transfer model of arthritis, COX-1 derived prostaglandins, in particular PGI2, make a striking contribution to the initiation and perpetuation of arthritis 26. In a collagen- induced arthritis model, COX-2- deletion suppresses considerably synovial inflammation and joint destruction, whereas arthritis in COX- 1–deficient mice is indistinguishable from controls 27.
COX-2 deletion also suppresses acute inflammation in the air pouch model. Here, the COX-2 inhibitor, NS-398, administered six hours after carageenan treatment, reduced PG production in wild-type mice to levels comparable to those seen in COX-2-knockout mice, and was also effective during the early stages of inflammation 28. When compared to wild-type mice, the deficiency of COX-2 reduces the level of PGE2 production by approximately 75%, while the deficiency of COX-1 reduces the PGE2 level by 25% during this early stage. By day 7 following carrageenan treatment, higher numbers of inflammatory cells were present in the pouch fluid of COX-2-knockout mice, and little resolution of inflammation was apparent compared to wild-type or COX-1-knockout mice 28. These findings indicate that both COX isoforms contribute to prostaglandin production during inflammation and also that COX-2-derived prostaglandins appear to be important in both the acute inflammatory process and in the resolution phase. A contribution of COX-2 to both phases of inflammation was also reported in other models. Gilroy et al. 29 reported that COX-2 expression and PGE2 levels increased transiently early in the course of carrageenan-induced pleurisy in rats. Later in the response, COX-2 was induced again to even greater levels and generated anti-inflammatory prostaglandins, such as PGD2 and 15-deoxy-Δ12–14-PGJ2 (15d-PGJ2) but only low levels of the proinflammatory PGE2. Further support for an anti-inflammatory role of COX-2 in this model was the finding that late administration of the COX-2–selective inhibitor, NS-398, exacerbates the inflammatory response. Furthermore, Wallace et al. 30 observed that in the paw carrageenan model, the resultant inflammation resolves within 7 days in wild-type mice but is unaltered over this period in COX-2–deficient mice. An anti-inflammatory role of COX-2 derived prostanoids has also been reported in models of inflammatory colitis and allergic airway disease 31. Thus, COX-2 appears to have a dual role in the inflammatory process, initially contributing to the onset of inflammation and later helping to resolve the process. While COX-2 does play a role in supporting resolution of this process in some models of inflammation, it is unclear which products of the enzyme might contribute in which settings to this effect. This is exemplified by the case of 15d-PGJ2. Long touted as an endogenous ligand to peroxisome proliferator-activated receptor gamma (PPARγ), it remains to be established that endogenous concentration sufficient to subserve this function are formed in any model of resolving inflammation. Erroneously elevated levels have been reported using a variety of immunoassays, but the concentrations of bound and free compound documented to be formed by quantitative mass spectrometry fall far short of the EC50 for PPARγ activation 32.
In the case of atherosclerosis, deletion or inhibition of COX-2 has been shown variously to retard, accelerate or leave unaltered atherogenesis in mouse models 33. This may reflect an impact postnatally of disruption of the many roles of the enzyme in development in COX-2 knockouts; differences in timing of interventions with COX-2 inhibitors and a failure in most cases to define biochemically the selectivity for inhibition of COX-2 of the drug regimen deployed. In contrast, COX-1 deletion markedly attenuated lesion development in the Apolipoprotein E (ApoE) knockout mouse, as does inhibition of COX-1 and COX-2 together in low-density lipoprotein receptor-knockout model 34. Thus, products of COX-1 – like TxA2 – promote atherogenesis while there is more ambiguity around the role of COX-2. Nevertheless, deletion of the PGI receptor, the receptor for the major COX-2 product, PGI2, fosters the initiation and early development of atheroscelrosis in hyperlipidemic mice 35.
What is prostaglandin used for?
Synthetic prostaglandins are used:
- To induce childbirth (parturition) or abortion (PGE2 or PGF2, with or without mifepristone, a progesterone antagonist)
- Prostaglandins placed in your vagina next to your cervix will often ripen, or soften, the cervix, and contractions may even begin. Your baby’s heart rate will be monitored for a few hours. If labor does not begin, you may be allowed to leave the hospital and walk around.
- To prevent closure of patent ductus arteriosus in newborns with particular cyanotic heart defects (PGE1)
- To prevent and treat peptic ulcers (PGE)
- As a vasodilator in severe Raynaud’s phenomenon or ischemia of a limb
- In pulmonary hypertension
- In treatment of glaucoma (as in bimatoprost ophthalmic solution, a synthetic prostamide analog with ocular hypotensive activity) (PGF2α)
- To treat erectile dysfunction or in penile rehabilitation following surgery (PGE1 as alprostadil).
- To treat egg binding in small birds
- Von Euler US (1935). “Über die spezifische blutdrucksenkende Substanz des menschlichen Prostata- und Samenblasensekrets”. Wien Klin Wochenschr. 14 (33): 1182–3. doi:10.1007/BF01778029.
- Nicolaou, K. C.; E. J. Sorensen (1996). Classics in Total Synthesis. Weinheim, Germany: VCH. p. 65. ISBN 3-527-29284-5.
- Blackwell KA, Raisz LG, Pilbeam CC. Prostaglandins in bone: bad cop, good cop? Trends Endocrinol Metab. 2010;21(5):294–301
- Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):986–1000.
- Lisowska B, Kosson D, Domaracka K. Lights and shadows of NSAIDs in bone healing: the role of prostaglandins in bone metabolism. Drug Design, Development and Therapy. 2018;12:1753-1758. doi:10.2147/DDDT.S164562. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6014392/
- Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: Structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–182.
- Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte LB, Lipsky PE. Cyclooxygenase in biology and disease. FASEB J. 1998;12:1063–1073
- Smyth EM, Grosser T, Wang M, Yu Y, FitzGerald GA. Prostanoids in health and disease. J Lipid Res. 2009;50:S423–428.
- Tilley SL, Coffman TM, Koller BH. Mixed messages: modulation of inflammation and immune responses by prostaglandins and thromboxanes. J Clin Invest. 2001;108:15–23
- Ricciotti E, FitzGerald GA. Prostaglandins and Inflammation. Arteriosclerosis, thrombosis, and vascular biology. 2011;31(5):986-1000. doi:10.1161/ATVBAHA.110.207449. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3081099/
- Nelson, Randy F. (2005). An introduction to behavioral endocrinology (3rd ed.). Sunderland, Mass: Sinauer Associates. p. 100. ISBN 0-87893-617-3
- Lenz KM, Nugent BM, Haliyur R, McCarthy MM. Microglia are essential to masculinization of brain and behavior. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33(7):2761-2772. doi:10.1523/JNEUROSCI.1268-12.2013. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3727162/
- Hormonal and pheromonal control of spawning in goldfish. https://www.researchgate.net/publication/226044617_Hormonal_and_pheromonal_control_of_spawning_in_goldfish
- Lethaby A, Duckitt K, Farquhar C. Non‐steroidal anti‐inflammatory drugs for heavy menstrual bleeding. Cochrane Database of Systematic Reviews 2013, Issue 1. Art. No.: CD000400. DOI: 10.1002/14651858.CD000400.pub3
- Wright, Jason and Solange Wyatt. The Washington Manual Obstetrics and Gynecology Survival Guide. Lippincott Williams & Wilkins, 2003.
- Nonsteroid anti-inflammatory therapy suppresses the development of proliferative vitreoretinopathy more effectively than a steroid one. Tikhonovich, M.V., Erdiakov, A.K. & Gavrilova, S.A. Int Ophthalmol (2018) 38: 1365. https://doi.org/10.1007/s10792-017-0594-3
- Yuan C, Sidhu RS, Kuklev DV, Kado Y, Wada M, Song I, Smith WL. Cyclooxygenase Allosterism, Fatty Acid-mediated Cross-talk between Monomers of Cyclooxygenase Homodimers. J Biol Chem. 2009;284:10046–10055
- Yu Y, Fan J, Chen XS, Wang D, Klein-Szanto AJ, Campbell RL, FitzGerald GA, Funk CD. Genetic model of selective COX2 inhibition reveals novel heterodimer signaling. Nat Med. 2006;12:699–704.
- Role of prostacyclin in the cardiovascular response to thromboxane A2. Science. 2002 Apr 19;296(5567):539-41. https://www.ncbi.nlm.nih.gov/pubmed/11964481
- Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol. 1971;231:232–235
- Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol. 1971;231:232–235.
- McAdam BF, Mardini IA, Habib A, Burke A, Lawson JA, Kapoor S, FitzGerald GA. Effect of regulated expression of human cyclooxygenase isoforms on eicosanoid and isoeicosanoid production in inflammation. J Clin Invest. 2000;105:1473–1482.
- Schonbeck U, Sukhova GK, Graber P, Coulter S, Libby P. Augmented expression of cyclooxygenase-2 in human atherosclerotic lesions. Am J Pathol. 1999;155:1281–1291
- Langenbach R, Morham SG, Tiano HF, Loftin CD, Ghanayem BI, Chulada PC, Mahler JF, Lee CA, Goulding EH, Kluckman KD, Kim HS, Smithies O. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell. 1995;83:483–492
- Dinchuk JE, Car BD, Focht RJ, Johnston JJ, Jaffee BD, Covington MB, Contel NR, Eng VM, Collins RJ, Czerniak PM, Gorry SA, Trzaskos JM. Renal abnormalities and an altered inflammatory response in mice lacking cyclooxygenase II. Nature. 1995;378:406–409
- Chen M, Boilard E, Nigrovic PA, Clark P, Xu D, Fitzgerald GA, Audoly LP, Lee DM. Predominance of cyclooxygenase 1 over cyclooxygenase 2 in the generation of proinflammatory prostaglandins in autoantibody-driven K/BxN serum-transfer arthritis. Arthritis Rheum. 2008;58:1354–1365.
- Ochi T, Ohkubo Y, Mutoh S. Role of cyclooxygenase-2, but not cyclooxygenase-1, on type II collagen-induced arthritis in DBA/1J mice. Biochem Pharmacol. 2003;66:1055–1060.
- Langenbach R, Loftin C, Lee C, Tiano H. Cyclooxygenase knockout mice-Models for elucidating isoform-specific functions. Biochem Pharmacol. 1999;58:1237–1246
- Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA. Inducible cyclooxygenase may have anti-inflammatory properties. Nat Med. 1999:698–701
- Wallace JL, Mcknight W, Reuter BK, Vergnolle N. NSAID-induced gastric damage in rats: requirement for inhibition of both cyclooxygenase 1 and 2. Gastroenterology. 2000;119:706–714.
- Morteau O, Morham SG, Sellon R, Dieleman LA, Langenbach R, Smithies O, Sartor RB. Impaired mucosal defense to acute colonic injury in mice lacking cyclooxygenase-1 or cyclooxygenase-2. J Clin Invest. 2000;105:469–478.
- Bell-Parikh LC, Ide T, Lawson JA, McNamara P, Reilly M, FitzGerald GA. Biosynthesis of 15-deoxy-delta12,14-PGJ2 and the ligation of PPAR gamma. J Clin Invest. 2003;112:945–955.
- Tilley SL, Coffman TM, Koller BH. Mixed messages: modulation of inflammation and immune responses by prostaglandins and thromboxanes. J Clin Invest. 2001;108:15–23.
- McClelland S, Gawaz M, Kennerknecht E, Konrad CS, Sauer S, Schuerzinger K, Massberg S, Fitzgerald DJ, Belton O. Contribution of cyclooxygenase-1 to thromboxane formation, platelet-vessel wall interactions and atherosclerosis in the ApoE null mouse. Atherosclerosis. 2009;202:84–91.
- Egan KM, Lawson JA, Fries S, Koller B, Rader DJ, Smyth EM, et al. COX-2-derived prostacyclin confers atheroprotection on female mice. Science. 2004;306:1954–1957





{kind=link}